
Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know?





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
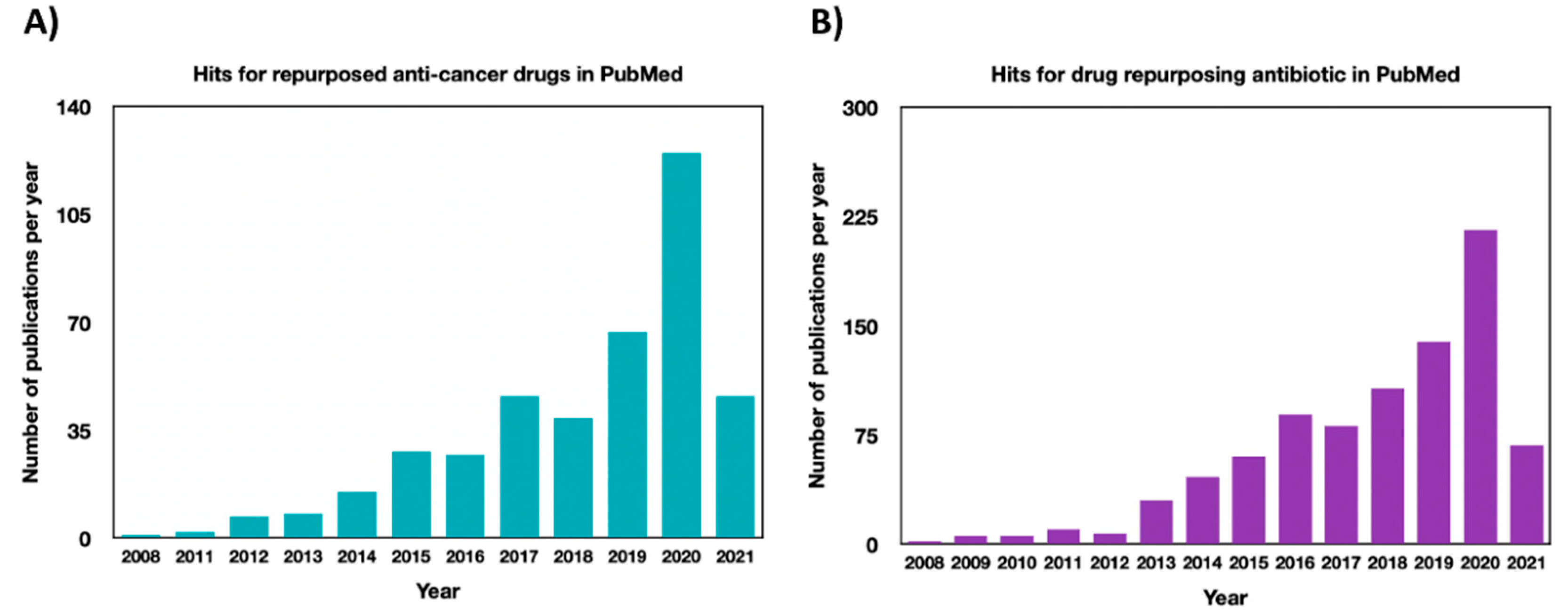
1. Introduction
Repurposing Drugs in Oncology
2. Repurposing of Antimicrobial Agents
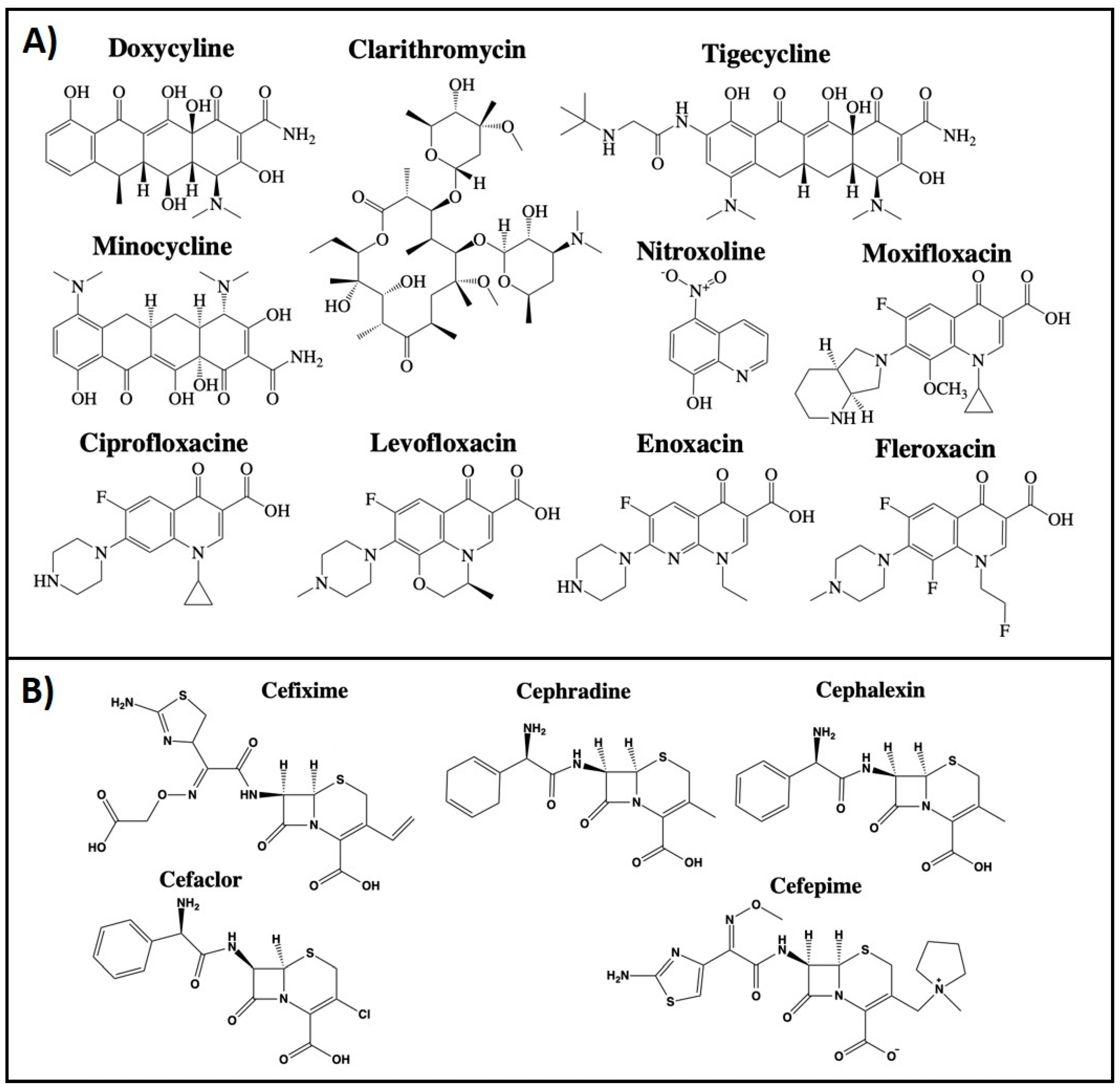
2.1. Antibiotics
2.1.1. Clarithromycin
2.1.2. Doxycycline
2.1.3. Minocycline
2.1.4. Tigecycline
2.1.5. Nitroxoline
2.1.6. Cephalosporins
2.1.7. Fluoroquinolones
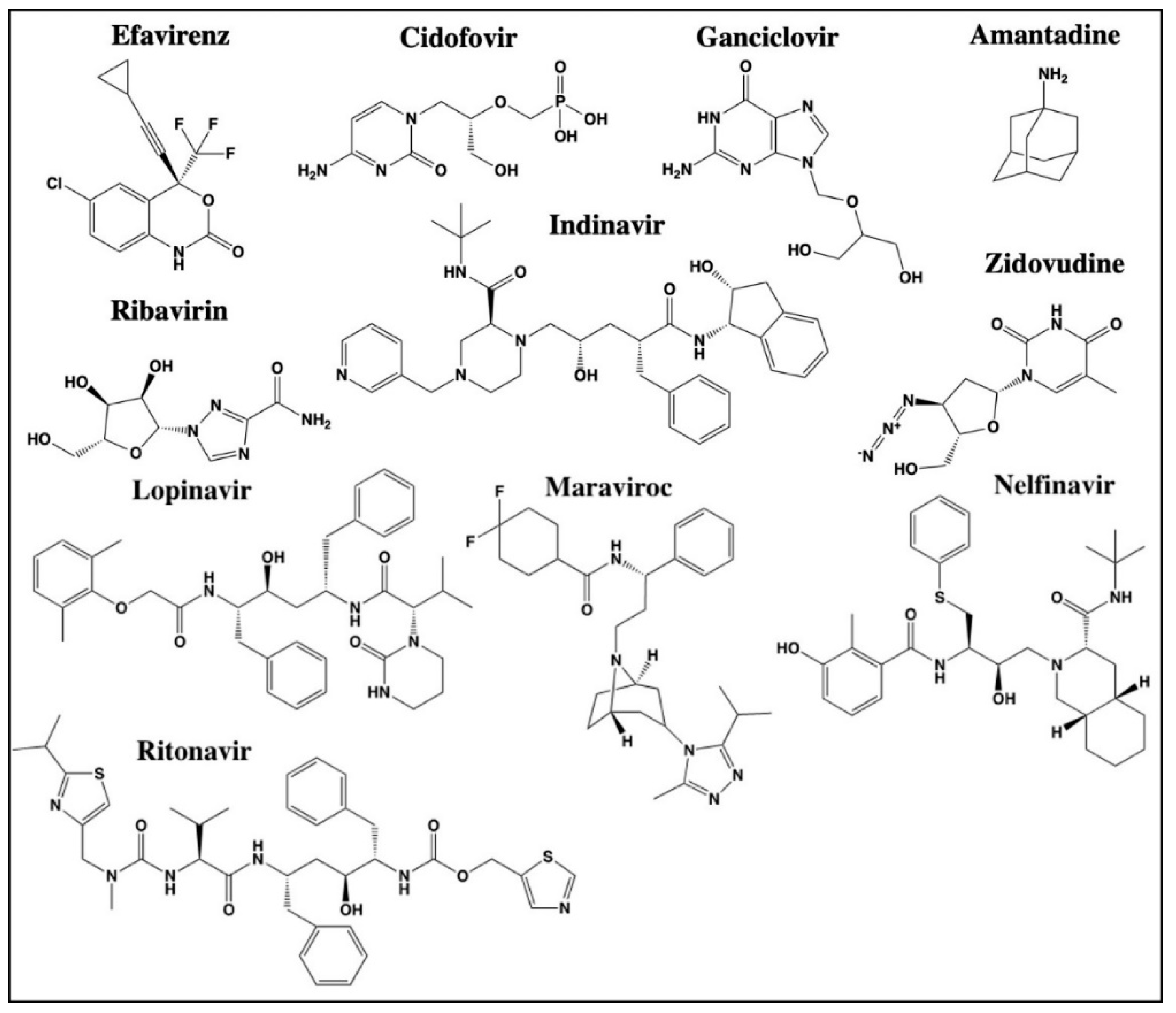
2.2. Antivirals
2.2.1. Ganciclovir
2.2.2. Lopinavir
2.2.3. Indinavir
2.2.4. Cidofovir
2.2.5. Efavirenz
2.2.6. Maraviroc
2.2.7. Nelfinavir
2.2.8. Ritonavir
2.2.9. Ribavirin
2.2.10. Zidovudine
2.2.11. Amantadine
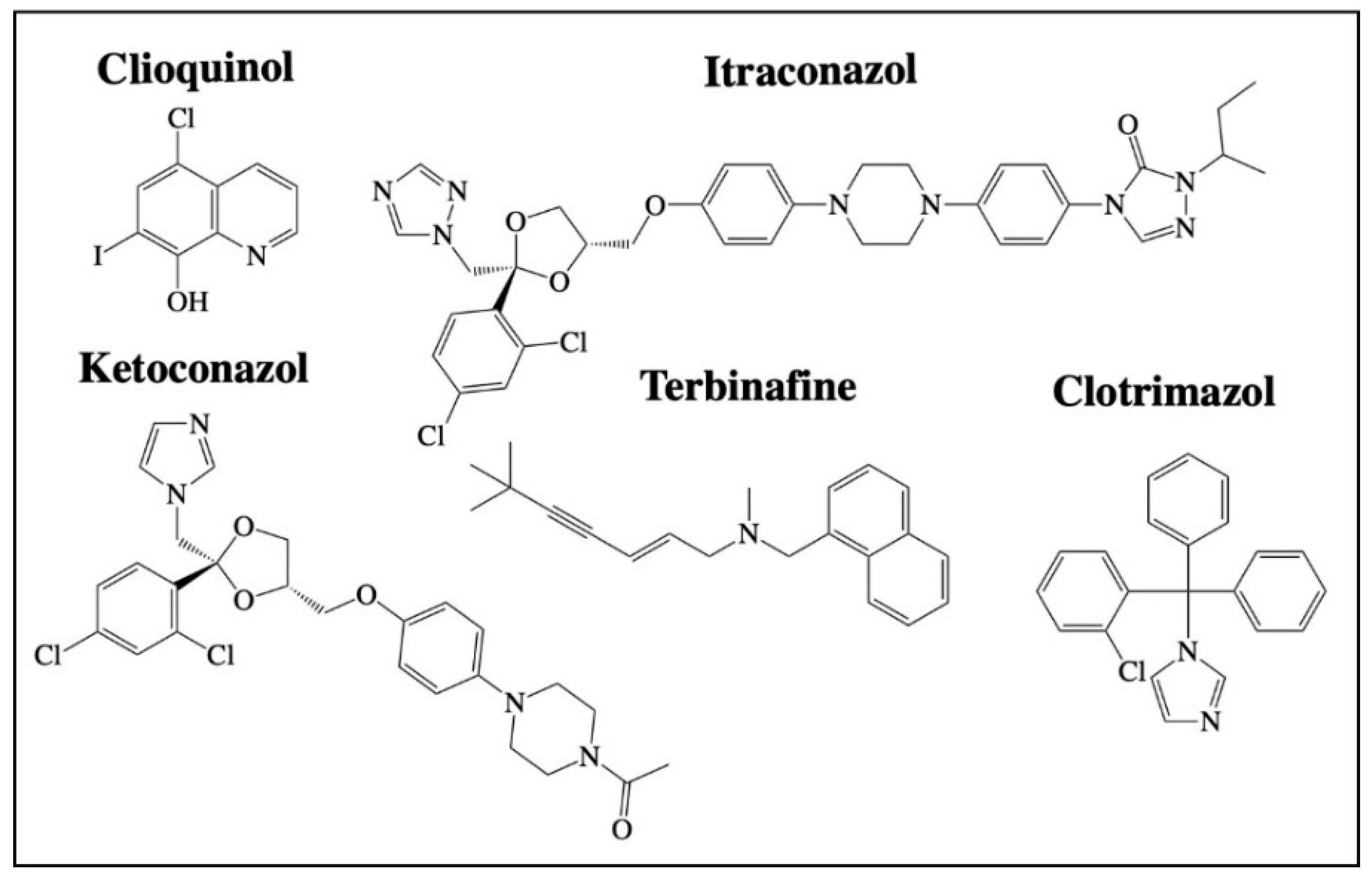
2.3. Antifungals
2.3.1. Itraconazole
2.3.2. Ketoconazole
2.3.3. Clioquinol
2.3.4. Clotrimazole
2.3.5. Terbinafine
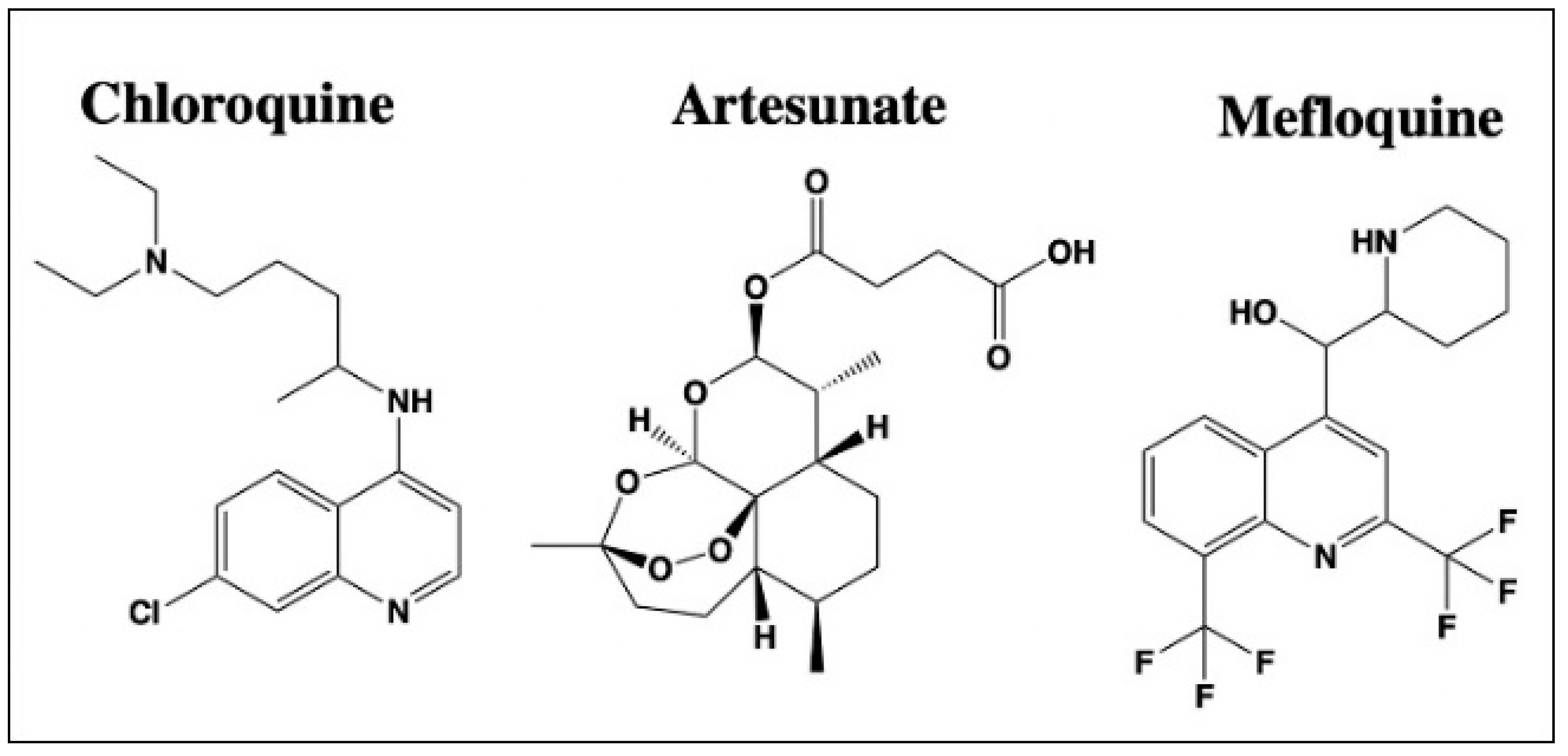
2.4. Anti-Malarial Drugs
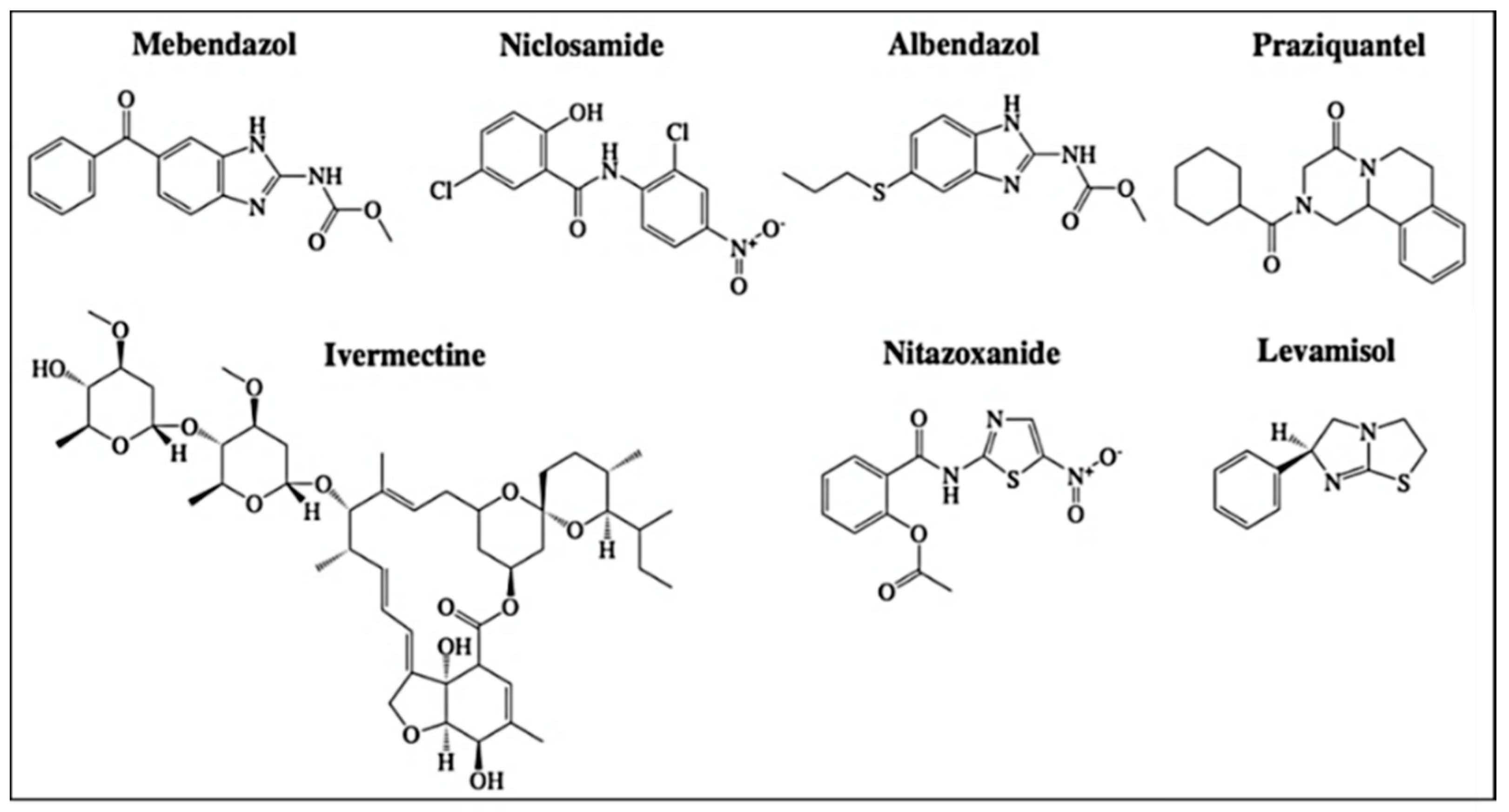
2.5. Anthelmintic Agents
2.5.1. Mebendazole, Niclosamide, Albendazole, and Ivermectin
2.5.2. Ivermectin
2.5.3. Nitazoxanide
2.5.4. Praziquantel
2.5.5. Levamisole
2.5.6. Pyrvinium
3. Drug Repurposing Pros and Cons
4. Conclusions and Future Prospects
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2017, 175, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Szabo, C. Inventing new therapies without reinventing the wheel: The power of drug repurposing. Br. J. Pharmacol. 2018, 175, 165–167. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repur-posing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Khanna, I. Drug discovery in pharmaceutical industry: Productivity challenges and trends. Drug Discov. Today 2012, 17, 1088–1102. [Google Scholar] [CrossRef]
- Palve, V.; Liao, Y.; Rix, L.L.R.; Rix, U. Turning liabilities into opportunities: Off-target based drug repurposing in cancer. Semin. Cancer Biol. 2021, 68, 209–229. [Google Scholar] [CrossRef]
- Boguski, M.S.; Mandl, K.D.; Sukhatme, V.P. Repurposing with a Difference. Science 2009, 324, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Aubé, J. Drug Repurposing and the Medicinal Chemist. ACS Med. Chem. Lett. 2012, 3, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Carley, D.W. Drug repurposing: Identify, develop and commercialize new uses for existing or abandoned drugs. Part I. IDrugs 2005, 8, 306–309. [Google Scholar] [PubMed]
- Carley, D.W. Drug repurposing: Identify, develop and commercialize new uses for existing or abandoned drugs. Part II. IDrugs 2005, 8, 310–313. [Google Scholar] [PubMed]
- Gelijns, A.C.; Rosenberg, N.; Moskowitz, A.J. Pancreatitis and mutations of the cystic fibrosis gene. N. Engl. J. Med. 1998, 339, 687–698. [Google Scholar]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by re-purposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nature re-views. Drug Dis. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.C.; Ekins, S.; Williams, A.J.; Tropsha, A. A bibliometric review of drug repurposing. Drug Discov. Today 2018, 23, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Norton, L.; Albanell, J.; Kim, Y.M.; Mendelsohn, J. Recombinant humanized an-ti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xeno-grafts. Cancer Res. 1998, 58, 2825–2831. [Google Scholar] [PubMed]
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Müller, M.; Druker, B.J.; Lydon, N.B. In-hibition of the Abl pro-tein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56, 100–104. [Google Scholar] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Pegram, M.D.; Lipton, A.; Hayes, D.F.; Weber, B.L.; Baselga, J.M.; Tripathy, D.; Baly, D.; Baughman, S.A.; Twaddell, T.; Glaspy, J.A.; et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chem-otherapy treatment. J. Clin. Oncol. 1998, 16, 2659–2671. [Google Scholar] [CrossRef]
- A Engelman, J.; Settleman, J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr. Opin. Genet. Dev. 2008, 18, 73–79. [Google Scholar] [CrossRef]
- Lovly, C.; Shaw, A.T. Molecular Pathways: Resistance to Kinase Inhibitors and Implications for Therapeutic Strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.D.; Chen, W.Y. Hiding in plain view: The potential for commonly used drugs to re-duce breast cancer mortality. Breast Cancer Res. 2012, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Mestres, J.; Gregori-Puigjané, E.; Valverde, S.; Solé, R.V. Data completeness—the Achilles heel of drug-target networks. Nat. Biotechnol. 2008, 26, 983–984. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Moulder, S.L.; Ueno, N.T.; Wheler, J.J.; Meric-Bernstam, F.; Kurzrock, R.; Janku, F. Challenges and perspective of drug repurposing strategies in early phase clinical trials. Oncoscience 2015, 2, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Gns, H.S.; Gr, S.; Murahari, M.; Krishnamurthy, M. An update on Drug Repurposing: Re-written saga of the drug’s fate. Biomed. Pharmacother. 2019, 110, 700–716. [Google Scholar] [CrossRef] [PubMed]
