Simple Summary
Chronic lymphocytic leukemia harbors a high degree of genetic variability and interpatient heterogeneity. Some of the genetic alterations have an impact on the disease’s prognosis and evolution, but few data exist about the response to new approved targeted therapies in patients carrying recurrent mutations other than TP53. In this review, we present the knowledge about the impact of these new genetic alterations in the treatment response together with the possibility to use new actionable targets.
Abstract
Chronic lymphocytic leukemia (CLL) is characterized by a high degree of genetic variability and interpatient heterogeneity. In the last decade, novel alterations have been described. Some of them impact on the prognosis and evolution of patients. The approval of BTK inhibitors, PI3K inhibitors and Bcl-2 inhibitors has drastically changed the treatment of patients with CLL. The effect of these new targeted therapies has been widely analyzed in TP53-mutated cases, but few data exist about the response of patients carrying other recurrent mutations. In this review, we describe the biological pathways recurrently altered in CLL that might have an impact on the response to these new therapies together with the possibility to use new actionable targets to optimize treatment responses.
1. Introduction
In the last decade, genomic and epigenomic studies have unravel novel alterations that play an important role in the prognosis and evolution of chronic lymphocytic leukemia (CLL) [1,2,3,4,5,6], revealing CLL’s genetic and interpatient heterogeneity. The behavior of the disease is influenced by microenvironmental signals that regulate the proliferation and survival of CLL cells [7]. Two major molecular CLL subgroups have been identified according to the mutational status of the immunoglobulin (Ig) heavy-chain variable region (IGHV) genes. Those harboring unmutated IGHV genes (U-CLL, ≥98% identity with the germline) originate from B cells that have not experienced the germinal center and those with mutated IGHV genes (M-CLL, <98% identity with the germline) originate from post-germinal center B cells [8,9,10]. Furthermore, approximately one third of CLL cases present virtually identical Ig rearrangements, known as stereotypes [11]. Some of these subsets have prognostic value [12] and recently a single point mutation in IGLV3-21 (R110-mutated IGLV3-21) has been associated with an aggressive biological subtype of CLL [13,14].
The mutational landscape of CLL is very heterogeneous, being NOTCH1, SF3B1, TP53 and ATM the genes that are mutated in more than 5% of cases [2,5]. Recurrent mutations can be grouped in 8 main cellular pathways: DNA damage response (ATM, TP53, POT1); Notch signaling (NOTCH1, FBXW7); RNA splicing and metabolism (SF3B1, U1, XPO1, DDX3X, RPS15); B-cell receptor (BCR) and Toll-like receptor (TLR) signaling (MYD88, PAX5, KLHL6, BCOR, TLR2, IKZF3); MAPK-ERK pathway (BRAF, KRAS, NRAS, EGR2); NF-κB signaling (BIRC3, NFKB2, NFKBIE, TRAF2, TRAF3); chromatin modification (CHD2, SETD2, KMT2D, ASXL1) and cell cycle (ATM, TP53, CCND2, CDKN1B, CDKN2A) [7]. Furthermore, the number of driver alterations affects the clinical behavior, being the worst prognosis in patients with higher number of driver aberrations [2,15]. The heterogeneity between patients may be influenced by: (a) the cell of origin: U-CLL present more driver mutations than M-CLL and some mutations appear mainly in one of the two major molecular subgroups (e.g., MYD88 and PAX5 in M-CLL and U1, NOTCH1, POT1 in U-CLL) [2,5]; (b) the age of CLL patients (e.g., young patients carry more frequently MYD88 mutations) [1,16], (c) the presence of subclonal mutations that may be acquired during disease evolution [5,15,17] and (d) the course of the disease: some mutations impact in the need of treatment (SF3B1, POT1, ATM) and others (TP53, BIRC3, MAP2K1, NOTCH1) are more frequent at relapse after chemoimmunotherapy [7,18,19]. At epigenetic level, few patients carry mutations in chromatin remodeler genes [2,5]. CLL cells are hypomethylated and most of the differences observed between U-CLL and M-CLL are related to different cell of origin. The most important changes are restricted to few transcription factor binding sites and enhancers controling important genes implicated in CLL pathogenesis such as BCR and NF-κB signaling [20].
The treatment of patients with CLL has evolved in the last years from conventional chemotherapeutic agents (e.g., fludarabine, fludarabine plus cyclophosphamide (FC), chlorambucil or bendamustine) combined with anti-CD20 antibodies (rituximab or obinutuzumab), to novel targeted agents. The targeted drugs currently approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) are the Bruton’s tyrosine kinase (BTK) inhibitors ibrutinib and acalabrutinib, the Bcl-2 inhibitor venetoclax [21] and the phosphatidylinositol 3-kinase (PI3K) inhibitors idelalisib and duvelisib [22]. These drugs have shown higher clinical responses compared to standard chemoimmunotherapy and all of them seem to act in a TP53 independent manner. Due to the prognostic and therapeutic impact of TP53 alterations (17p deletion and TP53 mutation) in CLL, the response of TP53-mutated cases to the new agents has been widely analyzed (summarized in Table 1) and recently reviewed [23]. On the contrary, few data exist about the effect of other recurrent mutations in the response to the new approved targeted therapies (Table 1). In this way, the effect of other mutated genes relevant for the DNA damage response pathway such as ATM, located at 11q region, a region frequently deleted in CLL cases, has not been widely explored, although it is accepted that 11q alterations are associated with unfavorable prognosis [24]. Another gene located at the minimal deleted 11q region is BIRC3. BIRC3 encodes c-IAP2, a member of the human Inhibitors of Apoptosis Proteins (IAPs) family [25] that acts as a negative regulator of non-canonical NF-κB signaling [26]. BIRC3 deletions coexist with ATM deletions, both have prognostic value in CLL [27,28,29], being patients with a biallelic alteration (deletion and mutation) those with a significantly shorter time to first treatment (TTFT) [30]. BIRC3 alterations are associated with fludarabine-chemoresistance and adverse prognosis [29], but few information exist about the response to new therapies (Table 1). Mutations in key genes of the alternative NF-kB pathway, such as BIRC3, confer resistance to BTK inhibitors in mantle cell lymphoma (MCL) [31,32], that can be overcome with the addition of NIK inhibitors [33]. Furthermore, IAP antagonists, such as SMAC mimetics, have showed activity in CLL cells but not specifically in cases with BIRC3 alterations [34].
In addition, many other mutations have shown a prognostic value, which impacts on the patient’s outcome, regardless of the treatment received. But only TP53 and NOTCH1 have a predictive impact, as it has been demonstrated in comparative trials that some treatments have different effect in the presence of these mutations [18,35,36] (Table 1). All this knowledge together with the characterization of the molecular effects of recurrent mutations, foster the development of direct target inhibitors leading to a precision medicine [19,37]. However, the design of direct and specific inhibitors is challenging. In particular, it is very difficult to directly target TP53 due to its structure and nuclear localization [38]. In this context, molecules able to restore the physiological function of TP53, or strategies to potentiate TP53 function by using compounds that bind to MDM2 in the p53-binding pocket have been developed [23]. In CLL, patients with subclonal TP53 mutations might benefit from treatment with these compounds [39]. In addition, other proteins are able to regulate TP53 expression, for example, the XPO1 inhibitor selinexor, which enhances p53 nuclear retention and induces the transcription of TP53 target genes in CLL cells [40]. Other strategies include the use of inhibitors of the checkpoint kinase 1 (CHK1), taking into account that TP53-mutated cells lack the G1/S checkpoints and are more vulnerable to this inhibition [41] or the use of compounds inducing reactive oxygen species (ROS) irrespective of p53 status [42]. In this review, we will focus on altered genes/pathways that play a role in the response of new approved therapies in CLL, such as the TLR, MAPK and Notch signaling pathways as well as RNA splicing that can be tackled with new agents to improve the efficacy of current therapy for CLL.

Table 1.
Effect of the main recurrent mutations on the response to different treatment strategies in CLL.
Table 1.
Effect of the main recurrent mutations on the response to different treatment strategies in CLL.
