
Transforming Growth Factor-β and Oxidative Stress in Cancer: A Crosstalk in Driving Tumor Transformation



{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Transforming Growth Factor β (TGF-β) in Cancer Metabolism
3. Oxidative Stress in Cancer Metabolism
3.1. Redox Homeostasis in Cancer Metabolism
NOX in Cancer Metabolism
3.2. Antioxidant Systems in Cancer Metabolism
3.2.1. Superoxide Dismutase (SOD) in Cancer Metabolism
3.2.2. Catalase (CAT) in Cancer Metabolism
3.2.3. Reduced Glutathione (GSH) in Cancer Metabolism
3.3. ROS as Secondary Messengers in Cancer Metabolism
3.3.1. ROS and NF-kB
3.3.2. ROS and HIF-1α
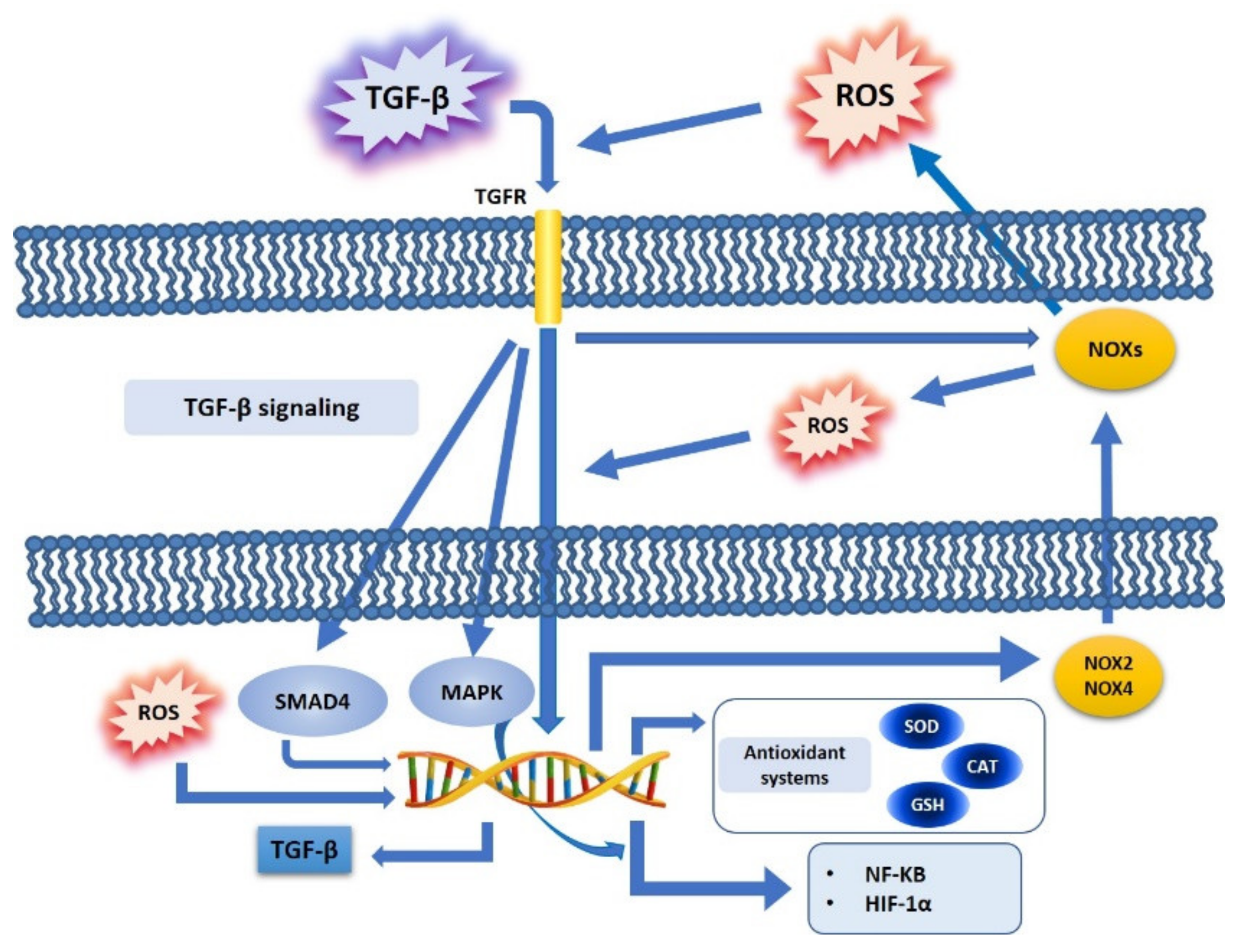
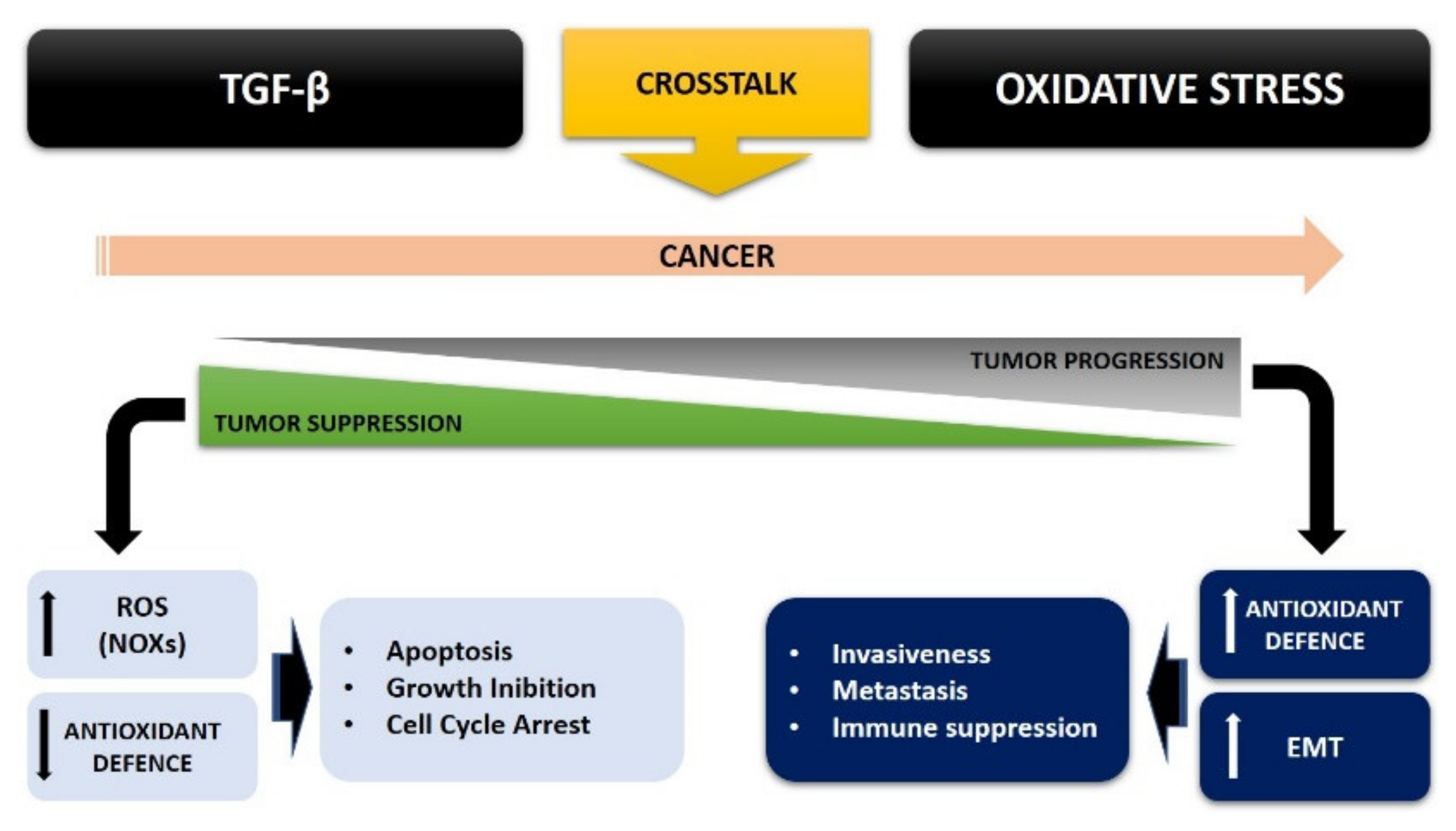
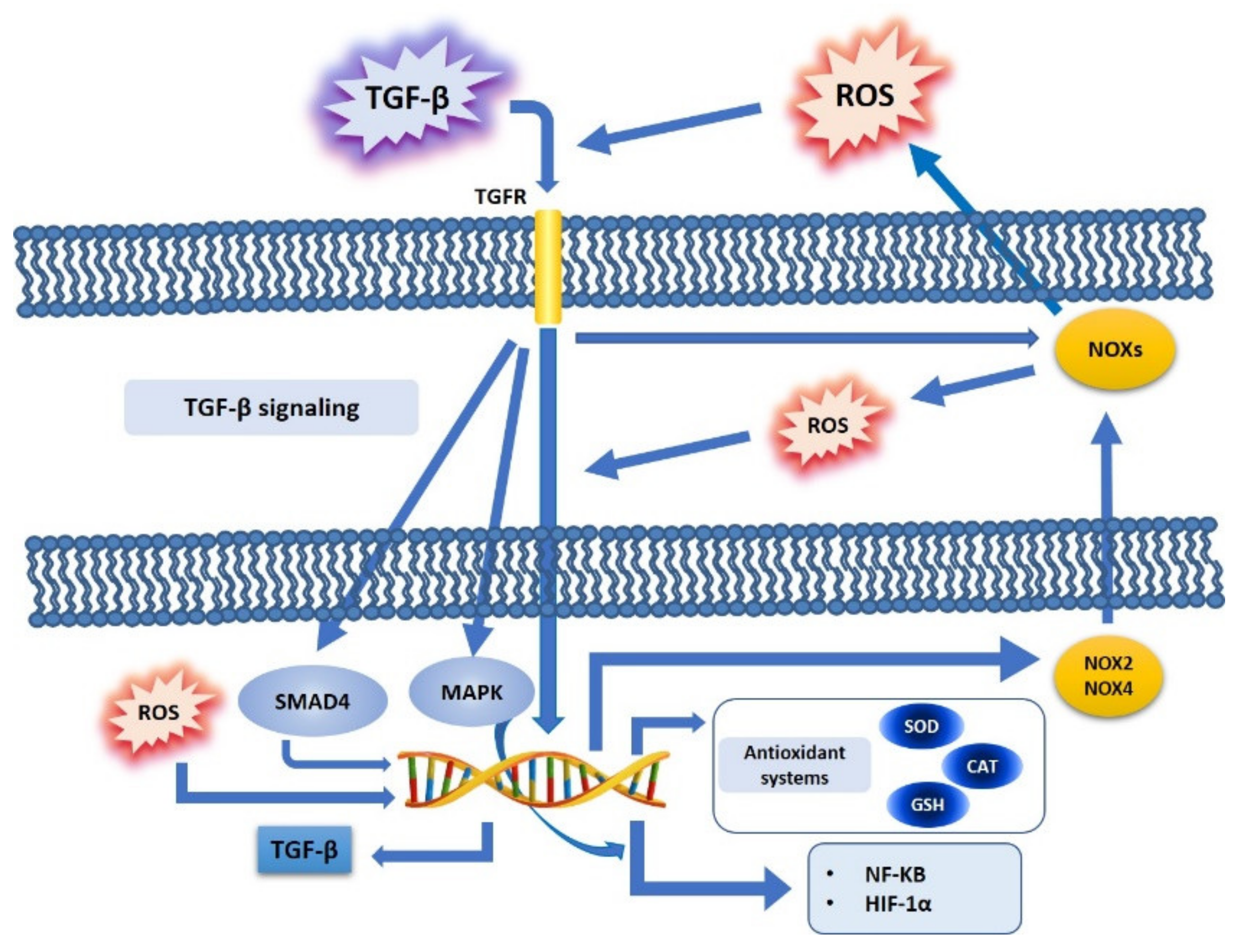
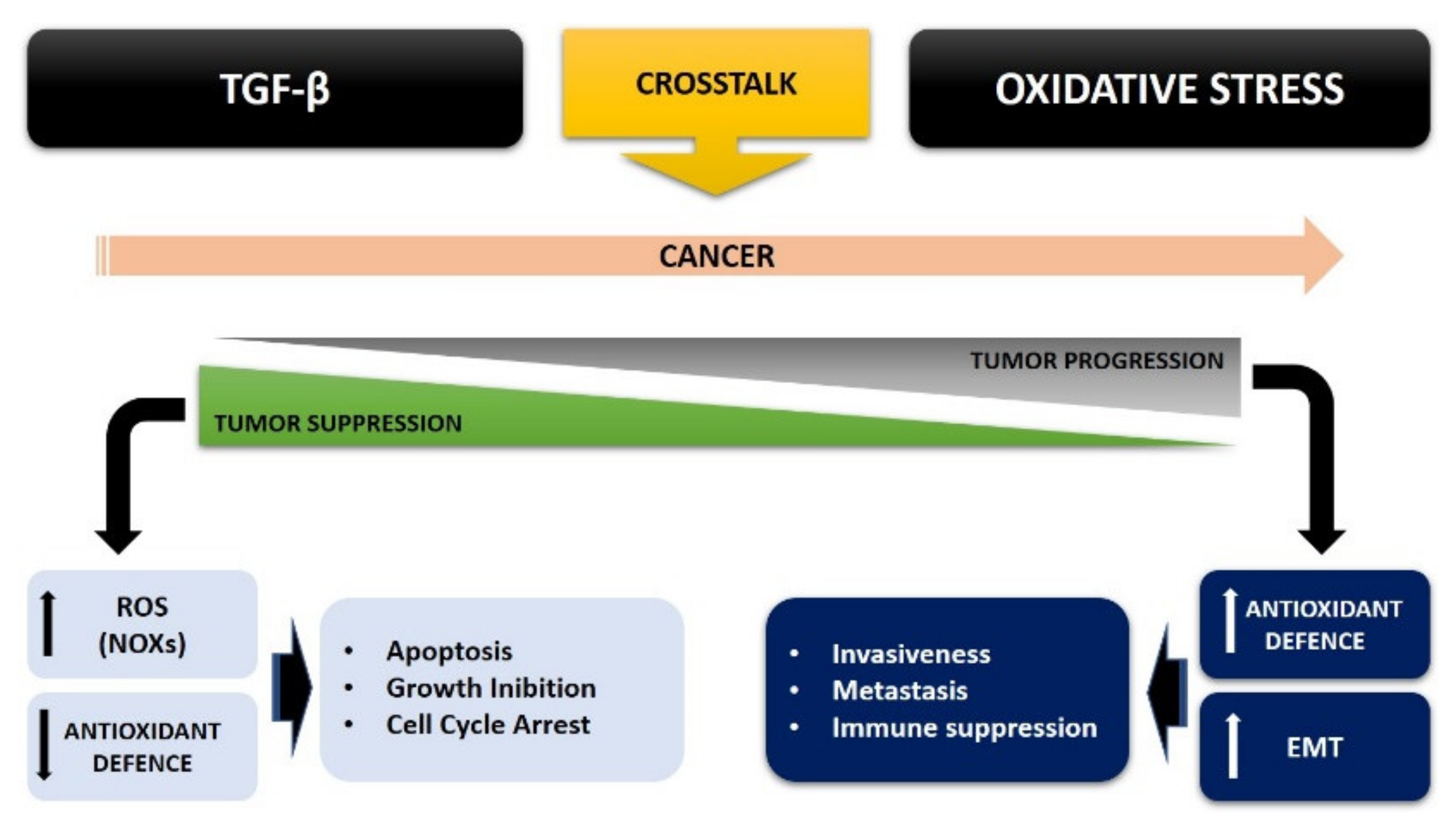
4. TGF-β and Oxidative Stress Crosstalk in Cancer Cell Metabolism
4.1. TGF-β, ROS, and NOX Family
4.2. TGF-β, ROS, and Antioxidant Systems
4.3. TGF-β, ROS, and Redox-Sensitive Factors
4.4. TGF-β, ROS, and the Epithelial Mesenchymal Transition (EMT)
4.5. TGF-β and ROS Crosstalk in Cancer: A Possible Therapeutic Approach
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Kang, Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor β in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ren, J.; Ten Dijke, P. Targeting TGFβ signal transduction for cancer therapy. Signal. Transduct. Target. 2021, 6, 8. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β tumor suppression through a lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming growth factor-β signaling pathway in colorectal cancer and its tumor microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, M.L.; Nair, R.; Sebastian, P.; Karunagaran, D. Changes in expression, and/or mutations in TGF-beta receptors (GF-beta RI and TGF-beta RII) and Smad 4 in human ovarian tumors. J. Cancer Res. Clin. Oncol. 