- Ilmer, M.; Westphalen, C.B.; Niess, H.; D’Haese, J.G.; Angele, M.K.; Werner, J.; Renz, B.W. Repurposed Drugs in Pancreatic Ductal Adenocarcinoma: An Update. Cancer J. 2019, 25, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Rica Capistrano, I.; Crispino, S.; Gyawali, B.; Rooman, I.; van Nuffel, A.M.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The repur-posing drugs in oncology database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef]
- Murray, J.C.; Levy, B. Repurposed Drugs Trials by Cancer Type: Lung Cancer. Cancer J. 2019, 25, 127–133. [Google Scholar] [CrossRef]
- Bouche, G.; Pantziarka, P.; Meheus, L. Beyond aspirin and metformin: The untapped potential of drug repurposing in oncology. Eur. J. Cancer 2017, 72, S121–S122. [Google Scholar] [CrossRef]
- Tsavaris, N.; Kosmas, C.; Vadiaka, M.; Kanelopoulos, P.; Boulamatsis, D. Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br. J. Cancer 2002, 87, 21–27. [Google Scholar] [CrossRef]
- Koenig, H.; Patel, A. Biochemical basis for fluorouracil neurotoxicity. The role of Krebs cycle inhibition by fluoroacetate. Arch. Neurol. 1970, 23, 155–160. [Google Scholar] [CrossRef]
- Lalami, Y.; Paesmans, M.; Aoun, M.; Munoz-Bermeo, R.; Reuss, K.; Cherifi, S.; Alexopoulos, C.G.; Klastersky, J. A prospective randomised evaluation of G-CSF or G-CSF plus oral antibi-otics in chemotherapy-treated patients at high risk of developing febrile neutropenia. Support Care Cancer 2004, 12, 725–730. [Google Scholar] [CrossRef]
- Luyt, C.-E.; Bréchot, N.; Trouillet, J.-L.; Chastre, J. Antibiotic stewardship in the intensive care unit. Crit. Care 2014, 18, 480. [Google Scholar] [CrossRef]
- Champney, W.S.; Burdine, R. Azithromycin and clarithromycin inhibition of 50S ribosomal subunit formation in Staphy-lococcus aureus cells. Curr. Microbiol. 1998, 36, 119–123. [Google Scholar] [CrossRef]
- Peters, D.H.; Clissold, S.P. Clarithromycin. A review of its antimicrobial activity, pharmacoki-netic properties and therapeutic potential. Drugs 1992, 44, 117–164. [Google Scholar] [CrossRef]
- Mikasa, K.; Sawaki, M.; Kita, E.; Hamada, K.; Teramoto, S.; Sakamoto, M.; Maeda, K.; Konishi, M.; Narita, N. Significant survival benefit to patients with advanced non-small-cell lung cancer from treatment with clarithromycin. Chemotherapy 1997, 43, 288–296. [Google Scholar] [CrossRef]
- Saad, A.S.; Shaheen, S.M.; Elhamamsy, M.H.; Badary, O.A. An open-label randomized con-trolled phase II study of clar-ithromycin (CL) plus CVP in patients (pts) with previously untreated stage III/IV indolent non Hodgkin lymphoma (NHL). JCO 2014, 32, e19510. [Google Scholar] [CrossRef]
- Hamada, K.; Mikasa, K.; Yunou, Y.; Kurioka, T.; Majima, T.; Narita, N.; Kita, E. Adjuvant effect of clarithromycin on chemo-therapy for murine lung cancer. Chemotherapy 2000, 46, 49–61. [Google Scholar] [CrossRef]
- Moriya, S.; Che, X.-F.; Komatsu, S.; Abe, A.; Kawaguchi, T.; Gotoh, A.; Inazu, M.; Tomoda, A.; Miyazawa, K. Macrolide anti-biotics block autophagy flux and sensitize to bortezomib via en-doplasmic reticulum stress-mediated CHOP induction in myeloma cells. Int. J. Oncol. 2013, 42, 1541–1550. [Google Scholar] [CrossRef]
- Sassa, K.; Mizushima, Y.; Fujishita, T.; Oosaki, R.; Kobayashi, M. Therapeutic effect of clar-ithromycin on a transplanted tumor in rats. Antimicrob. Agents Chemother. 1999, 43, 67–72. [Google Scholar] [CrossRef]
- Nakamura, N.; Kikukawa, Y.; Takeya, M.; Mitsuya, M.; Hata, H. Clarithromycin attenuates autophagy in myeloma cells. Int. J. Oncol. 2010, 37, 815–820. [Google Scholar]
- Coleman, M.; Leonard, J.; Lyons, L.; Pekle, K.; Nahum, K.; Pearse, R.; Niesvizky, R.; Michaeli, J. BLT-D (clarithromycin Biaxin, low-dose thalidomide, and dexamethasone) for the treatment of myeloma and Waldenström’s macroglobulinemia. Leuk. Lymphoma 2002, 43, 1777–1782. [Google Scholar] [CrossRef]
- Schafranek, L.; Leclercq, T.M.; White, D.L.; Hughes, T.P. Clarithromycin enhances da-satinib-induced cell death in chronic myeloid leukemia cells, by inhibition of late stage au-tophagy. Leuk. Lymphoma 2013, 54, 198–201. [Google Scholar] [CrossRef]
- Komatsu, S.; Miyazawa, K.; Moriya, S.; Takase, A.; Naito, M.; Inazu, M.; Kohno, N.; Itoh, M.; Tomoda, A. Clarithromycin enhances bortezomib-induced cytotoxicity via endoplasmic retic-ulum stress-mediated CHOP (GADD153) induction and autophagy in breast cancer cells. Int. J. Oncol. 2012, 40, 1029–1039. [Google Scholar] [CrossRef]