| Mutation | Drug/s | Trial | Response to the Treatment in the Mutated Group | Ref. | |
|---|---|---|---|---|---|
| TP53 | FCR vs. FC | CLL8 | Worse PFS and OS with both treatments | [43] | |
| Lenalidomide | Worse OR and PFS in TN and R/R | [44] | |||
| Ibrutinib | Shorter PFS, OS | [45,46] | |||
| Ibrutinib vs. ofatumumab | RESONATE Phase III | Shorter PFS with ibrutinib | [47] | ||
| Ofatumumab vs. OfIde | Similar PFS with OfIde | [48] | |||
| IdeR followed by idelalisib | Similar PFS | [49] | |||
| Duvelisib | DUO extension | Similar PFS | [50] | ||
| Venetoclax | Shorter duration of response | [51] | |||
| VenOb | Shorter PFS | [52] | |||
| (Bendamustine) + VenOb | CLL2-BAG | Lower MRD negativity rates | [53] | ||
| VenR vs. BR | MURANO Phase III | Higher MRD positivity rates at EOT | [54] | ||
| Chl vs. OfChl | COMPLEMENT 1 | Worse PFS and OS with both treatments | [24] | ||
| ObChl vs. IbrOb | iLLUMINATE | Similar PFS to overall population with IbrOb | [55] | ||
| ObChl vs. VenOb | CLL14 | Lower ORR with ObChl, independent prognostic factor for PFS with both treatments | [56] | ||
| ObChl vs. acalabrutinib vs. ObAca | ELEVATE-TN | PFS benefit with ObAca | [57] | ||
| Acalabrutinib | ACE-CL-001 | Similar OR rate regardless del(17p) status | [58] | ||
| Acalabrutinib vs. IdeR vs. BR | ASCEND | PFS benefit with acalabrutinib | [59] | ||
| MYD88 | Chl vs. OfChl | COMPLEMENT 1 | No effect in PFS | [24] | |
| MAPKs | Fludarabine vs. FC vs. Chl | CLL4 | Independent markers of poor OS | [60] | |
| PI3K inhibitors | Mutations enriched in non-responder subgroup | [61] | |||
| BRAF | VenR vs. BR | MURANO Phase III | Higher MRD positivity rates at EOCT and at EOT | [54] | |
| KRAS | Lenalidomide | Worse OR in TN and R/R | [44] | ||
| Chl vs. ChlR vs. ObChl | CLL11 | Non response to chemoimmunotherapy | [62] | ||
| NOTCH1 | FCR vs. FC | CLL8 | No benefit from the addition of rituximab to FC | [43] | |
| Chl vs. OfChl | COMPLEMENT 1 | Reduced ofatumumab efficacy | [24] | ||
| Ibrutinib vs. ofatumumab | RESONATE Phase III | Reduced ofatumumab efficacy | [63] | ||
| Ibrutinib | Shorter PFS and OS | [64] | |||
| Venetoclax | Shorter duration of response | [51] | |||
| VenR vs. BR | MURANO Phase III | Higher MRD positivity rates at EOT | [54] | ||
| ObChl vs. VenOb | CLL14 | Shorter PFS with ObChl, not statistically significant with VenOb | [56] | ||
| SF3B1 | FCR vs. FC | CLL8 | Worse PFS with both treatments | [43] | |
| Lenalidomide | Worse OS and PFS in R/R | [44] | |||
| Chl vs. OfChl | COMPLEMENT 1 | Worse PFS in both arms (also in low VAF patients) | [24] | ||
| Ibrutinib vs. ofatumumab | RESONATE Phase III | Trend to shorter PFS not statistically significant | [47] | ||
| Ibrutinib | Mutation enriched in postreatment samples | [65] | |||
| Ibrutinib vs. acalabrutinib | In patients with RT, SF3B1 mutations were more frequent than BTK mutations | [66] | |||
| Venetoclax | Shorter duration of response | [51] | |||
| ObChl vs. VenOb | CLL14 | Independent prognostic factor for PFS with ObChl | [56] | ||
| BIRC3 | Fludarabine vs. FC vs. Chl | CLL4 | Shorter PFS and OS | [60] | |
| FCR | Shorter PFS | [29] | |||
| Chl vs. OfChl | COMPLEMENT 1 | Similar PFS and OS | [24] | ||
| Ibrutinib vs. ofatumumab | RESONATE Phase III | Similar PFS | [63] | ||
| Ibrutinib | Mutation enriched in postreatment samples | [65] | |||
| VenR vs. BR | MURANO Phase III | Higher MRD positivity rates at EOCT, shorter PFS with BR | [54,67] | ||
| ObChl vs. VenOb | CLL14 | Independent prognostic factor for PFS with ObChl | [56] | ||
BR: bendamustine + rituximab, Chl: chlorambucil, ChlR: chlorambucil + rituximab, DOR: duration of response, EOCT: end of combination therapy, EOT: end of treatment, FC: fludarabine + cyclophosphamide, FCR: fludarabine + cyclophosphamide + rituximab, GClb: obinutuzumab + chlorambucil, IbrOb: ibrutinib + obinutuzumab, IdeR: idelalisib + rituximab, MRD: minimal residual disease, ObAca: obinutuzumab + acalabrutinib, ObChl: obinutuzumab + chlorambucil, OfChl: ofatumumab + chlorambucil, OfIde: ofatumumab + idelalisib, OS: overall survival, OR: overall response, PFS: progression free survival, R/R: relapsed/refractory patients, RT: Richter transformation, TN: treatment naïve, VAF: variant allele frequency, VenOb: venetoclax + obinutuzumab, VenR: venetoclax + rituximab.
2. TLR Signaling
Toll-like receptors (TLRs) are part of the innate immune system capable to identify pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [68]. PAMPs and DAMPs triggers TLRs transduction and initiate innate and adaptive immune responses to eliminate pathogens and repair the damaged tissues. TLR signaling is necessary together with BCR recognition and T-cell interaction for B-cell activation [69,70].
A total of ten TLRs are expressed in human immune and non-immune cells. They are located in the cell surface (e.g., TLR1, TLR2 and TLR4-6) or in endolysosomes (TLR3 and TLR7-9) [71]. When TLRs recognize some PAMPs or DAMPs, a cell signaling cascade is initiated throughout the recruitment of an adaptor protein. There are two types of adaptor proteins: myeloid differentiation factor 88 (MyD88) and TIR-domain containing adaptor molecule (TRIF). MyD88 forms the Myddosome together with interleukin 1 receptor associated kinase 4 (IRAK4) and kinase 1 and 2 (IRAK1/2) [72]. The Myddosome produces pro-inflammatory cytokines (e.g., IL-1, IL-6, IL-12 and TNF) and via TNF receptor-associated factor 6 (TRAF6), activates different molecular pathways.
The most relevant are: (1) the nuclear factor kappa- B cells (NF-κB) [73] and the Janus kinase/signal transducer and activator of transcription 3 (JAK-STAT3) pathways, involved in the activation, expansion and survival of cells and cytokine secretion; (2) the mitogen-activated protein kinase (MAPKs) pathway, which favors the expression of pro-inflammatory genes and (3) the interferon regulatory factor 5 (IRF5) pathway [74] that together with NF-κB pathway, promotes also the production of pro-inflammatory cytokines (Figure 1). Aside from MyD88-dependent signal transduction, TLR3 and TLR4 use the TRIF adaptor protein. TLR3 binds directly to TRIF, whereas TLR4 needs the adaptor TRAM (TRIF-related adaptor molecule). TLR3 and 4 bind to TRAF3 and TRAF6 leading to the activation of type I interferon genes and NF-κB pathway, respectively [75]. Abnormal TLR activation impairs the immune homeostasis and contributes to the onset of several inflammatory and autoimmune diseases and also some tumor malignancies [76,77]. In CLL, the BCR and TLRs from B cells recognize autoantigens and bacterial components [73]. This activation could explain, at least partially, why CLL patients have severe infections and autoimmune complications [78].
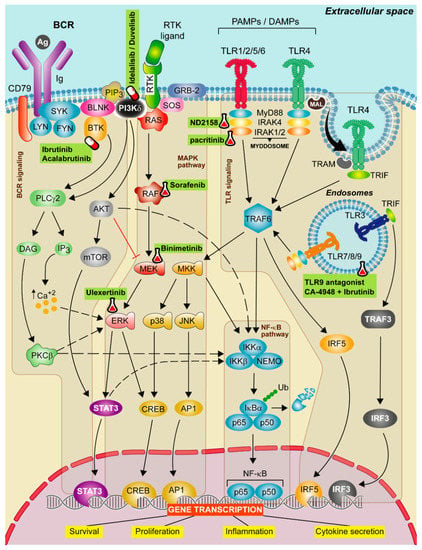
Figure 1.
Therapeutic strategies targeting TLR, BCR, and MAPK signaling in CLL. Schematic representation of the main activation events in the B-cell receptor (BCR), mitogen-activated protein kinase (MAPK) and Toll-like receptor (TLR) signaling pathways. BCR signaling activation iniciates when the antigen binds to the receptor leading to CD79 phosphorylation through LYN and SYK tyrosine kinases forming a signalosome that includes the BLNK, the BTK and the PI3Kδ. These proteins transduce signals to calcium signaling modulator PLCγ2. PLCγ2 degrades the PIP2 into IP3 and DAG, releasing calcium from endoplasmic reticulum (ER) and promoting the activation of the PKCβ. Then, PKCβ activates the NF-κB pathway and ERK signaling. The canonical MAPK pathway is activated after a ligand binds to the RTK. Then, RAS protein recruits adaptor proteins, such as GRB-2 and SOS and promotes the formation of RAF dimers. This induces the MEK-ERK cascade concluding with the translocation of STAT3 and CREB transcription factors to the nucleus. In contrast, the non-canonical MAPK pathway is activated by TRAF6, then MKK is phosphorylated and this leads to the activation of NF-κB pathway and to p38 and JNK phosphorylation that promote the translocation of CREB and AP1 transcriptions factors to the nucleus. All these events favor cell survival, cell proliferation and cytokine secretion signals. Finally, TLR signaling pathway is activated by TLRs located on either the cell membrane or endolysosomal membranes. These receptors are capable to detect PAMPs which initiate the downstream signaling, mediated by two main adaptor proteins: MyD88 and/or TIR domain containing adapter molecule (TRIF) dependent pathways. MyD88 is recruited to the TIR domain of TLRs inducing IRAK1/2 and IRAK4, jointly with MyD88, to form the Myddosome. Aside from MyD88-dependent signal transduction, TLR3 and TLR4 use the TRIF adaptor protein. TLR3 binds directly to TRIF whereas TLR4 needs TRAM. TLR3 and 4 bind to TRAF3 which induce the expression of IRF3, promoting the synthesis of type I interferons (IFNs). All these complexes activate TRAF6 leading to the activation of NF-κB, IRF5 and MAPK pathways promoting the gene transcription of cell survival, proliferation, inflammation and proinflammatory cytokine production. Targeted drugs are highlighted in green. Pill icon: approved drugs and Erlenmeyer icon: preclinical studies or clinical trials.
2.1. Recurrent Mutations and Alterations in TLR Pathway
Gain of function mutations in TLR pathway produce an increase on cell proliferation, cell survival and cytokine production in absence of cognate ligands, which results in a better supportive tumor microenvironment [79,80]. Plasma from CLL patients contains high levels of unmethylated DNA, which can trigger antilogous TLR9 activation, with the promotion of CLL cell activation and trafficking to lymphoid tissues [81].
In CLL, MYD88 is mutated in 2% to 5% of cases [1,16]. The most frequent mutation is L265P, the typical mutation described in other lymphoid malignancies [82], but about 15% of the mutated CLL cases harbor other MYD88 somatic mutations (V147L, S243N, and S219C) [16]. Additionally, sporadic mutations in IRAK1 and in TLRs (TLR2, TLR6) are found [16]. MYD88 mutations are enriched among M-CLL cases. Patients harboring MYD88 mutations are predominantly male and young, they do not present concomitant high risk mutations (e.g., TP53, NOTCH1, SF3B1, ATM or BIRC3) or adverse cytogenetics (del17p, del11q) [16]. CLL cases with MYD88 mutations are enriched in NF-κB and STAT3 gene expression signatures and have high basal cytokine secretion [83]. Furthermore, MYD88 mutations induce a de novo chromatin activation enriched in genes from the NF-κB pathway [20]. MYD88 mutations are predominantly clonal and the clinical impact of this mutation is controversial [16,80,84,85].