2010, 136, 351–361. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Z.; Tan, J.; Dong, H.; Zhang, X. Multispectral imaging reveals hyperactive TGF-beta signaling in colorectal cancer. Cancer Biol. Ther. 2018, 19, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-β suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120. [Google Scholar] [CrossRef]
- Brooks, S.A.; Lomax-Browne, H.J.; Carter, T.M.; Kinch, C.E.; Hall, D.M.S. Molecular interactions in cancer cell metastasis. Acta Histochem. 2010, 112, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.; Schumacker, P. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The two faces of reactive oxygen species in cancer. Ann. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Athreya, K.; Xavier, M.F. Antioxidants in the Treatment of Cancer. Nutr. Cancer 2017, 69, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- Waghela, B.N.; Vaidya, F.U.; Agrawal, Y.; Santra, M.K.; Mishra, V.; Pathak, C. Molecular insights of NADPH oxidases and its pathological consequences. Cell Biochem. Funct. 2021, 39, 218–234. [Google Scholar] [CrossRef]
- Helfinger, V.; Freiherr von Gall, F.; Henke, N.; Kunze, M.M.; Schmid, T.; Rezende, F.; Heidler, J.; Wittig, I.; Radeke, H.H.; Marschall, V.; et al. Genetic deletion of Nox4 enhances cancerogen-induced formation of solid tumors. Proc. Natl. Acad. Sci. USA 2021, 118, e2020152118. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; López-Luque, J.; Fernando, J.; Sánchez, A.; Fernández, M.; Navarro, E.; Fabregat, I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef]