- Komatsu, S.; Moriya, S.; Che, X.-F.; Yokoyama, T.; Kohno, N.; Miyazawa, K. Combined treatment with SAHA, bortezomib, and clarithromycin for concomitant targeting of aggresome formation and intracellular proteolytic pathways enhances ER stress-mediated cell death in breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 437, 41–47. [Google Scholar] [CrossRef]
- Yatsunami, J.; Turuta, N.; Wakamatsu, K.; Hara, N.; Hayashi, S. Clarithromycin is a potent inhibitor of tumor-induced an-giogenesis. Res. Exp. Med. 1997, 197, 189–197. [Google Scholar] [CrossRef]
- Nakajima, M.; Welch, D.R.; Wynn, D.M.; Tsuruo, T.; Nicolson, G.L. Serum and plasma M(r) 92,000 progelatinase levels correlate with spontaneous metastasis of rat 13762NF mammary adenocarcinoma. Cancer Res. 1993, 53, 5802–5807. [Google Scholar]
- Welch, D.R.; Fabra, A.; Nakajima, M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc. Natl. Acad. Sci. USA 1990, 87, 7678–7682. [Google Scholar] [CrossRef]
- Chukwudi, C.U. rRNA Binding Sites and the Molecular Mechanism of Action of the Tetracy-clines. Antimicrob. Agents Chemother. 2016, 60, 4433–4441. [Google Scholar] [CrossRef]
- Doxycyclin. Available online: https://www.gelbe-liste.de/wirkstoffe/Doxycyclin_30#Pharmakologie (accessed on 18 March 2021).
- Lambs, L.; Venturim, M.; Révérend, B.D.-L.; Kozlowski, H.; Berthon, G. Metal ion-tetracycline interactions in biological fluids: Part 8. Potentiometric and spectroscopic studies on the formation of Ca(II) and Mg(II) complexes with 4-dedimethylamino-tetracycline and 6-desoxy-6-dem. J. Inorg. Biochem. 1988, 33, 193–209. [Google Scholar] [CrossRef]
- Onoda, T.; Ono, T.; Dhar, D.K.; Yamanoi, A.; Fujii, T.; Nagasue, N. Doxycycline inhibits cell proliferation and invasive po-tential: Combination therapy with cyclooxygenase-2 inhibitor in human colorectal cancer cells. J. Lab. Clin. Med. 2004, 143, 207–216. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Li, L.; Li, C. Doxycycline inhibits proliferation and induces apoptosis of both human papillomavirus positive and negative cervical cancer cell lines. Can. J. Physiol. Pharmacol. 2016, 94, 526–533. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, L.; Zhang, F.; Vlashi, E. Doxycycline inhibits the cancer stem cell phenotype and epithelial-to-mesenchymal transition in breast cancer. Cell Cycle 2017, 16, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an Inhibitor of Mitochondrial Biogenesis, Effectively Reduces Cancer Stem Cells (CSCs) in Early Breast Cancer Patients: A Clinical Pilot Study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.; Sotgia, F.; Lisanti, M. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Fiorillo, M.; Chadwick, A.; Ozsvari, B.; Reeves, K.J.; Smith, D.L.; Clarke, R.; Howell, S.; Cappello, A.R.; Martinez-Outschoorn, U.; et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget 2015, 6, 14005–14025. [Google Scholar] [CrossRef]
- Garrido-Mesa, N.; Zarzuelo, A.; Gálvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Minocyclin. Available online: https://www.gelbe-liste.de/wirkstoffe/Minocyclin_1823 (accessed on 22 March 2021).
- Nelson, M.L. Chemical and Biological Dynamics of Tetracyclines. Adv. Dent. Res. 1998, 12, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Pourgholami, M.H.; Mekkawy, A.H.; Badar, S.; Morris, D.L. Minocycline inhibits growth of epi-thelial ovarian cancer. Gynecologic Oncol. 2012, 125, 433–440. [Google Scholar] [CrossRef]
- Ataie-Kachoie, P.; Badar, S.; Morris, D.L.; Pourgholami, M.H. Minocycline targets the NF-κB Nexus through suppression of TGF-β1-TAK1-IκB signaling in ovarian cancer. Mol. Cancer Res. 2013, 11, 1279–1291. [Google Scholar] [CrossRef]
- Ataie-Kachoie, P.; Morris, D.L.; Pourgholami, M.H. Minocycline suppresses interleukine-6, its receptor system and signaling pathways and impairs migration, invasion and adhesion ca-pacity of ovarian cancer cells: In vitro and in vivo studies. PLoS ONE 2013, 8, e60817. [Google Scholar] [CrossRef]
- Niu, G.; Liao, Z.; Cai, L.; Wei, R.; Sun, L. The Combined Effects of Celecoxib and Minocycline Hydrochloride on Inhibiting the Osseous Metastasis of Breast Cancer in Nude Mice. Cancer Biotherapy Radiopharm. 2008, 23, 469–476. [Google Scholar] [CrossRef]
- Markovic, D.; Vinnakota, K.; van Rooijen, N.; Kiwit, J.; Synowitz, M.; Glass, R.; Kettenmann, H. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain, Behav. Immun. 2011, 25, 624–628. [Google Scholar] [CrossRef]
- Liu, W.-T.; Lin, C.-H.; Hsiao, M.; Gean, P.-W. Minocycline inhibits the growth of glioma by in-ducing autophagy. Autophagy 2011, 7, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Tigecycline. Available online: https://www.gelbe-liste.de/wirkstoffe/Tigecyclin_49119 (accessed on 19 March 2021).