2.2. Targeting TLR Pathway
Two major strategies have been described for TLRs inhibition: blocking the binding site of TLR ligands to its receptor interfering in the intracellular signaling pathway, which can be achieved by small molecule inhibitors, monoclonal antibodies, oligonucleotides, lipid-A analogs, microRNAs, and nano-inhibitors (Figure 1) [86]; or by inhibiting IRAK proteins from the Myddosome with small molecules. There is an ongoing clinical trial targeting the TLR pathway, using IRAK4 inhibitor (CA-4948) alone or in combination with ibrutinib in relapsed/refractory (R/R) hematologic malignancies (NCT03328078) [87].
ND2158 is a small molecular IRAK4 inhibitor, which exhibited robust activity in diffuse large B-cell lymphoma activated B-cell subtype presenting MYD88 mutations [79]. In CLL cells, ND2158 decreases cell viability independently of MYD88 mutational status and inhibits tumor proliferation promoted by the TLR agonists, resulting in blockade of NF-κB and STAT3 signaling. Furthermore, ND2158 decreases the release of inflammatory factors from monocytes, reducing their tumor protective activity. The effect of the compound has been tested in the Eµ-TCL1 adoptive transfer mouse model that mimics human CLL disease and the tumor microenvironment [88,89]. In this context, ND2158 induces a slowed down leukemia progression, a decrease in monocytes number, cytokine secretion and cytotoxic T-cell (CD8+) activity and expansion. The decrease of CD8+ effector T cells is accompanied of a decreased expression of proliferation and activation markers (CD25, CD28 and CD137) and increased expression of exhaustion markers (PD-1, TIGIT and LAG3). This exhaustion phenotype may reduce their antitumor activity [59].
TLR9 antagonists have also been used to inhibit TLR signaling (Figure 1), showing a completely blockage when these inhibitors are used in combination with ibrutinib [57]. In this way, ibrutinib inhibits BCR signaling efficaciously but partially the TLR signaling [67] being TLR9 signaling activation a common escape mechanism after treatment of CLL cells with ibrutinib and venetoclax [68].
In vitro studies have confirmed that the combination of ibrutinib with IRAK4 inhibitors show a superior antitumor activity that each compound alone [83,90]. A clinical trial combining ibrutinib and IRAK inhibitors is ongoing (NCT03328078).
3. MAPK Signaling
Mitogen-activated protein kinases (MAPKs) are a group of proteins that participate in the regulation of proliferation, differentiation, migration and survival of cells. The canonical activation of this pathway starts when a ligand (e.g., a cytokine, hormone or growth/differentiation factor) binds to the extracellular portion of a receptor tyrosine kinase (RTK), with the consequent phosphorylation and the activation of retrovirus-associated DNA sequences (RAS) proteins. Then, RAS activates a member of the serine-threonine kinase RAF family (BRAF), that facilitates the phosphorylation of the mitogen-activated ERK kinase (MEK) and MEK activates extracellular signal-regulated kinase (ERK), the most important kinase in the cascade that activates different transcription factors [91]. The MAPK pathway is also activated upon BCR ligation, being the magnitude of MAPK signaling activation a direct readout of BCR signaling [92]. BCR is also able to regulate phospholipase C gamma, which causes calcium mobilization; and the PI3K activation [93] (Figure 1).
3.1. Recurrent Mutations and Alterations in MAPK Pathway
The MAPK pathway is altered in around half of neoplasms with different frequencies across tumor types [94]. In CLL, mutations in this pathway are detected in 5–8% of cases depending on disease stage [2,5]. These mutated cases present an increased expression of MAPK pathway genes and high levels of phosphorylated ERK, a known surrogate marker of MAPK pathway activation [95]. The most frequent mutated gene is BRAF, detected in 2% of CLL cases [5,95,96]. Most of these mutations cluster around the kinase domain [97], but they are different to the typical V600E mutation seen in other malignancies [98]. In addition, other mutations in this pathway have been reported: upstream BRAF (KITLG, KIT, PTPN11, GNB1, NRAS and KRAS) and downstream BRAF (MAP2K1 alias Mek1 and MAP2K2 alias Mek2) [95].
Mutations in MAPK pathway are related to adverse biological features such as high expression of CD38, CD49d and ZAP-70, U-CLL [95] and trisomy 12 [99]. Patients carrying mutations in this pathway have a 5-year TTFT similar to patients with mutations associated with worse outcome (TP53, ATM or BIRC3), while patients presenting concomitant mutations of TP53, ATM or BIRC3 and in genes of the MAPK pathway have the worst TTFT [95]. Subclonal BRAF mutations also have a prognostic impact on TTFT [15] and are associated with an adverse overall survival (OS) [100]. In the CLL4 trial, mutations in BRAF, KRAS and NRAS presented a reduced OS in both FC and rituximab and FC (FCR) treatments [60]. BRAF mutations are associated with refractoriness to fludarabine [101] and KRAS mutations with resistance to chlorambucil-based chemotherapy or anti CD20-chemoimmunotherapy [62].
Regarding the new targeted therapies (Table 1), mutations in the MAPK pathway are mediators of primary resistance to PI3K inhibitors [61]. Furthermore, activation of ERK1/2 has been associated with ibrutinib resistance [102], raising the possibility that patients with mutations in this pathway might have reduced sensitivity to ibrutinib. Although overexpression of BRAF in lymphoma cells shows resistance to venetoclax, more venetoclax resistant CLL samples are needed to confirm these results [103]. In this way, lower minimal residual disease rates were seen in patients treated with venetoclax plus rituximab [54].
In addition, recurrent mutations in EGR2 have been described in about 8% of advanced-stage patients with CLL and are related to a poor outcome [97,104]. After BCR stimulation, EGR2 is activated via ERK phosphorylation [105]. Consequently, EGR2 mutations participate in the dysregulation of BCR signaling [97].
3.2. Targeting MAPK Pathway
V600 BRAF inhibitors (vemurafenib and dabrafenib) and MEK inhibitors (trametinib, selumetinib, cobimetinib and binimetinib) are used in the clinics for patients with mutations in MAPK pathway [106]. In CLL, sorafenib, a multikinase inhibitor, induces cell death independently of BRAF status, whereas incubation of BRAF mutated CLL cells with PLX4720, the vemurafenib progenitor [96], vemurafenib or dabrafenib [95] fails to induce cytotoxicity probably because these BRAF inhibitors are specific for V600 mutation, and this specific mutation is rare in CLL. In contrast, ulixertinib, an ERK inhibitor [107], decreases ERK phosphorylation in MAPK-mutated CLL cases [95]. Furthermore, it has been reported that the MEK1/2 inhibitor binimetinib is effective alone or in combination with venetoclax [108] and that CLL cells carrying trisomy 12 are sensitive to MEK and ERK inhibitors [109].
4. Notch Signaling
Notch pathway is a well evolutionary conserved signaling cascade involved in cell-fate decisions during development and the maintenance of self-renewal. Notch ligands expressed on the surface of a signal-sending cell interact with Notch receptors, which are expressed on the surface of a signal-receiving cell. There are four mammalian Notch receptors (Notch1–4) and two groups of ligands, Jagged (Jagged 1 and Jagged 2) and Delta-like (DLL1, DLL3, and DLL4) [110]. Ligand-receptor binding induces sequential cleavages by metalloproteases and γ-secretase, leading to the nuclear translocation of the intracellular Notch domain (NICD), where displaces co-repressors, recruits co-activators and finally activates the expression of several genes related to cell differentiation, proliferation and survival (Figure 2). Among the genes under direct Notch transcriptional control, note the transcriptional repressors HES1 and HEY1, and the oncogene MYC, which contributes to the Notch-mediated transformation process of some tumors [111,112]. Physiologically, it is well-known that Notch signaling regulates T-cell commitment of common lymphoid progenitors at the expenses of B-cell differentiation. However, Notch also plays a role in B-cell development, both in the early B-cell development as well as in the differentiation toward marginal zone B cells [113].
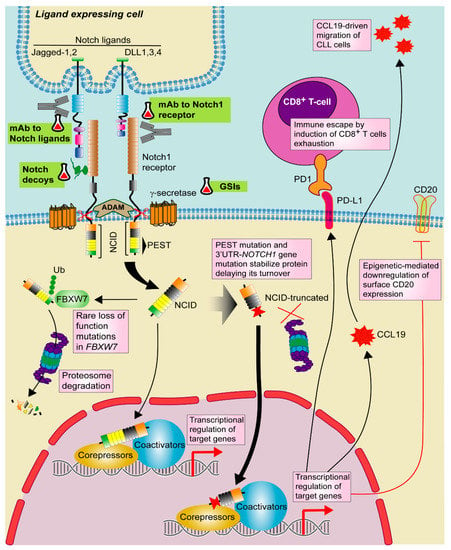
Figure 2.
Targeting Notch1 pathway in CLL. Notch1 signaling is activated by one of the Notch ligands (DLL1,3,4 or Jagged-1,2) that binds to the Notch1 receptor on a contacting cell, with subsequent induction of a series of proteolytic events mediated by ADAM-metalloproteases and γ-secretases. These events promote the release of active Notch intracellular domain (NICD). NICD translocates to the nucleus where activates the transcription of target genes together with several transcriptional regulators. Notch1 signaling is shut down by phosphorylation of NICD and subsequent poly-ubiquitination (Ub) by F-box containing protein (FBXW7) that acts as a signal for proteasomal degradation. In CLL, recurrent mutations in NOTCH1 PEST domain and 3′UTR stabilize the protein and delay its turnover. Although rare, loss of function mutations in FBXW7 has been also described. In CLL, Notch1 signaling modulates CCL19-driven migration and immune escape by PD-1/PD-L1 axis as well as T-cell exhaustion. Notch1 also induces CD20 downregulation through an epigenetic mechanism. Targeted drugs are highlighted in green. Pill icon: approved drugs and Erlenmeyer icon: preclinical studies or clinical trials. GSIs: γ-secretase inhibitors, mAb: monoclonal antibody.
4.1. NOTCH Mutations in CLL
NOTCH1 activating mutations are one of the most recurrent alterations in CLL, accounting for 10–15% of patients at diagnosis [1,114]. NOTCH1-mutated patients present an adverse prognosis, high risk of transformation and poor outcome [1,115,116]. Thus, NOTCH1 mutations prevalence increases to 20% in chemorefractory patients and up to 30% in CLL with Richter transformation (RT) [115,116,117].