- Cecerska-Heryć, E.; Surowska, O.; Heryć, R.; Serwin, N.; Napiontek-Balińska, S.; Dołęgowska, B. Are antioxidant enzymes essential markers in the diagnosis and monitoring of cancer patients—A review. Clin. Biochem. 2021, 93, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.-T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular superoxide dismutase and its role in cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef]
- Islam, M.O.; Bacchetti, T.; Ferretti, G. Alterations of Antioxidant Enzymes and Biomarkers Nitro-oxidative Stress in Tissues of Bladder Cancer. Oxid. Med. Cell Longev. 2019, 2019, 2730896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.H.; Park, J.-H.; Chang, S.-G. Expression of antioxidant enzymes (catalase, superoxide dismutase, and glutathione peroxidase) in human bladder cancer. Korean J. Urol. 2007, 48, 921–926. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancer cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NF-kappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decades long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-𝛽-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, F.; Kaneko, E.; Sugimoto, T.; Ishijima, T.; Wakamatsu, M.; Yuasa, A.; Sampei, R.; Mori, K.; Nose, K.; Shibanuma, M. A mitochondrial thioredoxin-sensitive mechanism regulates TGF-𝛽-mediated gene expression associated with epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2014, 443, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGF𝛽 activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Tochhawng, L.; Deng, S.; Pervaiz, S.; Yap, C.T. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013, 13, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Vijayachandra, K.; Higgins, W.; Lee, J.; Glick, A. Induction of p16ink4a and p19ARF by TGF𝛽1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol. Carcinog. 