- Beabout, K.; Hammerstrom, T.G.; Perez, A.M.; Magalhaes, B.D.F.; Prater, A.G.; Clements, T.; Arias, C.A.; Saxer, G.; Shamoo, Y. The Ribosomal S10 Protein Is a General Target for Decreased Tigecycline Susceptibility. Antimicrob. Agents Chemother. 2015, 59, 5561–5566. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Investig. 2016, 126, 3739–3757. [Google Scholar] [CrossRef]
- Xu, Z.; Yan, Y.; Li, Z.; Qian, L.; Gong, Z. The Antibiotic Drug Tigecycline: A Focus on its Promising Anticancer Properties. Front. Pharmacol. 2016, 7, 473. [Google Scholar] [CrossRef]
- Hu, H.; Dong, Z.; Tan, P.; Zhang, Y.; Liu, L.; Yang, L.; Liu, Y.; Cui, H. Antibiotic drug tigecycline inhibits melanoma progression and metastasis in a p21CIP1/Waf1-dependent manner. Oncotarget 2015, 7, 3171–3185. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, W.; Huang, Q.; Wang, J.; Wang, Y.; Li, H.; Fu, X. Tigecycline as a dual inhibitor of retinoblastoma and angio-genesis via inducing mitochondrial dysfunctions and oxidative damage. Sci. Rep. 2018, 8, 11747. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Zhang, Y.; Wang, W.; Wu, J.; Yang, Q.; Xu, W.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Inhibition of autophagy enhances the antitumour activity of tigecycline in multiple myeloma. J. Cell. Mol. Med. 2018, 22, 5955–5963. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, Y. Inhibition of mitochondrial translation as a therapeutic strategy for human ovarian cancer to overcome chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378. [Google Scholar] [CrossRef]
- Zhong, X.; Zhao, E.; Tang, C.; Zhang, W.; Tan, J.; Dong, Z.; Ding, H.-F.; Cui, H. Antibiotic drug tigecycline reduces neuro-blastoma cells proliferation by inhibiting Akt activation in vitro and in vivo. Tumour Biol. 2016, 37, 7615–7623. [Google Scholar] [CrossRef]
- Ren, A.; Qiu, Y.; Cui, H.; Fu, G. Tigecycline exerts an antitumoral effect in oral squamous cell carcinoma. Oral Dis. 2015, 21, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef]
- Norberg, E.; Lako, A.; Chen, P.-H.; A Stanley, I.; Zhou, F.; Ficarro, S.B.; Chapuy, B.; Chen, L.; Rodig, S.; Shin, D.; et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ. 2016, 24, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Song, M.; Zhou, M.; Hu, Y. Antibiotic tigecycline enhances cisplatin activity against human hepatocellular carcinoma through inducing mitochondrial dysfunction and oxidative damage. Biochem. Biophys. Res. Commun. 2017, 483, 17–23. [Google Scholar] [CrossRef]
- Tang, C.; Yang, L.; Jiang, X.; Xu, C.; Wang, M.; Wang, Q.; Zhou, Z.; Xiang, Z.; Cui, H. Antibiotic drug tigecycline inhibited cell proliferation and induced autophagy in gastric cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, N.; He, B.; Pan, C.; Lan, Y.; Zhou, H.; Liu, X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017, 36, 1–14. [Google Scholar] [CrossRef]
- Andrea, A.D.; Gritti, I.; Nicoli, P.; Giorgio, M.; Doni, M.; Conti, A.; Bianchi, V.; Casoli, L.; Sabò, A.; Mironov, A.; et al. The mitochondrial translation machinery as a therapeutic target in Myc-driven lymphomas. Oncotarget 2016, 7, 72415–72430. [Google Scholar] [CrossRef]
- Li, H.; Jiao, S.; Li, X.; Banu, H.; Hamal, S.; Wang, X. Therapeutic effects of antibiotic drug tigecycline against cervical squamous cell carcinoma by inhibiting Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 2015, 467, 14–20. [Google Scholar] [CrossRef]
- Fu, X.; Liu, W.; Huang, Q.; Wang, Y.; Li, H.; Xiong, Y. Targeting mitochondrial respiration se-lectively sensitizes pediatric acute lymphoblastic leukemia cell lines and patient samples to standard chemotherapy. Am. J. Cancer Res. 2017, 7, 2395–2405. [Google Scholar]
- Jia, X.; Gu, Z.; Chen, W.; Jiao, J. Tigecycline targets nonsmall cell lung cancer through inhi-bition of mitochondrial function. Fundam. Clin. Pharmacol. 2016, 30, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Oran, A.R.; Adams, C.M.; Zhang, X.-Y.; Gennaro, V.J.; Pfeiffer, H.K.; Mellert, H.S.; Seidel, H.E.; Mascioli, K.; Kaplan, J.; Gaballa, M.R.; et al. Multi-focal control of mitochondrial gene expression by oncogenic MYC provides potential therapeutic targets in cancer. Oncotarget 2016, 7, 72395–72414. [Google Scholar] [CrossRef] [PubMed]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Nitroxolin. Available online: https://www.gelbe-liste.de/wirkstoffe/Nitroxolin_822 (accessed on 22 March 2021).
- Wijma, R.A.; Huttner, A.; Koch, B.C.P.; Mouton, J.W.; Muller, A.E. Review of the pharmacoki-netic properties of nitrofurantoin and nitroxoline. J. Antimicrobe. Chemother. 2018, 73, 2916–2926. [Google Scholar] [CrossRef]
- Pelletier, C.; Prognon, P.; Bourlioux, P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob. Agents Chemother. 1995, 39, 707–713. [Google Scholar] [CrossRef]
- Kresken, M.; Körber-Irrgang, B. In vitro activity of nitroxoline against Escherichia coli urine isolates from outpatient de-partments in Germany. Antimicrob. Agents Chemother. 2014, 58, 7019–7020. [Google Scholar] [CrossRef]
- Shim, J.S.; Matsui, Y.; Bhat, S.; Nacev, B.A.; Xu, J.; Bhang, H.-E.C.; Dhara, S.; Han, K.C.; Chong, C.R.; Pomper, M.G.; et al. Effect of Nitroxoline on Angiogenesis and Growth of Human Bladder Cancer. J. Natl. Cancer Inst. 2010, 102, 1855–1873. [Google Scholar] [CrossRef]
- Jiang, H.; Taggart, J.E.; Zhang, X.; Benbrook, D.M.; Lind, S.E.; Ding, W.-Q. Nitroxoline (8-hydroxy-5-nitroquinoline) is more a potent anti-cancer agent than clioquinol (5-chloro-7-iodo-8-quinoline). Cancer Lett. 2011, 312, 11–17. [Google Scholar] [CrossRef]
- Zhang, Q.I.; Wang, S.; Yang, D.; Pan, K.; Li, L.; Yuan, S. Preclinical pharmacodynamic evaluation of antibiotic nitroxoline for anticancer drug repurposing. Oncol. Lett. 2016, 11, 3265–3272. [Google Scholar] [CrossRef]
- Sosič, I.; Mirković, B.; Arenz, K.; Stefane, B.; Kos, J.; Gobec, S. Development of new cathepsin B inhibitors: Combining bioisosteric replacements and structure-based design to explore the structure-activity relationships of nitroxoline derivatives. J. Med. Chem. 2013, 56, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Lazovic, J.; Guo, L.; Nakashima, J.; Mirsadraei, L.; Yong, W.; Kim, H.J.; Ellingson, B.; Wu, H.; Pope, W.B. Nitroxoline induces apoptosis and slows glioma growth in vivo. Neuro-Oncology 2014, 17, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-L.; Hsu, L.-C.; Leu, W.-J.; Chen, C.-S.; Guh, J.-H. Repurposing of nitroxoline as a potential anticancer agent against human prostate cancer—A crucial role on AMPK/mTOR signaling pathway and the interplay with Chk2 activation. Oncotarget 2015, 6, 39806–39820. [Google Scholar] [CrossRef]
- Chapman, T.M.; Perry, C.M. Cefepime: A review of its use in the management of hospitalized patients with pneumonia. Am. J. Respir. Med. Drugs Devices Other Interv. 2003, 2, 75–107. [Google Scholar] [CrossRef]
- Efimova, E.V.; Mauceri, H.J.; Golden, D.W.; Labay, E.; Bindokas, V.P.; Darga, T.E.; Chakraborty, C.; Barreto-Andrade, J.C.; Crawley, C.; Sutton, H.G.; et al. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res. 2010, 70, 6277–6282. [Google Scholar] [CrossRef]
- Tanrisever, B.; Santella, P.J. Cefadroxil. A review of its antibacterial, pharmacokinetic and therapeutic properties in comparison with cephalexin and cephradine. Drugs 1986, 32 (Suppl. S3), 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Jacobs, M.R.; Fritsche, T.R. Review of the spectrum and potency of orally administered cephalosporins and amoxicillin/clavulanate. Diagn. Microbiol. Infect. Dis. 2007, 57, 5S–12S. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Miick, C. Characterization of penicillin-binding protein 2 of Staphylococcus aureus: Deacylation reaction and identification of two penicillin-binding peptides. Antimicrob. Agents Chemother. 1992, 36, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Klastersky, J.; Daneau, D.; Weerts, D. Cephradine. Antibacterial activity and clinical effectiveness. Chemotherapy 1973, 18, 191–204. [Google Scholar] [CrossRef]
- Markham, A.; Brogden, R.N. Cefixime. A review of its therapeutic efficacy in lower respiratory tract infections. Drugs 1995, 49, 1007–1022. [Google Scholar] [CrossRef]
- Labay, E.; Mauceri, H.J.; Efimova, E.V.; Flor, A.C.; Sutton, H.G.; Kron, S.J.; Weichselbaum, R.R. Repurposing cephalosporin antibiotics as pro-senescent radiosensitizers. Oncotarget 2016, 7, 33919–33933. [Google Scholar] [CrossRef]
- Zhang, Z.; Bi, C.; Fan, Y.; Wang, H.; Bao, Y. Cefepime, a fourth-generation cephalosporin, in complex with manganese, inhibits proteasome activity and induces the apoptosis of human breast cancer cells. Int. J. Mol. Med. 2015, 36, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L. Molecular Mechanisms of DNA Gyrase Inhibition by Quinolone Antibacterials. Charact. Porous Solids III 1994, 29A, 285–304. [Google Scholar] [CrossRef]
- Davis, R.; Markham, A.; Balfour, J.A. Ciprofloxacin. An updated review of its pharmacology, therapeutic efficacy and tolerability. Drugs 1996, 51, 1019–1074. [Google Scholar] [CrossRef] [PubMed]
- Bourikas, L.A.; Kolios, G.; Valatas, V.; Notas, G.; Drygiannakis, I.; Pelagiadis, I.; Manousou, P.; Klironomos, S.; Mouzas, I.A.; Kouroumalis, E. Ciprofloxacin decreases survival in HT-29 cells via the induction of TGF-beta1 secretion and enhances the anti-proliferative effect of 5-fluorouracil. Br. J. Pharmacol. 2009, 157, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Suresh, N.; Nagesh, H.N.; Sekhar, K.V.G.C.; Kumar, A.; Shirazi, A.N.; Parang, K. Synthesis of novel ciprofloxacin analogues and evaluation of their anti-proliferative effect on human cancer cell lines. Bioorg. Med. Chem. Lett. 2013, 23, 6292–6295. [Google Scholar] [CrossRef]
- Ebisuno, S.; Inagaki, T.; Kohjimoto, Y.; Ohkawa, T. The cytotoxic effects of fleroxacin and ciprofloxacin on transitional cell carcinoma in Vitro. Cancer 1997, 80, 2263–2267. [Google Scholar] [CrossRef]
- Beberok, A.; Rzepka, Z.; Respondek, M.; Rok, J.; Sierotowicz, D.; Wrześniok, D. GSH depletion, mitochondrial membrane breakdown, caspase-3/7 activation and DNA fragmentation in U87MG glioblastoma cells: New insight into the mechanism of cytotoxicity induced by fluoroquinolones. Eur. J. Pharmacol. 2018, 835, 94–107. [Google Scholar] [CrossRef]
- Beberok, A.; Wrześniok, D.; Minecka, A.; Rok, J.; Delijewski, M.; Rzepka, Z.; Respondek, M.; Buszman, E. Ciprofloxacin-mediated induction of S-phase cell cycle arrest and apoptosis in COLO829 melanoma cells. Pharmacol. Rep. PR 2018, 70, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Beberok, A.; Wrześniok, D.; Rok, J.; Rzepka, Z.; Respondek, M.; Buszman, E. Ciprofloxacin triggers the apoptosis of human triple-negative breast cancer MDA-MB-231 cells via the p53/Bax/Bcl-2 signaling pathway. Int. J. Oncol. 2018, 52, 1727–1737. [Google Scholar] [CrossRef]
- Sousa, E.; Graça, I.; Baptista, T.; Vieira, F.Q.; Palmeira, C.; Henrique, R.; Jerónimo, C. Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing. Epigenetics 2013, 8, 548–558. [Google Scholar] [CrossRef]
- Felicetti, T.; Cecchetti, V.; Manfroni, G. Modulating microRNA Processing: Enoxacin, the Progenitor of a New Class of Drugs. J. Med. Chem. 2020, 63, 12275–12289. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Pyles, H.M.; Diaz-Cruz, E.S.; Barton, C.E. Enoxacin and Epigallocatechin Gallate (EGCG) Act Synergistically to Inhibit the Growth of Cervical Cancer Cells in Culture. Molecules 2019, 24, 1580. [Google Scholar] [CrossRef]
- Mukherjee, P.; Mandal, E.R.; Das, S.K. Evaluation of Antiproliferative Activity of Enoxacin on a Human Breast Cancer Cell Line. Int. J. Hum. Genet. 2005, 5, 57–63. [Google Scholar] [CrossRef]
- Yadav, V.; Talwar, P. Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: An underestimated truth. Biomed. Pharmacother. 2019, 111, 934–946. [Google Scholar] [CrossRef]
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 27 May 2021).