Most of the mutations affect the PEST domain and result in a more stable truncated protein, with a delayed turnover that favors Notch1 signaling activation [1]. An additional hotspot in the non-coding 3′UTR of NOTCH1 has been identified in ~3% of CLL patients, which induces the loss of the PEST domain by aberrant splicing [2]. Although rare, loss of function mutations in FBXW7, a ubiquitin ligase implicated in Notch1 turnover, are also found (Figure 2) [3,4,118]. Furthermore, activation of Notch1 has been described independently of the presence of NOTCH mutations [119].
Autocrine and paracrine mechanisms of Notch activation have been described in CLL [120]. However, NOTCH1 mutations need the crosstalk between tumor CLL cells and surrounding cells expressing Notch ligands to trigger and sustain Notch signaling. In particular, lymph nodes would represent a specific niche for Notch activation in CLL because stromal and histiocytic cells express Notch ligands, such as DLL4 [121]. This link between Notch1 signaling and microenvironment may trigger a more aggressive behavior characterized by an increase on cell proliferation, chemotaxis and angiogenesis. In this context, Notch1 signaling regulates CLL cells migration through CCL19 and the overexpression of a genetic MYC-signature [122,123]. Accordingly, DLL stimulation triggers the expression of protumor target genes in NOTCH1-mutated CLL cells, together with an increase in cell proliferation, migration and angiogenesis [121].
Furthermore, Notch1 signaling promotes an immune escape mechanism in mutated CLL cells through the transcriptional regulation of HLA class-II genes and PD-L1. In particular, Notch1 up-regulates PD-L1 and impairs T-cell activation (Figure 2) [124]. A recent work showed that constitutive activation of Akt is common in NOTCH1-mutated CLL cells, suggesting a possible biomarker for RT [125]. Akt initiates CLL transformation via induction of Notch1 signaling between CLL cells and microenvironmental DLL1-expressing T cells.
NOTCH1 mutations have been related to reduced benefits from anti-CD20-based chemoimmunotherapy strategies, both with rituximab [43] and with ofatumumab [24,43]. Although the effects of NOTCH1 mutation in the pathogenesis of CLL seem mostly related to the transcriptional regulation of protumor target genes, the indirect alteration of the epigenetic environment has also been described. Thus, the resistance of NOTCH1-mutated CLL patients to the anti-CD20 rituximab could be likely due to the downregulation of surface CD20 expression by a HDAC-mediated epigenetic mechanism (Figure 2) [126].
Regarding the clinical impact of NOTCH1 mutations in the response to the new targeted agents, the treatment with ibrutinib as a single agent has showed that NOTCH1 mutation is strongly associated with lower redistribution lymphocytosis and impaired nodal shrinkage, leading to partial responses, subsequent relapses, shorter progression free survival (PFS) and OS [64]. Importantly, patients who develop RT under ibrutinib treatment have frequent adverse genomic alterations such as TP53 and NOTCH1 mutations [63]. In the case of venetoclax, NOTCH1 mutation is correlated with shorter duration of response, but not probability of response [51]. Although a trend for adverse PFS and lower undetectable minimal residual disease is found in NOTCH1-mutated CLL cases [24,54], further validation is necessarily required in other cohorts before drawing conclusions about the effect of NOTCH1 mutation in venetoclax-based regimens (Table 1).
4.2. Targeting Notch in CLL
Several strategies have been proposed for blocking Notch in hematological malignancies: targeting extracellular NOTCH1, the γ-secretase complex, Notch trafficking or Notch degradation [127]. Among them, γ-secretase inhibitors (GSIs) (Figure 2) are the most extensively evaluated drugs in different malignancies. It is known that in T-cell acute lymphoblastic leukemia (T-ALL), where more than 50% of patients have activating NOTCH1 mutations, GSIs efficiently inhibit Notch1 oncogenic protein [128]. In CLL, they have been tested in vitro as a single agent or combined with chemotherapy [120,129]. GSI treatment abrogates drug-induced apoptosis resistance reported in NOTCH1-mutated CLL cells [120,129]. In addition, GSI PF-03084014 combined with fludarabine downregulates angiogenesis and CXCL12-mediated migration and invasion in NOTCH1-mutated CLL cells [129].
Despite promising preclinical results, the non-selectivity and the undesired gastrointestinal toxicity of GSIs promoted the development of direct strategies to block Notch1 [130], such as antibodies against the specific Notch receptors (Figure 2) [131,132,133]. Brontictuzumab (OMP-52M51) is a monoclonal antibody against human Notch1 with promising antitumor efficacy in T-ALL xenograft models [134]. In CLL and MCL cells with NOTCH1 mutations, OMP-52M51 efficiently inhibits Notch stimulation and cell proliferation induced by DLL ligands [121,135]. Although a phase I study showed efficacy of brontictuzumab in solid tumors [136], the clinical development of this antibody has been stopped. Novel strategies such as antibodies targeting Notch ligands have also been investigated (Figure 2) [133]. In particular, dual targeting of DLL4 and VEGF induces antitumor responses in solid malignancies [137] and could be of potential interest in NOTCH-mutated B lymphoid malignancies. At preclinical level, other approaches have shown antitumor efficacy through Notch inhibition, such as the use of Notch decoys for either DLL-mediated or Jagged-mediated signaling as well as the use of natural products with ability to modulate Notch signaling (Figure 2) [133,138]. In CLL, another strategy that has been recently proposed for the therapeutic use of Notch targeted agents is its combination with ibrutinib. At biological level, Notch1 and BCR pathways are functionally linked, being NOTCH1-mutated CLL cells more responsive to BCR signaling [139]. This strategy could be particularly relevant in patients with RT, a subgroup with limited therapeutic options [117].
5. RNA Splicing
The spliceosome removes non-coding fragments (introns) from messenger RNA precursors (pre-mRNA) through two catalytic steps necessary for the expression of most eukaryotic genes (Figure 3A). Alternative splicing is key for the regulated generation of different mRNA transcripts and protein variants from a single gene. The spliceosome is a dynamic and complex molecular machinery that consists of more than 150 polypeptides and five small nuclear ribonucleoproteins (snRNP: U1, U2, U4, U5 and U6) each of which is composed of one small nuclear RNA (snRNA) and some associated proteins [140].
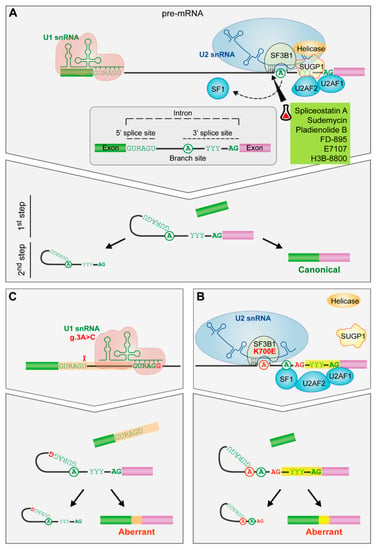
Figure 3.
The impact of spliceosome mutations in the spliceosome assembly. (A) Spliceosome assembly of U1 and U2 snRNPs. The spliceosome removes the internal sequences (introns) of the pre-mRNA and splices together the remaining fragments (exons) after the recognition of conserved sequences found at the 5′ and 3′ end of the intron and the branch site (an adenosine residue). U1 snRNP recognizes the 5′splice site by base pairing interactions. Then, SF1, U2AF2 and U2AF1 recognize the branch site, the polypyrimidine tract and the AG dinucleotide of the 3′ splice site, respectively. Afterwards, SF3B1 helps in the recognition of the branch site by U2 snRNA. Finally, the subsequent recruitment of other snRNPs will drive to the splicing of the intron. (B) SF3B1 mutations. SF3B1 mutations disrupt interactions with SUGP1 spliceosomal protein and contribute to an upstream alternative 3′ splice site usage through the use of an alternative branch site. (C) U1 snRNA mutation. The A > C change in the third position favors the C-G base pairing between the third position of the U1 snRNA and the 5′splice site leading to the formation of aberrant 5′ splice sites. Targeted drugs are highlighted in green. Erlenmeyer icon: preclinical studies or clinical trials. R: purine, Y: pyrimidine.
U1 snRNP takes part in the first step of spliceosome assembly, recognizing the 5′ splice site through base pairing interactions involving the 5′ end of U1 snRNA and sequences at the 5′ end of the intron. Then, U2 snRNP binds to the branch site -an intronic adenosine involved in the first chemical step of the splicing reaction- through base pairing interactions between U2 snRNA and nucleotides flanking the branch site adenosine. This helix, characterized by the bulge out of the branch site adenosine, is recognized by SF3B1 protein, a key component of U2 snRNP, involving a conformational change that ultimately facilitates the approaching of the branch site adenosine to the 5′ splice site and the first catalytic step of the splicing reaction [141,142] (Figure 3A).
5.1. Recurrent Splicing Factor Mutations
Mutations in the spliceosome components have a role in cancer [143]. In CLL, mutations in SF3B1 and U1 have been described [3,144]. SF3B1 mutation disrupts interactions with SUGP1 spliceosomal protein, which is involved in the branch site recognition complex, contributing to the activation of an upstream alternative 3′ splice site through the use of an alternative branch site [145,146,147,148] (Figure 3B).
Approximately 50% of the aberrant RNA transcripts undergo nonsense mediated decay, resulting in downregulation of canonical transcripts [145,149]. In addition, full-length transcript analyses have shown downregulation of retained introns (i.e., enhanced splicing) in SF3B1 mutated patients [150].
SF3B1 is mutated in 8–21% of CLL patients [2,5]. Most of the mutations are missense and they are localized in the highly conserved C-terminal domain of SF3B1, composed of multiple HEAT repeats, being K700E the most frequently mutated site (50% of reported cases) [151]. SF3B1 mutations are associated with advanced stages, male sex, high leukocyte counts, elevated B2 microglobulin levels, high CD38 expression, U-CLL subgroup, intermediate CLL epigenetic subgroup, stereotyped BCR subset 2 and R110-mutated IGLV3-21 subgroup, as well as with 11q deletion and fludarabine resistance [11,13,152,153,154,155].