2009, 48, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zhaoa, H.; Weib, J.; Sun, J. Roles of TGF-β signaling pathway in tumor microenvirionment and cancer therapy. Int. Immunopharmacol. 2020, 89, 107101. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernández, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Tobar, N.; Villar, V.; Santibanez, J.F. ROS-NFκΒ mediates TGF-β1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol. Cell Biochem. 2010, 340, 195–202. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Rada, B.; Korzeniowska, A.; Leto, T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic. Biol. Med. 2012, 53, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraga, R.; Kato, M.; Miyagawa, S.; Kamata, T. Nox4-derived ROS signaling contributes to TGF-𝛽-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer. Res. 2013, 33, 4431–4438. [Google Scholar]
- Kim, Y.M.; Cho, M. Activation of NADPH oxidase subunit NCF4 induces ROS-mediated EMT signaling in HeLa cells. Cell Signal. 2014, 26, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.H.; Zhu, B.M.; Riedlinger, G.; Kang, K.; Hennighausen, L. The liver-specific tumor suppressor STAT5 controls expression of the reactive oxygen species-generating enzyme NOX4 and the proapoptotic proteins PUMA and BIM in mice. Hepatology 2012, 56, 2375–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.K.; Park, K.S. Upregulation of mitochondrial Nox4 mediates TGF-beta-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Renal. Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Bertran, E.; Caja, L.; Carmona-Cuenca, I.; Murillo, M.M.; Fabregat, I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-beta-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim. Biophys. Acta 2009, 1793, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, G. Central Signaling Elements of Intercellular Reactive Oxygen/Nitrogen Species-dependent Induction of Apoptosis in Malignant Cells. Anticancer Res. 2017, 37, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Arsalane, K.; Dubois, C.M.; Muanza, T.; Bégin, R.; Boudreau, F.; Asselin, C.; Cantin, A.M. Transforming growth factor-𝛽1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: Transcriptional effect on the GSH rate-limiting enzyme 𝛾-glutamylcysteine synthetase. Am. J. Respir. Cell Mol. Biol. 1997, 17, 599–607. [Google Scholar] [CrossRef]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.-H.; Huang, C.; Feng, Z.-Z.; Lv, X.-H.; Qiu, Z.-J. Hypoxia-induced snail expression through transcriptional regulation by HIF-1α in pancreatic cancer cells. Dig. Dis. Sci. 2013, 58, 3503–3515. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Derynck, R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxid. Med. Cell Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef]
- Gorowiec, M.R.; Borthwick, L.A.; Parker, S.M.; Kirby, J.A.; Saretzki, G.C.; Fisher, A.J. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Cannito, S.; Novo, E.; Compagnone, A.; Valfrè di Bonzo, L.; Busletta, C.; Zamara, E.; Paternostro, C.; Povero, D.; Bandino, A.; Bozzo, F.; et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 2008, 29, 2267–2278. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Han, C.; Huang, S.; Chen, S.; Luo, L.; Liu, Y. JNK1/β-catenin axis regulates H2O2-induced epithelial-to-mesenchymal transition in human lens epithelial cells. Biochem. Biophys. Res. Commun. 