- Viruses That Can Lead to Cancer. Available online: https://www.cancer.org/cancer/cancer-causes/infectious-agents/infections-that-can-lead-to-cancer/viruses.html (accessed on 27 May 2021).
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 19 May 2021).
- Crumpacker, C.S. Ganciclovir. N. Engl. J. Med. 1996, 335, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Eastham, J.A.; Chen, S.-H.; Sehgal, I.; Yang, G.; Timme, T.L.; Hall, S.J.; Woo, S.L.C.; Thompson, T.C. Prostate Cancer Gene Therapy: Herpes Simplex Virus Thymidine Kinase Gene Transduction Followed by Ganciclovir in Mouse and Human Prostate Cancer Models. Hum. Gene Ther. 1996, 7, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Satoh, T.; Li, R.; Shalev, M.; Gdor, Y.; Aguilar-Cordova, E.; Frolov, A.; Wheeler, T.M.; Miles, B.J.; Rauen, K.; et al. Biological response determinants in HSV-tk + ganciclovir gene therapy for prostate cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 13, 716–728. [Google Scholar] [CrossRef]
- Shao, D.; Zeng, Q.; Fan, Z.; Li, J.; Zhang, M.; Zhang, Y.; Li, O.; Chen, L.; Kong, X.; Zhang, H. Monitoring HSV-TK/ganciclovir cancer suicide gene therapy using CdTe/CdS core/shell quantum dots. Biomaterials 2012, 33, 4336–4344. [Google Scholar] [CrossRef] [PubMed]
- Määttä, A.-M.; Tenhunen, A.; Pasanen, T.; Meriläinen, O.; Pellinen, R.; Mäkinen, K.; Alhava, E.; Wahlfors, J. Non-small cell lung cancer as a target disease for herpes simplex type 1 thymidine kinase-ganciclovir gene therapy. Int. J. Oncol. 2004, 24, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, R.S.; Goa, K.L. Lopinavir/ritonavir: A review of its use in the management of HIV infection. Drugs 2003, 63, 769–802. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, V.; Plosker, G.L. Lopinavir/ritonavir: A review of its use in the management of HIV infection. Drugs 2006, 66, 1275–1299. [Google Scholar] [CrossRef] [PubMed]
- Brower, E.T.; Bacha, U.M.; Kawasaki, Y.; Freire, E. Inhibition of HIV-2 Protease by HIV-1 Protease Inhibitors in Clinical Use. Chem. Biol. Drug Des. 2008, 71, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Lopinavir. Available online: https://www.gelbe-liste.de/wirkstoffe/Lopinavir_44754 (accessed on 18 May 2021).
- Okubo, K.; Isono, M.; Asano, T.; Sato, A. Lopinavir-Ritonavir Combination Induces Endoplasmic Reticulum Stress and Kills Urological Cancer Cells. Anticancer Res. 2019, 39, 5891–5901. [Google Scholar] [CrossRef] [PubMed]
- Marima, R.; Hull, R.; Dlamini, Z.; Penny, C. Efavirenz and Lopinavir/Ritonavir Alter Cell Cycle Regulation in Lung Cancer. Front. Oncol. 2020, 10, 1693. [Google Scholar] [CrossRef] [PubMed]
- Paskas, S.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Al-Abed, Y.; He, M.; Rakocevic, S.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Lopinavir-NO, a nitric oxide-releasing HIV protease inhibitor, suppresses the growth of melanoma cells in vitro and in vivo. Investig. New Drugs 2019, 37, 1014–1028. [Google Scholar] [CrossRef]
- Piscitelli, S.C.; Burstein, A.H.; Chaitt, D.; Alfaro, R.M.; Falloon, J. Indinavir concentrations and St John’s wort. Lancet 2000, 355, 547–548. [Google Scholar] [CrossRef]
- Plosker, G.L.; Noble, S. Indinavir: A review of its use in the management of HIV infection. Drugs 1999, 58, 1165–1203. [Google Scholar] [CrossRef]
- Acosta, E.P.; Henry, K.; Baken, L.; Page, L.M.; Fletcher, C.V. Indinavir Concentrations and Antiviral Effect. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1999, 19, 708–712. [Google Scholar] [CrossRef]
- Pollak, E.B.; Parmar, M. Indinavir; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lee, A.; Saito, E.; Ekins, S.; McMurtray, A. Extracellular binding of indinavir to matrix metalloproteinase-2 and the alpha-7-nicotinic acetylcholine receptor: Implications for use in cancer treatment. Heliyon 2019, 5, e02526. [Google Scholar] [CrossRef]
- Sharma, S.; Baksi, R.; Agarwal, M. Repositioning of anti-viral drugs as therapy for cervical cancer. Pharmacol. Rep. 2016, 68, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V.; Palescandolo, E.; Spugnini, E.P.; Montesarchio, V.; De Luca, A.; Cardillo, I.; Cortese, G.; Baldi, A.; Chirianni, A. Evaluation of Antitumoral Properties of the Protease Inhibitor Indinavir in a Murine Model of Hepatocarcinoma. Clin. Cancer Res. 2006, 12, 2634–2639. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sgadari, C.; Barillari, G.; Toschi, E.; Carlei, D.; Bacigalupo, I.; Baccarini, S.; Palladino, C.; Leone, P.; Bugarini, R.; Malavasi, L.; et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat. Med. 2002, 8, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Sgadari, C.; Monini, P.; Barillari, G.; Ensoli, B. Use of HIV protease inhibitors to block Kaposi’s sarcoma and tumour growth. Lancet Oncol. 2003, 4, 537–547. [Google Scholar] [CrossRef]
- Monini, P.; Sgadari, C.; Toschi, E.; Barillari, G.; Ensoli, B. Antitumour effects of antiretroviral therapy. Nat. Rev. Cancer 2004, 4, 861–875. [Google Scholar] [CrossRef]
- Lea, A.P.; Bryson, H.M. Cidofovir. Drugs 1996, 52, 225–230, discussion 231. [Google Scholar] [CrossRef]
- Hadaczek, P.; Ozawa, T.; Soroceanu, L.; Yoshida, Y.; Matlaf, L.; Singer, E.; Fiallos, E.; James, C.D.; Cobbs, C.S. Cidofovir: A Novel Antitumor Agent for Glioblastoma. Clin. Cancer Res. 2013, 19, 6473–6483. [Google Scholar] [CrossRef]
- De Schutter, T.; Andrei, G.; Topalis, D.; Naesens, L.; Snoeck, R. Cidofovir selectivity is based on the different response of normal and cancer cells to DNA damage. BMC Med. Genom. 2013, 6, 18. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, X.; Chen, W.; Gao, Z.; Liu, A.; Guo, J.; Yan, Z.; Dou, Y.; Wang, H.; Li, Y. Effects of cidofovir on human papillomavirus-positive cervical cancer cells xenografts in nude mice. Oncol. Res. 2010, 18, 519–527. [Google Scholar] [CrossRef]
- Deutsch, E.; Haie-Meder, C.; Bayar, M.A.; Mondini, M.; Laporte, M.; Mazeron, R.; Adam, J.; Varga, A.; Vassal, G.; Magné, N.; et al. Phase I trial evaluating the antiviral agent Cidofovir in combination with chemoradiation in cervical cancer patients. Oncotarget 2016, 7, 25549–25557. [Google Scholar] [CrossRef]
- Efavirenz. Available online: https://www.ratiopharm.de/assets/products/de/label/Efavirenz%20Teva%20600%20mg%20Filmtabletten%20-%203.pdf?pzn=6990133 (accessed on 23 May 2021).
- Adkins, J.C.; Noble, S. Efavirenz. Drugs 1998, 56, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Harrer, T.; Büttner, M.; Schwegler, M.; Erber, S.; Fietkau, R.; Distel, L.V. Cytotoxic effect of efavirenz is selective against cancer cells and associated with the cannabinoid system. Aids 2013, 27, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Erber, S.; Harrer, T.; Klinker, H.; Roth, T.; Parsch, H.; Fiebig, N.; Fietkau, R.; Distel, L.V. Efavirenz Has the Highest Anti-Proliferative Effect of Non-Nucleoside Reverse Transcriptase Inhibitors against Pancreatic Cancer Cells. PLoS ONE 2015, 10, e0130277. [Google Scholar] [CrossRef]
- Brüning, A.; Jückstock, J.; Kost, B.; Tsikouras, P.; Weissenbacher, T.; Mahner, S.; Mylonas, I. Induction of DNA damage and apoptosis in human leukemia cells by efavirenz. Oncol. Rep. 2016, 37, 617–621. [Google Scholar] [CrossRef]
- Hecht, M.; Harrer, T.; Körber, V.; Sarpong, E.O.; Moser, F.; Fiebig, N.; Schwegler, M.; Stürzl, M.; Fietkau, R.; Distel, L.V. Cytotoxic effect of Efavirenz in BxPC-3 pancreatic cancer cells is based on oxidative stress and is synergistic with ionizing radiation. Oncol. Lett. 2018, 15, 1728–1736. [Google Scholar] [CrossRef]
- Houédé, N.; Pulido, M.; Mourey, L.; Joly, F.; Ferrero, J.; Bellera, C.; Priou, F.; Lalet, C.; Laroche-Clary, A.; Raffin, M.C.; et al. A Phase II Trial Evaluating the Efficacy and Safety of Efavirenz in Metastatic Castration-Resistant Prostate Cancer. oncologist 2014, 19, 1227–1228. [Google Scholar] [CrossRef]
- Sikora, M.J.; Rae, J.M.; Johnson, M.D.; Desta, Z. Efavirenz directly modulates the oestrogen receptor and induces breast cancer cell growth. HIV Med. 2010, 11, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef]
- Wang, R.; Feng, W.; Wang, H.; Wang, L.; Yang, X.; Yang, F.; Zhang, Y.; Liu, X.; Zhang, D.; Ren, Q.; et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett. 2020, 469, 151–161. [Google Scholar] [CrossRef]
- Zi, J.; Yuan, S.; Qiao, J.; Zhao, K.; Xu, L.; Qi, K.; Xu, K.; Zeng, L. Treatment with the C-C chemokine receptor type 5 (CCR5)-inhibitor maraviroc suppresses growth and induces apoptosis of acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2017, 7, 869–880. [Google Scholar]
- Pervaiz, A.; Ansari, S.; Berger, M.R.; Adwan, H. CCR5 blockage by maraviroc induces cytotoxic and apoptotic effects in colorectal cancer cells. Med Oncol. 2015, 32, 1–10. [Google Scholar] [CrossRef]
- Huang, H.; Zepp, M.; Georges, R.B.; Jarahian, M.; Kazemi, M.; Eyol, E.; Berger, M.R. The CCR5 antagonist maraviroc causes remission of pancreatic cancer liver metastasis in nude rats based on cell cycle inhibition and apoptosis induction. Cancer Lett. 2020, 474, 82–93. [Google Scholar] [CrossRef]
- Lee, E.; Fertig, E.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, N.; Borghese, C.P.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits Hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2018, 104, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS—Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Pyrko, P.; Kardosh, A.; Wang, W.; Xiong, W.; Chen, T.C.; Schönthal, A.H. HIV-1 Protease Inhibitors Nelfinavir and Atazanavir Induce Malignant Glioma Death by Triggering Endoplasmic Reticulum Stress. Cancer Res. 2007, 67, 10920–10928. [Google Scholar] [CrossRef]
- Gupta, A.K.; Li, B.; Cerniglia, G.J.; Ahmed, M.S.; Hahn, S.M.; Maity, A. The HIV Protease Inhibitor Nelfinavir Downregulates Akt Phosphorylation by Inhibiting Proteasomal Activity and Inducing the Unfolded Protein Response. Neoplasia 2007, 9, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, S.; Gills, J.J.; Mercado-Matos, J.R.; LoPiccolo, J.; Wilson, W.; Hollander, M.C.; A Dennis, P. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012, 3, e353. [Google Scholar] [CrossRef]