SF3B1-mutated CLL patients present CD20 downregulation and, similarly to NOTCH1-mutated cells, they also present high levels of active intracytoplasmic Notch1 accompanied by a NOTCH1-related gene set enrichment and overexpression of a DVL2 isoform, involved in the Wnt pathway and Notch1 signaling repression [156,157]. Additionally, other gene set enrichments including “Cytokine-cytokine receptor interaction” and “Phosphatidylinositol signaling system” have been associated with SF3B1 mutations [158]. SF3B1-mutated patients have a shorter PFS [159] and OS [3,153]. If the subclonal architecture of the tumor is considered, SF3B1 mutations with a variant allele frequency (VAF) > 12% predict for shorter TTFT [15,17]. But even patients with a low VAF show a shorter PFS after chlorambucil or chlorambucil and ofatumumab treatment [24] (Table 1). One-third of CLL patients resistant to BTK inhibitors therapy develop RT with a dismal outcome [160] and in this group, SF3B1 mutations are more frequent than BTK mutations [24], the most common ones described in ibrutinib resistant patients [161].
Recently, an A > C mutation in the third position of U1 snRNA has been described, which changes the preferential A–U base-pairing between U1 snRNA and the 5′ splice site to C–G base-pairing and thus, creates novel splice junctions, some of them in known cancer drivers (Figure 3C). U1 mutation is present in 3.8% of CLL patients, is associated to U-CLL subtype and confers an adverse prognosis with a shorter TTFT [144]. Notably, none of the samples with U1 mutation shows SF3B1 mutations [144]. Although they appear at very low frequency, mutations in additional components of U2 snRNP (U2AF2, SRSF1, SRSF7, RBMX, and ZRSR2) and other splicing factors have been described in CLL, as well as mutations in genes involved in RNA transport and metabolism [162].
5.2. Targeting the Spliceosome in CLL
Small-molecule splicing modulators bind to SF3B1, preventing the recognition of the branch site [163,164] (Figure 3A). These bacterial fermentation products and synthetic derivatives show antitumor properties. FR901464 derivatives, such as spliceostatin A and sudemycin, cause more cytotoxicity in primary CLL samples than in healthy B lymphocytes inducing apoptosis by the switch of MCL1 splicing toward its proapoptotic isoform [165,166]. In an adoptive transfer mouse CLL model, sudemycin D6 decreases the number of CLL primary cells in the peripheral blood and in the spleen [165]. Importantly, the combination of sudemycin D1 with ibrutinib shows an enhanced in vitro cytotoxicity [165] and so do the combinations of spliceostatin A and ABT-199 (venetoclax) or spliceostatin A and ABT-263 [166].
Pladienolide B and its derivative FD-895 also induce more apoptosis in CLL cells than in healthy lymphocytes by inducing a pattern of intron retention [167]. E7107, derived from pladienolide B, reprograms apoptosis, decreases Mcl-1 dependence and increases Bcl-2 dependence in CLL. This compound sensitizes primary CLL cells to venetoclax treatment and reverts venetoclax resistance in CLL-like cells from Eµ-TCL1–based adoptive transfer murine model [168]. E7107 entered two phase I clinical trials in advanced solid tumors achieving stable disease (NCT00499499) and partial response (NCT00459823) as the best tumor response. These studies have been discontinued because two patients presented vision loss and another one bilateral optical neuritis [169,170]. H3B-8800, an orally available pladienolide B derivative, has preferential cytotoxic effect on spliceosome-mutant epithelial and myeloid malignancies [171] and also on CLL cells (manuscript in preparation). A phase I clinical trial (NCT02841540) evaluates H3B-8800 in patients with myelodysplastic syndromes, acute myeloid leukemia or chronic myelomonocytic leukemia. Preliminary results show dose-dependent target engagement and predictable pharmacokinetic profile and safety even with prolonged dosing. Though no complete or partial responses have been achieved, decreased red blood cell or platelet transfusion requirements have been observed in 14% of enrolled patients [172].
6. Conclusions
CLL is a heterogeneous disease both at molecular and clinical level. Current genomic studies have identified novel mutated genes affecting important biological pathways, including TLR, MAPK and Notch signaling as well as RNA splicing. Some of these alterations contribute to the development and progression of CLL through specific mechanisms of action that include their relationship with the tumor microenvironment. In recent years, data on the prognostic value of these mutations have been gradually emerging. Given the low frequency of these mutations, only through the efforts of the entire scientific community we will unravel the real prognostic and predictive impact of these mutations. The development of more specific targeted therapies and novel combination treatments will help to design personalized effective treatments and strategies to improve the outcome of patients with CLL.
Author Contributions
I.L.-O., H.P.-A., F.A., M.L.-G. and D.C. prepared the first draft and wrote, reviewed and edited the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The group has been supported by the Spanish Ministry of Science and Innovation through the Plan Estatal de Investigación Científica y Técnica y de Innovación [RTI2018-094584-B-I00] and was cofounded by the European Regional Development Fund (ERDF) and the CERCA program from Generalitat de Catalunya, Centro de Investigación Biomédica en Cáncer (CIBERONC) and Generalitat de Catalunya [2017 SGR 1009].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Juan Valcárcel and Sophie Bonnal for critical reading of the manuscript. I.L.-O. is a recipient of a Severo Ochoa PhD4MD Program Fellowship and H.P.-A. is a recipient of a predoctoral fellowship from the Spanish Ministry of Science, Innovation and Universities [FPU19/03110].
Conflicts of Interest
I.L.-O., H.P.-A., F.A., M.L.-G. have nothing to disclose. D.C. reports grants and personal fees from H3 Biomedicine, Novartis and Incyte, all outside of the submitted work.
References
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beá, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Heath, S.; Bibikova, M.; Queirós, A.C.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Nadeu, F.; Colomer, D.; Campo, E. Chronic lymphocytic leukemia: From molecular pathogenesis to novel therapeutic strategies. Haematologica 2020, 105, 2205–2217. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V Gene Mutation Status and CD38 Expression As Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Seifert, M.; Sellmann, L.; Bloehdorn, J.; Wein, F.; Stilgenbauer, S.; Dürig, J.; Küppers, R. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J. Exp. Med. 2012, 209, 2183–2198. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Agathangelidis, A.; Rosenquist, R.; Ghia, P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia 2017, 31, 282–291. [Google Scholar] [CrossRef]
- ten Hacken, E.; Gounari, M.; Ghia, P.; Burger, J.A. The importance of B cell receptor isotypes and stereotypes in chronic lymphocytic leukemia. Leukemia 2019, 33, 287–298. [Google Scholar] [CrossRef]
- Nadeu, F.; Royo, R.; Clot, G.; Duran-Ferrer, M.; Navarro, A.; Martin, S.; Lu, J.; Zenz, T.; Baumann, T.S.; Jares, P.; et al. IGLV3-21R110 identifies an aggressive biological subtype of chronic lymphocytic leukemia with intermediate epigenetics. Blood 2020, 137, 2935–2946. [Google Scholar] [CrossRef] [PubMed]
- Maity, P.C.; Bilal, M.; Koning, M.T.; Young, M.; Van Bergen, C.A.M.; Renna, V.; Nicolò, A.; Datta, M.; Gentner-Göbel, E.; Barendse, R.S.; et al. IGLV3-21*01 is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 4320–4327. [Google Scholar] [CrossRef]
- Nadeu, F.; Clot, G.; Delgado, J.; Martín-García, D.; Baumann, T.; Salaverria, I.; Beà, S.; Pinyol, M.; Jares, P.; Navarro, A.; et al. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia 2018, 32, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Trillos, A.; Pinyol, M.; Navarro, A.; Aymerich, M.; Jares, P.; Juan, M.; Rozman, M.; Colomer, D.; Delgado, J.; Giné, E.; et al. Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood 2014, 123, 3790–3796. [Google Scholar] [CrossRef]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martín-García, D.; Beà, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Dalla-Favera, R. Chronic lymphocytic leukaemia: From genetics to treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef]
- Moia, R.; Patriarca, A.; Schipani, M.; Ferri, V.; Favini, C.; Sagiraju, S.; Al Essa, W.; Gaidano, G. Precision medicine management of chronic lymphocytic leukemia. Cancers 2020, 12, 642. [Google Scholar] [CrossRef]
- Beekman, R.; Chapaprieta, V.; Russiñol, N.; Vilarrasa-Blasi, R.; Verdaguer-Dot, N.; Martens, J.H.A.; Duran-Ferrer, M.; Kulis, M.; Serra, F.; Javierre, B.M.; et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 868–880. [Google Scholar] [CrossRef]
- Fürstenau, M.; Eichhorst, B. Novel Agents in Chronic Lymphocytic Leukemia: New Combination Therapies and Strategies to Overcome Resistance. Cancers 2021, 13, 1336. [Google Scholar] [CrossRef]
- Guarente, V.; Sportoletti, P. Lessons, Challenges and Future Therapeutic Opportunities for PI3K Inhibition in CLL. Cancers 2021, 13, 1280. [Google Scholar] [CrossRef]
- Moia, R.; Boggione, P.; Mahmoud, A.M.; Kodipad, A.A.; Adhinaveni, R.; Sagiraju, S.; Patriarca, A.; Gaidano, G. Targeting p53 in chronic lymphocytic leukemia. Expert Opin. Ther. Targets 2020, 24, 1239–1250. [Google Scholar] [CrossRef]
- Tausch, E.; Beck, P.; Schlenk, R.F.; Jebaraj, B.M.C.J.; Dolnik, A.; Yosifov, D.Y.; Hillmen, P.; Offner, F.; Janssens, A.; Govind Babu, K.; et al. Prognostic and predictive role of gene mutations in chronic lymphocytic leukemia: Results from the pivotal phase III study COMPLEMENT1. Haematologica 2020, 105, 2440–2447. [Google Scholar] [CrossRef]