2019, 511, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Parri, M.; Chiarugi, P. EMT and oxidative stress: A bidirectional interplay affecting tumor malignancy. Antioxid. Redox. Signal. 2012, 16, 1248–1263. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Milisav, I. The Role of Antioxidants in Cancer, Friends or Foes? Curr. Pharm. Des. 2019, 24, 5234–5244. [Google Scholar] [CrossRef]
- Souto, E.B.; Severino, P.; Basso, R.; Santana, M.H. Encapsulation of antioxidants in gastrointestinal-resistant nanoparticulate carriers. Methods Mol. Biol. 2013, 1028, 37–46. [Google Scholar]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-[beta] signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Chin-Yap Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramundo, V.; Giribaldi, G.; Aldieri, E. Transforming Growth Factor-β and Oxidative Stress in Cancer: A Crosstalk in Driving Tumor Transformation. Cancers 2021, 13, 3093. https://doi.org/10.3390/cancers13123093
Ramundo V, Giribaldi G, Aldieri E. Transforming Growth Factor-β and Oxidative Stress in Cancer: A Crosstalk in Driving Tumor Transformation. Cancers. 2021; 13(12):3093. https://doi.org/10.3390/cancers13123093
Chicago/Turabian StyleRamundo, Valeria, Giuliana Giribaldi, and Elisabetta Aldieri. 2021. "Transforming Growth Factor-β and Oxidative Stress in Cancer: A Crosstalk in Driving Tumor Transformation" Cancers 13, no. 12: 3093. https://doi.org/10.3390/cancers13123093
APA StyleRamundo, V., Giribaldi, G., & Aldieri, E. (2021). Transforming Growth Factor-β and Oxidative Stress in Cancer: A Crosstalk in Driving Tumor Transformation. Cancers, 13(12), 3093. https://doi.org/10.3390/cancers13123093