- Gills, J.J.; LoPiccolo, J.; Tsurutani, J.; Shoemaker, R.H.; Best, C.J.; Abu-Asab, M.; Borojerdi, J.; Warfel, N.A.; Gardner, E.R.; Danish, M.; et al. Nelfinavir, A Lead HIV Protease Inhibitor, Is a Broad-Spectrum, Anticancer Agent that Induces Endoplasmic Reticulum Stress, Autophagy, and Apoptosis In vitro and In vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Gingelmaier, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol. Ther. 2009, 8, 226–232. [Google Scholar] [CrossRef] [PubMed]
- HIV Drug Shows Efficacy in Treating Mouse Models of HER2+ Breast Cancer. JNCI J. Natl. Cancer Inst. 2012, 104. [CrossRef]
- Xiang, T.; Du, L.; Pham, P.; Zhu, B.; Jiang, S. Nelfinavir, an HIV protease inhibitor, induces apoptosis and cell cycle arrest in human cervical cancer cells via the ROS-dependent mitochondrial pathway. Cancer Lett. 2015, 364, 79–88. [Google Scholar] [CrossRef]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir Down-regulates Hypoxia-Inducible Factor 1α and VEGF Expression and Increases Tumor Oxygenation: Implications for Radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.; Abreu, M.H.; Bartosch, C.; Ricardo, S. Recycling the Purpose of Old Drugs to Treat Ovarian Cancer. Int. J. Mol. Sci. 2020, 21, 7768. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Uchihara, J.-N.; Terashima, K.; Honda, M.; Sata, T.; Ito, M.; Fujii, N.; Uozumi, K.; Tsukasaki, K.; Tomonaga, M.; et al. Efficient intervention of growth and infiltration of primary adult T-cell leukemia cells by an HIV protease inhibitor, ritonavir. Blood 2006, 107, 716–724. [Google Scholar] [CrossRef]
- Pati, S.; Pelser, C.B.; Dufraine, J.; Bryant, J.L.; Reitz, M.S., Jr.; Weichold, F.F. Antitumorigenic effects of HIV protease inhibitor ritonavir: Inhibition of Kaposi sarcoma. Blood 2002, 99, 3771–3779. [Google Scholar] [CrossRef] [PubMed]
- Srirangam, A.; Milani, M.; Mitra, R.; Guo, Z.; Rodriguez, M.; Kathuria, H.; Fukuda, S.; Rizzardi, A.; Schmechel, S.; Skalnik, D.G.; et al. The human immunodeficiency virus protease inhibitor ritonavir inhibits lung cancer cells, in part, by inhibition of survivin. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 661–670. [Google Scholar] [CrossRef]
- Srirangam, A.; Mitra, R.; Wang, M.; Gorski, J.C.; Badve, S.; Baldridge, L.A.; Hamilton, J.; Kishimoto, H.; Hawes, J.; Li, L.; et al. Effects of HIV Protease Inhibitor Ritonavir on Akt-Regulated Cell Proliferation in Breast Cancer. Clin. Cancer Res. 2006, 12, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Batchu, R.B.; Gruzdyn, O.V.; Bryant, C.S.; Qazi, A.M.; Kumar, S.; Chamala, S.; Kung, S.T.; Sanka, R.S.; Puttagunta, U.S.; Weaver, D.W.; et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals 2014, 7, 46–57. [Google Scholar] [CrossRef]
- Kumar, S.; Bryant, C.S.; Chamala, S.; Qazi, A.; Seward, S.; Pal, J.; Steffes, C.P.; Weaver, D.W.; Morris, R.; Malone, J.M.; et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol. Cancer 2009, 8, 26. [Google Scholar] [CrossRef]
- Gaedicke, S.; Firat-Geier, E.; Constantiniu, O.; Lucchiari-Hartz, M.; Freudenberg, M.; Galanos, C.; Niedermann, G. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: Induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002, 62, 6901–6908. [Google Scholar]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.; Raje, N.S.; Rosen, S.T.; et al. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef]
- Ikezoe, T.; Daar, E.S.; Hisatake, J.; Taguchi, H.; Koeffler, H.P. HIV-1 protease inhibitors decrease proliferation and induce differentiation of human myelocytic leukemia cells. Blood 2000, 96, 3553–3559. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Hisatake, Y.; Takeuchi, T.; Ohtsuki, Y.; Yang, Y.; Said, J.W.; Taguchi, H.; Koeffler, H.P. HIV-1 protease inhibitor, ritonavir: A potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004, 64, 7426–7431. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.P.; Herrmann, E.; Sarrazin, C.; Zeuzem, S. Ribavirin mode of action in chronic hepatitis C: From clinical use back to molecular mechanisms. Liver Int. 2008, 28, 1332–1343. [Google Scholar] [CrossRef]
- Jin, J.; Xiang, W.; Wu, S.; Wang, M.; Xiao, M.; Deng, A. Targeting eIF4E signaling with ribavirin as a sensitizing strategy for ovarian cancer. Biochem. Biophys. Res. Commun. 2019, 510, 580–586. [Google Scholar] [CrossRef]
- Urtishak, K.A.; Wang, L.-S.; Culjkovic-Kraljacic, B.; Davenport, J.W.; Porazzi, P.; Vincent, T.L.; Teachey, D.T.; Tasian, S.K.; Moore, J.S.; Seif, A.E.; et al. Targeting EIF4E signaling with ribavirin in infant acute lymphoblastic leukemia. Oncogene 2018, 38, 2241–2262. [Google Scholar] [CrossRef]
- Pettersson, F.; Yau, C.; Dobocan, M.C.; Culjkovic-Kraljacic, B.; Retrouvey, H.; Retrouvay, H.; Puckett, R.; Flores, L.M.; Krop, I.E.; Rousseau, C.; et al. Ribavirin treatment effects on breast cancers overexpressing eIF4E, a biomarker with prognostic specificity for luminal B-type breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2874–2884. [Google Scholar] [CrossRef]
- Tan, K.; Culjkovic, B.; Amri, A.; Borden, K.L.B. Ribavirin targets eIF4E dependent Akt survival signaling. Biochem. Biophys. Res. Commun. 2008, 375, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Volpin, F.; Casaos, J.; Sesen, J.; Mangraviti, A.; Choi, J.; Gorelick, N.; Frikeche, J.; Lott, T.; Felder, R.; Scotland, S.J.; et al. Use of an anti-viral drug, Ribavirin, as an anti-glioblastoma therapeutic. Oncogene 2017, 36, 3037–3047. [Google Scholar] [CrossRef]
- Chen, J.; Xu, X.; Chen, J. Clinically relevant concentration of anti-viral drug ribavirin selectively targets pediatric osteosarcoma and increases chemosensitivity. Biochem. Biophys. Res. Commun. 2018, 506, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Ye, J.; Song, M.; Zhou, M.; Hu, Y. Ribavirin augments doxorubicin’s efficacy in human hepatocellular carcinoma through inhibiting doxorubicin-induced eIF4E activation. J. Biochem. Mol. Toxicol. 2018, 32, e22007. [Google Scholar] [CrossRef]
- Dai, D.; Chen, H.; Tang, J.; Tang, Y. Inhibition of mTOR/eIF4E by anti-viral drug ribavirin effectively enhances the effects of paclitaxel in oral tongue squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2017, 482, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Huq, S.; Casaos, J.; Serra, R.; Peters, M.; Xia, Y.; Ding, A.S.; Ehresman, J.; Kedda, J.N.; Morales, M.; Gorelick, N.L.; et al. Repurposing the FDA-Approved Antiviral Drug Ribavirin as Targeted Therapy for Nasopharyngeal Carcinoma. Mol. Cancer Ther. 2020, 19, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Kraljacic, B.C.; Arguello, M.; Amri, A.; Cormack, G.; Borden, K. Inhibition of eIF4E with ribavirin cooperates with common chemotherapies in primary acute myeloid leukemia specimens. Leukemia 2011, 25, 1197–1200. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, J.; He, Q.; Dong, Y.; Liu, Y. Azidothymidine inhibits cell growth and telomerase activity and induces DNA damage in human esophageal cancer. Mol. Med. Rep. 2017, 15, 4055–4060. [Google Scholar] [CrossRef]
- Langtry, H.D.; Campoli-Richards, D.M. Zidovudine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1989, 37, 408–450. [Google Scholar] [CrossRef]
- Li, H.; Song, T.; Xu, W.; Yu, Y.; Xin, X.; Hui, D. Effect of 3′-Azido-3′-Deoxythymidine (AZT) on Telomerase Activity and Proliferation of HO-8910 Cell Line of Ovarian Cancer. Int. J. Biomed. Sci. IJBS 2006, 2, 34–40. [Google Scholar]
- Brown, T.; Sigurdson, E.; Rogatko, A.; Broccoli, D. Telomerase inhibition using azidothymidine in the HT-29 colon cancer cell line. Ann. Surg. Oncol. 2003, 10, 910–915. [Google Scholar] [CrossRef]
- Fang, J.-L.; Beland, F. Long-Term Exposure to Zidovudine Delays Cell Cycle Progression, Induces Apoptosis, and Decreases Telomerase Activity in Human Hepatocytes. Toxicol. Sci. 2009, 111, 120–130. [Google Scholar] [CrossRef]
- Falchetti, A.; Franchi, A.; Bordi, C.; Mavilia, C.; Masi, L.; Cioppi, F.; Recenti, R.; Picariello, L.; Marini, F.; Del Monte, F.; et al. Azidothymidine Induces Apoptosis and Inhibits Cell Growth and Telomerase Activity of Human Parathyroid Cancer Cells in Culture. J. Bone Miner. Res. 2004, 20, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Mattson, D.M.; Ahmad, I.M.; Dayal, D.; Parsons, A.D.; Aykin-Burns, N.; Li, L.; Orcutt, K.P.; Spitz, D.R.; Dornfeld, K.J.; Simons, A.L. Cisplatin combined with zidovudine enhances cytotoxicity and oxidative stress in human head and neck cancer cells via a thiol-dependent mechanism. Free Radic. Biol. Med. 2009, 46, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, C.; Kato, M.; Kuroda, D.; Ohyanagi, H. Experimental Studies on Potentiation of the Antitumor Activity of 5-Fluorouracil with 3′-Azido-3-deoxythymidine for the Gastric Cancer Cell Line MKN28in vivo. Jpn. J. Cancer Res. 1997, 88, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.S.; Johnson, A.; Gan, Y.; Wientjes, M.G.; Au, J.L. Synergy Between 3′Azido-3′deoxythymidine and Paclitaxel in Human Pharynx FaDu Cells. Pharm. Res. 2003, 20, 957–961. [Google Scholar] [CrossRef]
- Zhou, F.-X.; Liao, Z.-K.; Dai, J.; Xiong, J.; Xie, C.-H.; Luo, Z.-G.; Liu, S.-Q.; Zhou, Y.-F. Radiosensitization effect of zidovudine on human malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Amantadin. Available online: https://www.gelbe-liste.de/wirkstoffe/Amantadin_21448 (accessed on 18 March 2021).
- Gianutsos, G.; Chute, S.; Dunn, J.P. Pharmacological changes in dopaminergic systems induced by long-term administration of amantadine. Eur. J. Pharmacol. 1985, 110, 357–361. [Google Scholar] [CrossRef]
- Scatton, B.; Cheramy, A.; Besson, M.; Glowinski, J. Increased synthesis and release of dopamine in the striatum of the rat after amantadine treatment. Eur. J. Pharmacol. 1970, 13, 131–133. [Google Scholar] [CrossRef]
- Jefferson, T.; Demicheli, V.; Rivetti, D.; Jones, M.; Di Pietrantonj, C.; Rivetti, A. Antivirals for influenza in healthy adults: Systematic review. Lancet 2006, 367, 303–313. [Google Scholar] [CrossRef]
- Uhnoo, I.; Linde, A.; Pauksens, K.; Lindberg, A.; Eriksson, M.; Norrby, R. Treatment and prevention of influenza: Swedish recommendations. Scand. J. Infect. Dis. 2003, 35, 3–11. [Google Scholar] [CrossRef]
- Itroconazol. Available online: https://www.gelbe-liste.de/wirkstoffe/Itraconazol_10331 (accessed on 25 March 2021).