- Liang, J.; Zhao, W.; Tong, P.; Li, P.; Zhao, Y.; Li, H.; Liang, J. Comprehensive molecular characterization of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med. Genom. 2020, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Frazzi, R. BIRC3 and BIRC5: Multi-faceted inhibitors in cancer. Cell Biosci. 2021, 11, 8. [Google Scholar] [CrossRef]
- Rose-Zerilli, M.J.J.; Forster, J.; Parker, H.; Parker, A.; Rodri, A.É.; Chaplin, T.; Gardiner, A.; Steele, A.J.; Collins, A.; Young, B.D.; et al. ATM mutation rather than BIRC3 deletion and/or mutation predicts reduced survival in 11q-deleted chronic lymphocytic leukemia: Data from the UK LRF CLL4 trial. Haematologica 2014, 99, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Fangazio, M.; Rasi, S.; Vaisitti, T.; Monti, S.; Cresta, S.; Chiaretti, S.; Del Giudice, I.; Fabbri, G.; Bruscaggin, A.; et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood 2012, 119, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Diop, F.; Moia, R.; Favini, C.; Spaccarotella, E.; De Paoli, L.; Bruscaggin, A.; Spina, V.; Terzi-Di-Bergamo, L.; Arruga, F.; Tarantelli, C.; et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2020, 105, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Raponi, S.; Del Giudice, I.; Ilari, C.; Cafforio, L.; Messina, M.; Cappelli, L.V.; Bonina, S.; Piciocchi, A.; Marinelli, M.; Peragine, N.; et al. Biallelic BIRC3 inactivation in chronic lymphocytic leukaemia patients with 11q deletion identifies a subgroup with very aggressive disease. Br. J. Haematol. 2019, 185, 156–159. [Google Scholar] [CrossRef]
- Rahal, R.; Frick, M.; Romero, R.; Korn, J.M.; Kridel, R.; Chan, F.C.; Meissner, B.; Bhang, H.E.; Ruddy, D.; Kauffmann, A.; et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 2014, 20, 87–92. [Google Scholar] [CrossRef]
- Colomer, D.; Campo, E. Unlocking new therapeutic targets and resistance mechanisms in mantle cell lymphoma. Cancer Cell 2014, 25, 7–9. [Google Scholar] [CrossRef][Green Version]
- Vidal-Crespo, A.; Rodriguez, V.; Matas-Céspedes, A.; Lee, E.; Rivas-Delgado, A.; Giné, E.; Navarro, A.; Beà, S.; Campo, E.; López-Guillermo, A.; et al. The Bruton tyrosine kinase inhibitor CC-292 shows activity in mantle cell lymphoma and synergizes with lenalidomide and NIK inhibitors depending on nuclear factor-κB mutational status. Haematologica 2017, 102, e447–e451. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, K.; Fu, M.; Onida, F.; Wierda, W.G.; Keating, M.J.; Gandhi, V. Reactivation of smac-mediated apoptosis in chronic lymphocytic leukemia cells: Mechanistic studies of smac mimetic. Oncotarget 2016, 7, 39458–39472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Condoluci, A.; Rossi, D. Genetic mutations in chronic lymphocytic leukemia: Impact on clinical treatment. Expert Rev. Hematol. 2019, 12, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ballman, K.V. Biomarker: Predictive or prognostic? J. Clin. Oncol. 2015, 33, 3968–3971. [Google Scholar] [CrossRef]
- Patriarca, A.; Gaidano, G. A step ahead toward precision medicine for chronic lymphocytic leukemia. Haematologica 2020, 105, 2352–2355. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer 2021, 148, 8–17. [Google Scholar] [CrossRef]
- Ciardullo, C.; Aptullahoglu, E.; Woodhouse, L.; Lin, W.Y.; Wallis, J.P.; Marr, H.; Marshall, S.; Bown, N.; Willmore, E.; Lunec, J. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica 2019, 104, 2429–2442. [Google Scholar] [CrossRef]
- Zhong, Y.; El-Gamal, D.; Dubovsky, J.A.; Beckwith, K.A.; Harrington, B.K.; Williams, K.E.; Goettl, V.M.; Jha, S.; Mo, X.; Jones, J.A.; et al. Selinexor suppresses downstream effectors of B-cell activation, proliferation and migration in chronic lymphocytic leukemia cells. Leukemia 2014, 28, 1158–1163. [Google Scholar] [CrossRef]
- Boudny, M.; Zemanova, J.; Khirsariya, P.; Borsky, M.; Verner, J.; Cerna, J.; Oltova, A.; Seda, V.; Mraz, M.; Jaros, J.; et al. Novel CHK1 inhibitor MU380 exhibits significant single-agent activity in TP53-mutated chronic lymphocytic leukemia cells. Haematologica 2019, 104, 2443–2455. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Managò, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Schnaiter, A.; Paschka, P.; Zenz, T.; Rossi, M.; Döhner, K.; Bühler, A.; Böttcher, S.; Ritgen, M.; Kneba, M.; et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: Results from the CLL8 trial. Blood 2014, 123, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Hu, B.; Wang, F.; Yan, Y.; Kim, E.; Vitale, C.; Patel, K.P.; Strati, P.; Gumbs, C.; Little, L.; et al. Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 2018, 131, 1820–1832. [Google Scholar] [CrossRef]
- O’Brien, S.; Furman, R.R.; Coutre, S.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Wierda, W.; Jones, J.; Zhao, W.; et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: A 5-year experience. Blood 2018, 131, 1910–1919. [Google Scholar] [CrossRef]
- Ahn, I.E.; Tian, X.; Ipe, D.; Cheng, M.; Albitar, M.; Tsao, L.C.; Zhang, L.; Ma, W.; Herman, S.E.M.; Gaglione, E.M.; et al. Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia Treated With Ibrutinib: Development and Validation of a Four-Factor Prognostic Model. J. Clin. Oncol. 2021, 39, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs. ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef]
- Jones, J.A.; Robak, T.; Brown, J.R.; Awan, F.T.; Badoux, X.; Coutre, S.; Loscertales, J.; Taylor, K.; Vandenberghe, E.; Wach, M.; et al. Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: An open-label, randomised phase 3 trial. Lancet Haematol. 2017, 4, e114–e126. [Google Scholar] [CrossRef]
- Sharman, J.P.; Coutre, S.E.; Furman, R.R.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.W.; et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J. Clin. Oncol. 2019, 37, 1391–1402. [Google Scholar] [CrossRef]
- Davids, M.S.; Kuss, B.J.; Hillmen, P.; Montillo, M.; Moreno, C.; Essell, J.; Lamanna, N.; Nagy, Z.; Tam, C.S.; Stilgenbauer, S.; et al. Efficacy and safety of duvelisib following disease progression on ofatumumab in patients with relapsed/refractory CLL or SLL in the DUO crossover extension study. Clin. Cancer Res. 2020, 26, 2096–2103. [Google Scholar] [CrossRef]
- Roberts, A.W.; Ma, S.; Kipps, T.J.; Coutre, S.E.; Davids, M.S.; Eichhorst, B.; Hallek, M.; Byrd, J.C.; Humphrey, K.; Zhou, L.; et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood 2019, 134, 111–122. [Google Scholar] [CrossRef]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.-M.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.; von Tresckow, J.; Bahlo, J.; Robrecht, S.; Langerbeins, P.; Al-Sawaf, O.; Engelke, A.; Fink, A.M.; Fischer, K.; Tausch, E.; et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): Primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018, 19, 1215–1228. [Google Scholar] [CrossRef]
- Kater, A.P.; Wu, J.Q.; Kipps, T.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Robak, T.; de la Serna, J.; et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations From the MURANO Phase III Study. J. Clin. Oncol. 2020, 38, 4042–4054. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 43–56. [Google Scholar] [CrossRef]
- Tausch, E.; Schneider, C.; Robrecht, S.; Zhang, C.; Dolnik, A.; Bloehdorn, J.; Bahlo, J.; Al-Sawaf, O.; Ritgen, M.; Fink, A.M.; et al. Prognostic and predictive impact of genetic markers in patients with CLL treated with obinutuzumab and venetoclax. Blood 2020, 135, 2402–2412. [Google Scholar] [CrossRef]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): A randomised, controlled, phase 3 trial. Lancet 2020, 395, 1278–1291. [Google Scholar] [CrossRef]
- Byrd, J.C.; Woyach, J.A.; Furman, R.R.; Martin, P.; O’Brien, S.M.; Brown, J.R.; Stephens, D.M.; Barrientos, J.C.; Devereux, S.; Hillmen, P.; et al. Acalabrutinib in treatment-naïve chronic lymphocytic leukemia: Mature results from phase II study demonstrating durable remissions and long-term tolerability. J. Clin. Oncol. 2020, 38, 8024. [Google Scholar] [CrossRef]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M.; Kaplan, P.; Kraychok, I.; Illes, A.; de la Serna, J.; et al. Ascend: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2020, 38, 2849–2861. [Google Scholar] [CrossRef]
- Blakemore, S.J.; Clifford, R.; Parker, H.; Antoniou, P.; Stec-Dziedzic, E.; Larrayoz, M.; Davis, Z.; Kadalyayil, L.; Colins, A.; Robbe, P.; et al. Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: Data from the randomised UK LRF CLL4 trial. Leukemia 2020, 34, 1760–1774. [Google Scholar] [CrossRef]
- Murali, I.; Kasar, S.; Naeem, A.; Tyekucheva, S.; Khalsa, J.K.; Thrash, E.M.; Itchaki, G.; Livitz, D.; Leshchiner, I.; Dong, S.; et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia (cll). Blood 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Herling, C.D.; Klaumünzer, M.; Rocha, C.K.; Altmüller, J.; Thiele, H.; Bahlo, J.; Kluth, S.; Crispatzu, G.; Herling, M.; Schiller, J.; et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood 2016, 128, 395–404. [Google Scholar] [CrossRef]
- Brown, J.R.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.E.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia 2018, 32, 83–91. [Google Scholar] [CrossRef]
- Del Poeta, G.; Biagi, A.; Laurenti, L.; Chiarenza, A.; Pozzo, F.; Innocenti, I.; Postorino, M.; Rossi, F.M.; Del Principe, M.I.; Bomben, R.; et al. Impaired nodal shrinkage and apoptosis define the independent adverse outcome of NOTCH1 mutated patients under ibrutinib therapy in chronic lymphocytic leukaemia. Haematologica 2020. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Gángó, A.; Alpár, D.; Galik, B.; Marosvári, D.; Kiss, R.; Fésüs, V.; Aczél, D.; Eyüpoglu, E.; Nagy, N.; Nagy, Á.; et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int. J. Cancer 2020, 146, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Jain, P.; Patel, K.P.; Routbort, M.; Bueso-Ramos, C.; Alhalouli, T.; Khoury, J.D.; Luthra, R.; Ferrajoli, A.; Keating, M.; et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor–resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer 2019, 125, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Jiang, Y.; Chyla, B.; Seymour, J.F. Response in patients with BIRC3-mutated relapsed/refractory chronic lymphocytic leukemia treated with fixed-duration venetoclax and rituximab. Haematologica 2020, 105, e382–e383. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Ruprecht, C.R.; Lanzavecchia, A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur. J. Immunol. 2006, 36, 810–816. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Onai, N.; Lanzavecchia, A. A role for toll-like receptors in acquired immunity: Up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood 2003, 101, 4500–4504. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- Balka, K.R.; Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Ntoufa, S.; Vilia, M.G.; Stamatopoulos, K.; Ghia, P.; Muzio, M. Toll-like receptors signaling: A complex network for NF-κB activation in B-cell lymphoid malignancies. Semin. Cancer Biol. 2016, 39, 15–25. [Google Scholar] [CrossRef]
- Cushing, L.; Winkler, A.; Jelinsky, S.A.; Lee, K.; Korver, W.; Hawtin, R.; Rao, V.R.; Fleming, X.M.; Lin, L.L. IRAK4 kinase activity controls Toll-like receptor–induced inflammation through the transcription factor IRF5 in primary human monocytes. J. Biol. Chem. 2017, 292, 18689–18698. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Drexler, S.K.; Foxwell, B.M. The role of Toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 506–518. [Google Scholar] [CrossRef]
- Dajon, M.; Iribarren, K.; Cremer, I. Toll-like receptor stimulation in cancer: A pro- and anti-tumor double-edged sword. Immunobiology 2017, 222, 89–100. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Understanding the Immunodeficiency in Chronic Lymphocytic Leukemia. Potential Clinical Implications. Hematol. Oncol. Clin. North Am. 2013, 27, 207–235. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L.; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201. [Google Scholar] [CrossRef]
- Baliakas, P.; Hadzidimitriou, A.; Agathangelidis, A.; Rossi, D.; Sutton, L.A.; Kminkova, J.; Scarfo, L.; Pospisilova, S.; Gaidano, G.; Stamatopoulos, K.; et al. Prognostic relevance of MYD88 mutations in CLL: The jury is still out. Blood 2015, 126, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.; Coulter, E.M.; Halliwell, E.; Profitos-Peleja, N.; Walsby, E.; Clark, B.; Phillips, E.H.; Burley, T.A.; Mitchell, S.; Devereux, S.; et al. TLR9 expression in Chronic Lymphocytic Leukemia identifies a pro-migratory subpopulation and novel therapeutic target. Blood 2021, 137, 3064–3078. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P mutation in lymphoid malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef]
- Giménez, N.; Schulz, R.; Higashi, M.; Aymerich, M.; Villamor, N.; Delgado, J.; Juan, M.; López-Guerra, M.; Campo, E.; Rosich, L.; et al. Targeting IRAK4 disrupts inflammatory pathways and delays tumor development in chronic lymphocytic leukemia. Leukemia 2020, 34, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Shuai, W.; Lin, P.; Strati, P.; Patel, K.P.; Routbort, M.J.; Hu, S.; Wei, P.; Khoury, J.D.; You, M.J.; Loghavi, S.; et al. Clinicopathological characterization of chronic lymphocytic leukemia with MYD88 mutations: L265P and non-L265P mutations are associated with different features. Blood Cancer J. 2020, 10, 86. [Google Scholar] [CrossRef]
- Improgo, M.R.; Tesar, B.; Klitgaard, J.L.; Magori-Cohen, R.; Yu, L.; Kasar, S.; Chaudhary, D.; Miao, W.; Fernandes, S.M.; Hoang, K.; et al. MYD88 L265P mutations identify a prognostic gene expression signature and a pathway for targeted inhibition in CLL. Br. J. Haematol. 2019, 184, 925–936. [Google Scholar]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Gummadi, V.R.; Boruah, A.; Ainan, B.R.; Vare, B.R.; Manda, S.; Gondle, H.P.; Kumar, S.N.; Mukherjee, S.; Gore, S.T.; Krishnamurthy, N.R.; et al. Discovery of CA-4948, an Orally Bioavailable IRAK4 Inhibitor for Treatment of Hematologic Malignancies. ACS Med. Chem. Lett. 2020, 11, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1 mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Eμ-TCL1 CLL mouse model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.S.; Roessner, P.M.; Yazdanparast, H.; Colomer, D.; Campo, E.; Kugler, S.; Yosifov, D.; Stilgenbauer, S.; Schmidt, M.; Gabriel, R.; et al. Control of chronic lymphocytic leukemia development by clonally-expanded CD8 + T-cells that undergo functional exhaustion in secondary lymphoid tissues. Leukemia 2019, 33, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Dadashian, E.L.; McAuley, E.M.; Liu, D.; Shaffer, A.L.; Young, R.M.; Iyer, J.R.; Kruhlak, M.J.; Staudt, L.M.; Wiestner, A.; Herman, S.E.M. TLR signaling is activated in lymph node–resident CLL cells and is only partially inhibited by ibrutinib. Cancer Res. 2019, 79, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Shukla, A.; Shukla, V.; Joshi, S.S. Regulation of MAPK signaling and implications in chronic lymphocytic leukemia. Leuk. Lymphoma 2018, 59, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Cooney, D.; Pradhan, M.; Mark Coggeshall, K. Convergence of signaling pathways on the activation of ERK in B cells. J. Biol. Chem. 2002, 277, 23420–23426. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Giménez, N.; Martínez-Trillos, A.; Montraveta, A.; Lopez-Guerra, M.; Rosich, L.; Nadeu, F.; Valero, J.G.; Aymerich, M.; Magnano, L.; Rozman, M.; et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica 2019, 104, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Jebaraj, B.M.C.; Kienle, D.; Bühler, A.; Winkler, D.; Döhner, H.; Stilgenbauer, S.; Zenz, T. BRAF mutations in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Mylonas, E.; Cosson, A.; Yoshida, K.; Della Valle, V.; Mouly, E.; Diop, M.; Scourzic, L.; Shiraishi, Y.; Chiba, K.; et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014, 4, 1088–1101. [Google Scholar] [CrossRef]
- Pisapia, P.; Pepe, F.; Iaccarino, A.; Sgariglia, R.; Nacchio, M.; Russo, G.; Gragnano, G.; Malapelle, U.; Troncone, G. BRAF: A Two-Faced Janus. Cells 2020, 9, 2549. [Google Scholar] [CrossRef]
- Vendramini, E.; Bomben, R.; Pozzo, F.; Benedetti, D.; Bittolo, T.; Rossi, F.M.; Dal Bo, M.; Rabe, K.G.; Pozzato, G.; Zaja, F.; et al. KRAS, NRAS, and BRAF mutations are highly enriched in trisomy 12 chronic lymphocytic leukemia and are associated with shorter treatment-free survival. Leukemia 2019, 33, 2111–2115. [Google Scholar] [CrossRef]
- Leeksma, A.C.; Taylor, J.; Wu, B.; Gardner, J.R.; He, J.; Nahas, M.; Gonen, M.; Alemayehu, W.G.; te Raa, D.; Walther, T.; et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia 2019, 33, 390–402. [Google Scholar] [CrossRef]
- Pandzic, T.; Larsson, J.; He, L.; Kundu, S.; Ban, K.; Akhtar-Ali, M.; Hellström, A.R.; Schuh, A.; Clifford, R.; Blakemore, S.J.; et al. Transposon mutagenesis reveals fludarabine resistance mechanisms in chronic lymphocytic leukemia. Clin. Cancer Res. 2016, 22, 6217–6227. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Guo, A.; Lu, P.; Ma, J.; Coleman, M.; Wang, Y.L. Functional characterization of BTK C481S mutation that confers ibrutinib resistance: Exploration of alternative kinase inhibitors. Leukemia 2015, 29, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef]
- Young, E.; Noerenberg, D.; Mansouri, L.; Ljungström, V.; Frick, M.; Sutton, L.A.; Blakemore, S.J.; Galan-Sousa, J.; Plevova, K.; Baliakas, P.; et al. EGR2 mutations define a new clinically aggressive subgroup of chronic lymphocytic leukemia. Leukemia 2017, 31, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Sanjo, H.; Pagès, G.; Kawano, Y.; Karasuyama, H.; Pouysségur, J.; Ogata, M.; Kurosaki, T. Erk Kinases Link Pre-B Cell Receptor Signaling to Transcriptional Events Required for Early B Cell Expansion. Immunity 2008, 28, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Lee Wong, D.J.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Crassini, K.; Shen, Y.; Stevenson, W.S.; Christopherson, R.; Ward, C.; Mulligan, S.P.; Best, O.G. MEK1/2 inhibition by binimetinib is effective as a single agent and potentiates the actions of Venetoclax and ABT-737 under conditions that mimic the chronic lymphocytic leukaemia (CLL) tumour microenvironment. Br. J. Haematol. 2018, 182, 360–372. [Google Scholar] [CrossRef]
- Dietrich, S.; Oleś, M.; Lu, J.; Sellner, L.; Anders, S.; Velten, B.; Wu, B.; Hüllein, J.; da Silva Liberio, M.; Walther, T.; et al. Drug-perturbation-based stratification of blood cancer. J. Clin. Investig. 2018, 128, 427–445. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef]
- Arruga, F.; Vaisitti, T.; Deaglio, S. The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies. Front. Oncol. 2018, 8, 550. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef]
- Villamor, N.; Conde, L.; Martínez-Trillos, A.; Cazorla, M.; Navarro, A.; Beà, S.; López, C.; Colomer, D.; Pinyol, M.; Aymerich, M.; et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia 2013, 27, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Spina, V.; Gaidano, G. Biology and treatment of Richter syndrome. Blood 2018, 131, 2761–2772. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and Other Novel Cancer Genes in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Fabbri, G.; Holmes, A.B.; Viganotti, M.; Scuoppo, C.; Belver, L.; Herranz, D.; Yan, X.J.; Kieso, Y.; Rossi, D.; Gaidano, G.; et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2017, 114, e2911–e2919. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Gizdic, B.; Serra, S.; Vaisitti, T.; Ciardullo, C.; Coscia, M.; Laurenti, L.; D’Arena, G.; Jaksic, O.; Inghirami, G.; et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia 2014, 28, 1060–1070. [Google Scholar] [CrossRef]