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Liu, M.; Wang, Q.; Shen, Y.; Mei, H.; Li, D.; Liu, W. Itraconazole exerts its anti-melanoma effect by suppressing Hedgehog, Wnt, and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 28510–28525. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Combination Chemotherapy with Itraconazole for Treating Metastatic Pancreatic Cancer in the Second-line or Additional Setting. Anticancer. Res. 2015, 35, 4191–4196. [Google Scholar] [PubMed]
- Pace, J.R.; DeBerardinis, A.M.; Sail, V.; Tacheva-Grigorova, S.K.; Chan, K.A.; Tran, R.; Raccuia, D.S.; Wechsler-Reya, R.J.; Hadden, M.K. Repurposing the Clinically Efficacious Antifungal Agent Itraconazole as an Anticancer Chemotherapeutic. J. Med. Chem. 2015, 59, 3635–3649. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Xu, J.; Lu, J.; Bhat, S.; Sullivan, J.D.J.; Liu, J.O. Inhibition of Angiogenesis by the Antifungal Drug Itraconazole. ACS Chem. Biol. 2007, 2, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Borgers, M.; Bossche, H.V.D.; De Brabander, M. The mechanism of action of the new antimycotic ketoconazole. Am. J. Med. 1983, 74, 2–8. [Google Scholar] [CrossRef]
- Ketoconazol. Available online: https://www.gelbe-liste.de/wirkstoffe/Ketoconazol_1697 (accessed on 26 March 2021).
- Lumholtz, I.B. Sygeplejerskens laegemiddel information; terapeutisk hivedgruppe: Benzodiazepiner. 3. Sygeplejersken 1975, 75, 12. [Google Scholar]
- Oates, J.A.; Wood, A.J.; Sonino, N. The Use of Ketoconazole as an Inhibitor of Steroid Production. N. Engl. J. Med. 1987, 317, 812–818. [Google Scholar] [CrossRef]
- Patel, V.; Liaw, B.; Oh, W. The role of ketoconazole in current prostate cancer care. Nat. Rev. Urol. 2018, 15, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Bok, R.A.; Small, E.J. The treatment of advanced prostate cancer with ketoconazole: Safety issues. Drug Saf. 1999, 20, 451–458. [Google Scholar] [CrossRef]
- Mahler, C.; Verhelst, J.; Denis, L. Ketoconazole and liarozole in the treatment of advanced prostatic cancer. Cancer 1993, 71, 1068–1073. [Google Scholar] [CrossRef]
- Bareggi, S.R.; Cornelli, U. Clioquinol: Review of its Mechanisms of Action and Clinical Uses in Neurodegenerative Disorders. CNS Neurosci. Ther. 2010, 18, 41–46. [Google Scholar] [CrossRef]
- Clioquinol. Available online: https://www.gelbe-liste.de/wirkstoffe/Clioquinol_1890 (accessed on 23 March 2021).
- Ding, W.-Q.; Liu, B.; Vaught, J.L.; Yamauchi, H.; Lind, S.E. Anticancer activity of the antibiotic clioquinol. Cancer Res. 2005, 65, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lou, J.R.; Ding, W.-Q. Clioquinol independently targets NF-kappaB and lysosome pathways in human cancer cells. Anticancer Res. 2010, 30, 2087–2092. [Google Scholar]
- Yu, H.; Zhou, Y.; Lind, S.E.; Ding, W.-Q. Clioquinol targets zinc to lysosomes in human cancer cells. Biochem. J. 2008, 417, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Crowley, P.; Gallagher, H. Clotrimazole as a pharmaceutical: Past, present and future. J. Appl. Microbiol. 2014, 117, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Clotrimazol. Available online: https://www.gelbe-liste.de/wirkstoffe/Clotrimazol_331 (accessed on 28 March 2021).
- Coelho, R.G.; Calaça, I.d.C.; Celestrini, D.d.M.; Correia, A.H.; Costa, M.A.S.M.; Sola-Penna, M. Clotrimazole disrupts glycolysis in human breast cancer without affecting non-tumoral tissues. Mol. Genet. Metab. 2011, 103, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Furtado, C.M.; Marcondes, M.C.; Sola-Penna, M.; de Souza, M.L.S.; Zancan, P. Clotrimazole preferentially inhibits human breast cancer cell proliferation, viability and glycolysis. PLoS ONE 2012, 7, e30462. [Google Scholar] [CrossRef]
- Meira, D.D.; Marinho-Carvalho, M.M.; Teixeira, C.A.; Veiga, V.F.; Da Poian, A.; Holandino, C.; de Freitas, M.S.; Sola-Penna, M. Clotrimazole decreases human breast cancer cells viability through alterations in cytoskeleton-associated glycolytic enzymes. Mol. Genet. Metab. 2005, 84, 354–362. [Google Scholar] [CrossRef]
- Penso, J.; Beitner, R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur. J. Pharmacol. 1998, 342, 113–117. [Google Scholar] [CrossRef]
- Penso, J.; Beitner, R. Clotrimazole decreases glycolysis and the viability of lung carcinoma and colon adenocarcinoma cells. Eur. J. Pharmacol. 2002, 451, 227–235. [Google Scholar] [CrossRef]
- Kadavakollu, S.; Stailey, C.; Kunapareddy, C.S.; White, S. Clotrimazole as a Cancer Drug: A Short Review. Med. Chem. 2014, 4, 722–724. [Google Scholar]
- Darkes, M.J.M.; Scott, L.J.; Goa, K.L. Terbinafine: A review of its use in onychomycosis in adults. Am. J. Clin. Dermatol. 2003, 4, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Terbafine. Available online: https://www.gelbe-liste.de/wirkstoffe/Terbinafin_16933 (accessed on 29 March 2021).
- Plantone, D.; Koudriavtseva, T. Current and Future Use of Chloroquine and Hydroxychloroquine in Infectious, Immune, Neoplastic, and Neurological Diseases: A Mini-Review. Clin. Drug Investig. 2018, 38, 653–671. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, J.; Farinotti, R. Clinical pharmacokinetics and metabolism of chloroquine. Focus on recent advancements. Clin. Pharmacokinet. 1996, 31, 257–274. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Coronado, L.M.; Nadovich, C.T.; Spadafora, C. Malarial hemozoin: From target to tool. Biochim. et Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 2032–2041. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Thomé, R.; Lopes, S.C.P.; Costa, F.T.M.; Verinaud, L. Chloroquine: Modes of action of an undervalued drug. Immunol. Lett. 2013, 153, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Silverman, E.; Bargman, J.M. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat. Rev. Nephrol. 2011, 7, 718–729. [Google Scholar] [CrossRef]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef]
- Loehberg, C.R.; Strissel, P.L.; Dittrich, R.; Strick, R.; Dittmer, J.; Dittmer, A.; Fabry, B.; Kalender, W.A.; Koch, T.; Wachter, D.L.; et al. Akt and p53 are potential mediators of reduced mammary tumor growth by cloroquine and the mTOR inhibitor RAD001. Biochem. Pharmacol. 2012, 83, 480–488. [Google Scholar] [CrossRef]
- Gopalakrishnan, A.M.; Kumar, N. Antimalarial Action of Artesunate Involves DNA Damage Mediated by Reactive Oxygen Species. Antimicrob. Agents Chemother. 2015, 59, 317–325. [Google Scholar] [CrossRef]
- Efferth, T.; Giaisi, M.; Merling, A.; Krammer, P.H.; Li-Weber, M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS ONE 2007, 2, e693. [Google Scholar] [CrossRef]
- Dell’Eva, R.; Pfeffer, U.; Vené, R.; Anfosso, L.; Forlani, A.; Albini, A.; Efferth, T. Inhibition of angiogenesis in vivo and growth of Kaposi’s sarcoma xenograft tumors by the anti-malarial artesunate. Biochem. Pharmacol. 2004, 68, 2359–2366. [Google Scholar] [CrossRef]
- Da Jeong, E.; Song, H.J.J.; Lim, S.; Lee, S.J.J.; Lim, J.E.; Nam, D.-H.; Joo, K.M.; Jeong, B.C.; Jeon, S.S.; Choi, H.Y.; et al. Repurposing the anti-malarial drug artesunate as a novel therapeutic agent for metastatic renal cell carcinoma due to its attenuation of tumor growth, metastasis, and angiogenesis. Oncotarget 2015, 6, 33046–33064. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Stein, H.A.; Turschner, S.; Toegel, I.; Mora, R.; Jennewein, N.; Efferth, T.; Eils, R.; Brady, N.R. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J. Biol. Chem. 2011, 286, 6587–6601. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, W.; Li, B.; Yao, Q.; Dong, J.; Cen, Y.; Pan, X.; Li, J.; Zheng, J.; Pang, X.; et al. Artesunate enhances radiosensitivity of human non-small cell lung cancer A549 cells via increasing NO production to induce cell cycle arrest at G2/M phase. Int. Immunopharmacol. 2011, 11, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-J.; Dai, H.-Q.; Huang, X.-W.; Feng, J.; Deng, J.-H.; Wang, Z.-X.; Yang, X.-M.; Liu, Y.-J.; Wu, Y.; Chen, P.-H.; et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol. Sin. 2021, 42, 301–310. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. New Trends for Antimalarial Drugs: Synergism between Antineo-plastics and Antimalarials on Breast Cancer Cells. Biomolecules 2020, 10. [Google Scholar]
- Li, Q.; Ni, W.; Deng, Z.; Liu, M.; She, L.; Xie, Q. Targeting nasopharyngeal carcinoma by artesunate through inhibiting Akt/mTOR and inducing oxidative stress. Fundam. Clin. Pharmacol. 2017, 31, 301–310. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.-C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernan-dez, I.S.; et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein syn-thesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef]
- Fujita, R.; Ishikawa, M.; Takayanagi, M.; Takayanagi, Y.; Sasaki, K. Enhancement of doxorubicin activity in multi-drug-resistant cells by mefloquine. Methods Find. Exp. Clin. Pharmacol. 2000, 22, 281–284. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Xue, R.; Zhao, J.; Di, M. Mefloquine effectively targets gastric cancer cells through phosphatase-dependent inhibition of PI3K/Akt/mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2016, 470, 350–355. [Google Scholar] [CrossRef]
- Li, Y.-H.; Yang, S.-L.; Zhang, G.-F.; Wu, J.-C.; Gong, L.-L.; Zhong, M.; Lin, R.-X. Mef-loquine targets β-catenin pathway and thus can play a role in the treatment of liver cancer. Microb. Pathog. 2018, 118, 357–360. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Han, K.; Li, S.; Xu, F.; Yang, Y. Antimalarial drug mefloquine inhibits nuclear factor kappa B signaling and induces apoptosis in colorectal cancer cells. Cancer Sci. 2018, 109, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Sukhai, M.A.; Prabha, S.; Hurren, R.; Rutledge, A.C.; Lee, A.Y.; Sriskanthadevan, S.; Sun, H.; Wang, X.; Skrtic, M.; Seneviratne, A.; et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J. Clin. Investig. 2012, 123, 315–328. [Google Scholar] [CrossRef]
- Sharma, N.; Thomas, S.; Golden, E.B.; Hofman, F.M.; Chen, T.C.; Petasis, N.; Schönthal, A.H.; Louie, S.G. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012, 326, 143–154. [Google Scholar] [CrossRef]
- Bai, S.H.; Ogbourne, S. Eco-toxicological effects of the avermectin family with a focus on abamectin and ivermectin. Chemosphere 2016, 154, 204–214. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, M.; Xu, W.; Yang, M.; Wang, B.; Gao, J.; Li, Y.; Tao, L. Avermectin Confers Its Cytotoxic Effects by Inducing DNA Damage and Mitochondria-Associated Apoptosis. J. Agric. Food Chem. 2016, 64, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.; Xu, W.; Gao, J.; Cao, H.; Yang, M.; Wang, B.; Hao, Y.; Tao, L. Cytotoxic effects of Avermectin on human HepG2 cells in vitro bioassays. Environ. Pollut. 2017, 220, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Martin, R. Modes of action of anthelmintic drugs. Vet. J. 1997, 154, 11–34. [Google Scholar] [CrossRef]
- Märtlbauer, E. Niclosamid; Thieme Gruppe: Stuttgart, Germany, 2008. [Google Scholar]
- Yin, J.; Park, G.; Lee, J.E.; Choi, E.Y.; Park, J.Y.; Kim, T.-H.; Park, N.; Jin, X.; Jung, J.-E.; Shin, D.; et al. DEAD-box RNA helicase DDX23 modulates glioma malignancy via elevating miR-21 biogenesis. Brain 2015, 138, 2553–2570. [Google Scholar] [CrossRef] [PubMed]
- Sharmeen, S.; Skrtic, M.; Sukhai, M.A.; Hurren, R.; Gronda, M.; Wang, X.; Fonseca, S.B.; Sun, H.; Wood, T.E.; Ward, R.; et al. The antiparasitic agent ivermectin induces chlo-ride-dependent membrane hyperpolarization and cell death in leukemia cells. Blood 2010, 116, 3593–3603. [Google Scholar] [CrossRef] [PubMed]
- Seth, C.; Mas, C.; Conod, A.; Mueller, J.; Siems, K.; Kuciak, M.; Borges, I.; Ruiz i Altaba, A. Long-Lasting WNT-TCF Response Blocking and Epigenetic Modifying Activities of With-anolide F in Human Cancer Cells. PLoS ONE 2016, 11, e0168170. [Google Scholar] [CrossRef]
- Nörenberg, W.; Sobottka, H.; Hempel, C.; Plötz, T.; Fischer, W.; Schmalzing, G.; Schaefer, M. Positive allosteric modulation by ivermectin of human but not murine P2X7 receptors. Br. J. Pharmacol. 2012, 167, 48–66. [Google Scholar] [CrossRef]
- Melotti, A.; Mas, C.; Kuciak, M.; Lorente-Trigos, A.; Borges, I.; Ruiz i Altaba, A. The river blindness drug Ivermectin and related macrocyclic lactones inhibit WNT-TCF pathway re-sponses in human cancer. EMBO Mol. Med. 2014, 6, 1263–1278. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, S.; Sun, Q.; Liu, B. Anthelmintic drug ivermectin inhibits angiogenesis, growth and survival of glioblastoma through inducing mitochondrial dysfunction and oxidative stress. Biochem. Biophys. Res. Commun. 2016, 480, 415–421. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Petrie, K.; Leibovitch, B.A.; Zeng, L.; Mezei, M.; Howell, L.; Gil, V.; Christova, R.; Bansal, N.; Yang, S.; et al. Selective Inhibition of SIN3 Corepressor with Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 1824–1836. [Google Scholar] [CrossRef]
- Draganov, D.; Gopalakrishna-Pillai, S.; Chen, Y.-R.; Zuckerman, N.; Moeller, S.; Wang, C.; Ann, D.; Lee, P.P. Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci. Rep. 2015, 5, 16222. [Google Scholar] [CrossRef]
- Dominguez-Gomez, G.; Chavez-Blanco, A.; Medina-Franco, J.L.; Saldivar-Gonzalez, F.; Flores-Torrontegui, Y.; Juarez, M.; Díaz-Chávez, J.; Gonzalez-Fierro, A.; Dueñas-González, A. Ivermectin as an inhibitor of cancer stem-like cells. Mol. Med. Rep. 2017, 17, 3397–3403. [Google Scholar] [CrossRef]
- Didier, A.; Loor, F. The abamectin derivative ivermectin is a potent P-glycoprotein inhibitor. Anti-Cancer Drugs 1996, 7, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, C.G.; Williamson, T.; Riggins, G.J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neuro-Oncol. 2018, 140, 529–538. [Google Scholar] [CrossRef]
- Fong, D.; Christensen, C.T.; Chan, M.M. Targeting Cancer Stem Cells with Repurposed Drugs to Improve Current Therapies. Recent Pat. Anti-Cancer Drug Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Rath, B. Repurposing of Anthelminthics as Anticancer Drugs. Oncomedicine 2018, 3, 1–8. [Google Scholar] [CrossRef]
- Rubin, J.; Mansoori, S.; Blom, K.; Berglund, M.; Lenhammar, L.; Andersson, C.; Loskog, A.; Fryknäs, M.; Nygren, P.; Larsson, R. Mebendazole stimulates CD14+ myeloid cells to en-hance T-cell activation and tumour cell killing. Oncotarget 2018, 9, 30805–30813. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.R.; Bai, R.-Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.; Riggins, G.J.; Bunz, F. Repurposing the Antihelmintic Mebendazole as a Hedgehog Inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pourgholami, M.H.; Wangoo, K.T.; Morris, D.L. Albendazole-cyclodextrin complex: En-hanced cytotoxicity in ovarian can-cer cells. Anticancer Res. 2008, 28, 2775–2779. [Google Scholar] [PubMed]
- Chu, S.W.L.; Badar, S.; Morris, D.L.; Pourgholami, M.H. Potent inhibition of tubulin polymerisation and proliferation of paclitaxel-resistant 1A9PTX22 human ovarian cancer cells by albendazole. Anticancer. Res. 2009, 29, 3791–3796. [Google Scholar]
- Noorani, L.; Stenzel, M.; Liang, R.; Pourgholami, M.H.; Morris, D.L. Albumin nanoparticles increase the anticancer efficacy of albendazole in ovarian cancer xenograft model. J. Nanobiotechnology 2015, 13, 1–12. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Szwajcer, M.; Chin, M.; Liauw, W.; Seef, J.; Galettis, P.; Morris, D.L.; Links, M. Phase I clinical trial to determine maximum tolerated dose of oral albendazole in pa-tients with advanced cancer. Cancer Chemother. Pharmacol. 2010, 65, 597–605. [Google Scholar] [CrossRef]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of Niclosamide as a Novel Anticancer Agent for Adrenocortical Carcinoma. Clin. Cancer Res. 2016, 22, 3458–3466. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-L.-O.; Son, A.-R.; Ahn, J.; Song, J.-Y. Niclosamide enhances ROS-mediated cell death through c-Jun activation. Biomed. Pharmacother. 2014, 68, 619–624. [Google Scholar] [CrossRef]
- Yu, X.; Liu, F.; Zeng, L.; He, F.; Zhang, R.; Yan, S.; Zeng, Z.; Shu, Y.; Zhao, C.; Wu, X.; et al. Niclosamide Exhibits Potent Anticancer Activity and Synergizes with Sorafenib in Human Renal Cell Cancer Cells. Cell. Physiol. Biochem. 2018, 47, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Chen, H.-N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Au-tophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Saravolatz, L.D. Nitazoxanide: A new thiazolide antiparasitic agent. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2005, 40, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Sisson, G.; Goodwin, A.; Raudonikiene, A.; Hughes, N.J.; Mukhopadhyay, A.K.; Berg, D.E.; Hoffman, P.S. Enzymes associat-ed with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob. Agents Chemother. 2002, 46, 2116–2123. [Google Scholar] [CrossRef]
- Müller, J.; Sidler, D.; Nachbur, U.; Wastling, J.; Brunner, T.; Hemphill, A. Thiazolides in-hibit growth and induce glutathi-one-S-transferase Pi (GSTP1)-dependent cell death in human colon cancer cells. Int. J. Cancer 2008, 123, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Senkowski, W.; Zhang, X.; Olofsson, M.H.; Isacson, R.; Höglund, U.; Gustafsson, M.; Nygren, P.; Linder, S.; Larsson, R.; Fryknäs, M. Three-Dimensional Cell Culture-Based Screening Identifies the Anthelmintic Drug Nitazoxanide as a Candi-date for Treatment of Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1504–1516. [Google Scholar] [CrossRef]
- Wang, X.; Shen, C.; Liu, Z.; Peng, F.; Chen, X.; Yang, G.; Zhang, D.; Yin, Z.; Ma, J.; Zheng, Z.; et al. Nitazoxanide, an antiprotozoal drug, inhib-its late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma. Cell Death Dis. 2018, 9, 1032. [Google Scholar] [CrossRef]
- Fan-Minogue, H.; Bodapati, S.; Solow-Cordero, D.; Fan, A.; Paulmurugan, R.; Massoud, T.F.; Felsher, D.W.; Gambhir, S.S. A c-Myc Activation Sensor-Based High-Throughput Drug Screening Identifies an Antineoplastic Effect of Nitazoxanide. Mol. Cancer Ther. 2013, 12, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, N.; Ehrisman, J. A functional perspective of nitazoxanide as a potential anti-cancer drug. Mutat. Res. 2014, 768, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L. Praziquantel. Parasitol. Res. 2003, 90, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Timson, D.J. The Mechanism of Action of Praziquantel: Six Hypotheses. Curr. Top. Med. Chem. 2018, 18, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Pax, R.; Bennett, J.L.; Fetterer, R. A benzodiazepine derivative and praziquantel: Effects on musculature of Schistosoma mansoni and Schistosoma japonicum. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1978, 304, 309–315. [Google Scholar] [CrossRef]
- Wu, Z.H.; Lu, M.-K.; Hu, L.Y.; Li, X. Praziquantel Synergistically Enhances Paclitaxel Efficacy to Inhibit Cancer Cell Growth. PLoS ONE 2012, 7, e51721. [Google Scholar] [CrossRef]
- Culetto, E.; Baylis, H.; Richmond, J.E.; Jones, A.K.; Fleming, J.T.; Squire, M.D.; Lewis, J.A.; Sattelle, D.B. The Caenorhabditis elegans unc-63 Gene Encodes a Levamisole-sensitive Nicotinic Acetylcholine Receptor α Subunit. J. Biol. Chem. 2004, 279, 42476–42483. [Google Scholar] [CrossRef]
- Rayes, D.; Flamini, M.; Hernando, G.; Bouzat, C. Activation of Single Nicotinic Receptor Channels from Caenorhabditis elegans Muscle. Mol. Pharmacol. 2007, 71, 1407–1415. [Google Scholar] [CrossRef]
- Hu, Y.; Xiao, S.-H.; Aroian, R.V. The new anthelmintic tribendimidine is an L-type (le-vamisole and pyrantel) nicotinic acetylcholine receptor agonist. PLoS Negl. Trop. Dis. 2009, 3, e499. [Google Scholar] [CrossRef] [PubMed]
- Renoux, G. The General Immunopharmacology of Levamisole. Drugs 1980, 20, 89–99. [Google Scholar] [CrossRef]
- Amery, W.; Gough, D. Levamisole and Immunotherapy: Some Theoretic and Practical Considerations and Their Relevance to Human Disease. Oncology 1981, 38, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Nageshwari, B.; Merugu, R. Effect of levamisole on expression of CD138 and interleu-kin-6 in human multiple myeloma cell lines. Indian J. Cancer 2017, 54, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Wang, C.; Wang, W.; Shang, Y.; Li, Y.; Ni, J.; Chen, S.-Z. Levamisole enhances DR4-independent apoptosis induced by TRAIL through inhibiting the activation of JNK in lung cancer. Life Sci. 2020, 257, 118034. [Google Scholar] [CrossRef] [PubMed]
- Wiegering, A.; Uthe, F.-W.; Hüttenrauch, M.; Mühling, B.; Linnebacher, M.; Krummenast, F.; Germer, C.-T.; Thalheimer, A.; Otto, C. The impact of pyrvinium pamoate on colon cancer cell viability. Int. J. Color. Dis. 2014, 29, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, L.; Hu, C.; Liang, S.; Fei, X.; Yan, N.; Zhang, Y.; Zhang, F. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int. J. Oncol. 2016, 48, 1175–1186. [Google Scholar] [CrossRef]
- Dattilo, R.; Mottini, C.; Camera, E.; Lamolinara, A.; Auslander, N.; Doglioni, G.; Muscolini, M.; Tang, W.; Planque, M.; Ercolani, C.; et al. Pyrvinium Pamoate Induces Death of Triple-Negative Breast Cancer Stem–Like Cells and Reduces Metastases through Effects on Lipid Anabolism. Cancer Res. 2020, 80, 4087–4102. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C.; Trigoso, C.I.; Llorente, A.R.; Del Castillo, P. DNA fluorescence induced by polymethine cation pyrvinium binding. J. Mol. Histol. 1991, 23, 548–552. [Google Scholar] [CrossRef]
- Deng, L.; Lei, Y.; Liu, R.; Li, J.; Yuan, K.; Li, Y.; Chen, Y.; Lu, Y.; Edwards, C.K., III; Huang, C.; et al. Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis. 2013, 4, e614. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Torchilin, V.; Patri, A.K.; Hrkach, J.; Stern, S.; Lee, R.; Nel, A.; Panaro, N.J.; Grodzinski, P. Best practices in cancer nanotechnology: Perspective from NCI nano-technology alliance. Clin. Cancer Res. 2012, 18, 3229–3241. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Shih, H.-P.; Zhang, X.; Aronov, A.M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indi-cations. Nat. Rev. Drug Discov. 2018, 17, 19–33. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology—Patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Hans Henri, P.K. Erklärung—Katastrophale Auswirkungen von COVID-19 auf Die Krebsversorgung; WHO: Geneva, Switzerland, 2021. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfab, C.; Schnobrich, L.; Eldnasoury, S.; Gessner, A.; El-Najjar, N. Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know? Cancers 2021, 13, 3193. https://doi.org/10.3390/cancers13133193
Pfab C, Schnobrich L, Eldnasoury S, Gessner A, El-Najjar N. Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know? Cancers. 2021; 13(13):3193. https://doi.org/10.3390/cancers13133193
Chicago/Turabian StylePfab, Christina, Luisa Schnobrich, Samir Eldnasoury, André Gessner, and Nahed El-Najjar. 2021. "Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know?" Cancers 13, no. 13: 3193. https://doi.org/10.3390/cancers13133193
APA StylePfab, C., Schnobrich, L., Eldnasoury, S., Gessner, A., & El-Najjar, N. (2021). Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know? Cancers, 13(13), 3193. https://doi.org/10.3390/cancers13133193