- López-Guerra, M.; Xargay-Torrent, S.; Fuentes, P.; Roldán, J.; González-Farré, B.; Rosich, L.; Silkenstedt, E.; García-León, M.J.; Lee-Vergés, E.; Giménez, N.; et al. Specific NOTCH1 antibody targets DLL4-induced proliferation, migration, and angiogenesis in NOTCH1-mutated CLL cells. Oncogene 2020, 39, 1185–1197. [Google Scholar] [CrossRef]
- Arruga, F.; Gizdic, B.; Bologna, C.; Cignetto, S.; Buonincontri, R.; Serra, S.; Vaisitti, T.; Gizzi, K.; Vitale, N.; Garaffo, G.; et al. Mutations in NOTCH1 PEST domain orchestrate CCL19-driven homing of chronic lymphocytic leukemia cells by modulating the tumor suppressor gene DUSP22. Leukemia 2017, 31, 1882–1893. [Google Scholar] [CrossRef]
- Pozzo, F.; Bittolo, T.; Vendramini, E.; Bomben, R.; Bulian, P.; Rossi, F.M.; Zucchetto, A.; Tissino, E.; Degan, M.; D’Arena, G.; et al. NOTCH1-mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin 1 and ribosome-associated components. Leukemia 2017, 31, 2407–2415. [Google Scholar] [CrossRef]
- Mangolini, M.; Maiques-Diaz, A.; Charalampopoulou, S.; Gerhard-Hartmann, E.; Bloehdorn, J.; Moore, A.; Lu, J.; Franklin, V.N.R.; Chilamakuri, C.S.R.; Moutsoupoulos, I.; et al. NOTCH1 drives immune-escape mechanisms in B cell malignancies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kohlhaas, V.; Blakemore, S.J.; Al-Maarri, M.; Nickel, N.; Pal, M.; Roth, A.; Hövelmeyer, N.; Schäfer, S.C.; Knittel, G.; Lohneis, P.; et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood 2021, 137, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, F.; Bittolo, T.; Arruga, F.; Bulian, P.; MacOr, P.; Tissino, E.; Gizdic, B.; Rossi, F.M.; Bomben, R.; Zucchetto, A.; et al. NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: Evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia 2016, 30, 182–189. [Google Scholar] [CrossRef]
- Sorrentino, C.; Cuneo, A.; Roti, G. Therapeutic targeting of notch signaling pathway in hematological malignancies. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019037. [Google Scholar]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris IV, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- López-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Montraveta, A.; Roldán, J.; Matas-Céspedes, A.; Villamor, N.; Aymerich, M.; López-Otín, C.; Pérez-Galán, P.; et al. The γ-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia 2015, 29, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of Notch signalling-are we there yet? Nat. Rev. Drug Discov. 2014, 13, 357–378. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; De Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Aste-Amézaga, M.; Zhang, N.; Lineberger, J.E.; Arnold, B.A.; Toner, T.J.; Gu, M.; Huang, L.; Vitelli, S.; Vo, K.T.; Haytko, P.; et al. Characterization of notch1 antibodies that inhibit signaling of both normal and mutated notch1 receptors. PLoS ONE 2010, 5, e9094. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Agnusdei, V.; Minuzzo, S.; Frasson, C.; Grassi, A.; Axelrod, F.; Satyal, S.; Gurney, A.; Hoey, T.; Seganfreddo, E.; Basso, G.; et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia 2014, 28, 278–288. [Google Scholar] [CrossRef]
- Silkenstedt, E.; Arenas, F.; Colom-Sanmartí, B.; Xargay-Torrent, S.; Higashi, M.; Giró, A.; Rodriguez, V.; Fuentes, P.; Aulitzky, W.E.; Van Der Kuip, H.; et al. Notch1 signaling in NOTCH1-mutated mantle cell lymphoma depends on Delta-Like ligand 4 and is a potential target for specific antibody therapy. J. Exp. Clin. Cancer Res. 2019, 38, 446. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Eckhardt, G.; Patnaik, A.; LoRusso, P.; Faoro, L.; Heymach, J.V.; Kapoun, A.M.; Xu, L.; Munster, P. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann. Oncol. 2018, 29, 1561–1568. [Google Scholar] [CrossRef]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig. New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- Gharaibeh, L.; Elmadany, N.; Alwosaibai, K.; Alshaer, W. Notch1 in cancer therapy: Possible clinical implications and challenges. Mol. Pharmacol. 2020, 98, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Bracciamà, V.; Vitale, N.; Vaisitti, T.; Gizzi, K.; Yeomans, A.; Coscia, M.; D’Arena, G.; Gaidano, G.; Allan, J.N.; et al. Bidirectional linkage between the B-cell receptor and NOTCH1 in chronic lymphocytic leukemia and in Richter’s syndrome: Therapeutic implications. Leukemia 2020, 34, 462–477. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Schmitzová, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Lührmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef]
- Bai, R.; Wan, R.; Yan, C.; Lei, J.; Shi, Y. Structures of the fully assembled Saccharomyces cerevisiae spliceosome before activation. Science 2018, 360, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.M.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome Sequencing Reveals Potential Mechanism of Cryptic 3′ Splice Site Selection in SF3B1-mutated Cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ali, A.M.; Lieu, Y.K.; Liu, Z.; Gao, J.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol. Cell 2019, 76, 82–95.e7. [Google Scholar] [CrossRef]
- Leeksma, A.C.; Derks, I.A.M.; Kasem, M.H.; Kilic, E.; de Klein, A.; Jager, M.J.; van de Loosdrecht, A.A.; Jansen, J.H.; Navrkalova, V.; Faber, L.M.; et al. The Effect of SF3B1 Mutation on the DNA Damage Response and Nonsense-Mediated mRNA Decay in Cancer. Front. Oncol. 2021, 10, 1–7. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1438. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, C.J. SF3B1 mutations in chronic lymphocytic leukemia. Blood 2013, 121, 4627–4634. [Google Scholar] [CrossRef]
- Rossi, D.; Bruscaggin, A.; Spina, V.; Rasi, S.; Khiabanian, H.; Messina, M.; Fangazio, M.; Vaisitti, T.; Monti, S.; Chiaretti, S.; et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: Association with progression and fludarabine-refractoriness. Blood 2011, 118, 6904–6908. [Google Scholar] [CrossRef] [PubMed]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Haferlach, T.; et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Hadzidimitriou, A.; Sutton, L.A.; Rossi, D.; Minga, E.; Villamor, N.; Larrayoz, M.; Kminkova, J.; Agathangelidis, A.; Davis, Z.; et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia 2015, 29, 329–336. [Google Scholar] [CrossRef]
- Queirós, A.C.; Villamor, N.; Clot, G.; Martinez-Trillos, A.; Kulis, M.; Navarro, A.; Penas, E.M.M.; Jayne, S.; Majid, A.; Richter, J.; et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2015, 29, 598–605. [Google Scholar] [CrossRef]
- Wang, L.; Brooks, A.N.; Fan, J.; Wan, Y.; Gambe, R.; Li, S.; Hergert, S.; Yin, S.; Freeman, S.S.; Levin, J.Z.; et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 750–763. [Google Scholar] [CrossRef]
- Pozzo, F.; Bittolo, T.; Tissino, E.; Vit, F.; Vendramini, E.; Laurenti, L.; D’Arena, G.; Olivieri, J.; Pozzato, G.; Zaja, F.; et al. SF3B1-mutated chronic lymphocytic leukemia shows evidence of NOTCH1 pathway activation including CD20 downregulation. Haematologica 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Lütge, A.; Lu, J.; Hüllein, J.; Walther, T.; Sellner, L.; Wu, B.; Rosenquist, R.; Oakes, C.C.; Dietrich, S.; Huber, W.; et al. Subgroup-specific gene expression profiles and mixed epistasis in chronic lymphocytic leukemia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, S.; Chen, S.; Chen, G.; Zhang, R.; Li, J.; Qu, J. SF3B1 mutation is a prognostic factor in chronic lymphocytic leukemia: A meta-analysis. Oncotarget 2017, 8, 69916–69923. [Google Scholar] [CrossRef]
- Jain, P.; Keating, M.; Wierda, W.; Estrov, Z.; Ferrajoli, A.; Jain, N.; George, B.; James, D.; Kantarjian, H.; Burger, J.; et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood 2015, 125, 2062–2067. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.J.; Rodríguez, D.; Villamor, N.; Kwarciak, A.; Tejedor, J.R.; Valcárcel, J.; López-Guillermo, A.; Martínez-Trillos, A.; Puente, X.S.; Campo, E.; et al. Frequent somatic mutations in components of the RNA processing machinery in chronic lymphocytic leukemia. Leukemia 2013, 27, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Peter, G.; Lührmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell 2018, 70, 265–273.e8. [Google Scholar] [CrossRef] [PubMed]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef]
- Xargay-Torrent, S.; López-Guerra, M.; Rosich, L.; Montraveta, A.; Roldán, J.; Rodríguez, V.; Villamor, N.; Aymerich, M.; Lagisetti, C.; Webb, T.R.; et al. The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget 2015, 6, 22734–22749. [Google Scholar] [CrossRef]
- Larrayoz, M.; Blakemore, S.J.; Dobson, R.C.; Blunt, M.D.; Rose-Zerilli, M.J.J.; Walewska, R.; Duncombe, A.; Oscier, D.; Koide, K.; Forconi, F.; et al. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 2016, 30, 351–360. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Kumar, D.; Villa, R.; La Clair, J.J.; Benner, C.; Sasik, R.; Jones, H.; Ghia, E.M.; Rassenti, L.Z.; Kipps, T.J.; et al. Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica 2015, 100, 945–954. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Valentin, R.; Regis, F.F.D.; Sun, J.; Yin, S.; Werner, L.; Deng, J.; Gruber, M.; Wong, J.; Zheng, M.; et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight 2018, 3, e121438. [Google Scholar] [CrossRef]
- Eskens, F.A.L.M.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; De Jonge, M.J.A.; Mizui, Y.; Wiemer, E.A.C.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).