
Epigenetic Deregulation of Apoptosis in Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
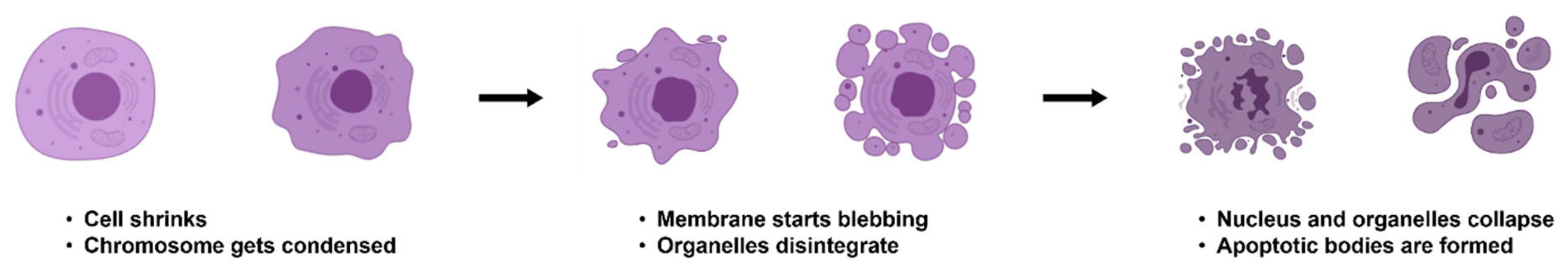
2. Mechanisms of Apoptosis
3. Hallmark of Cancer: Evasion of Apoptosis
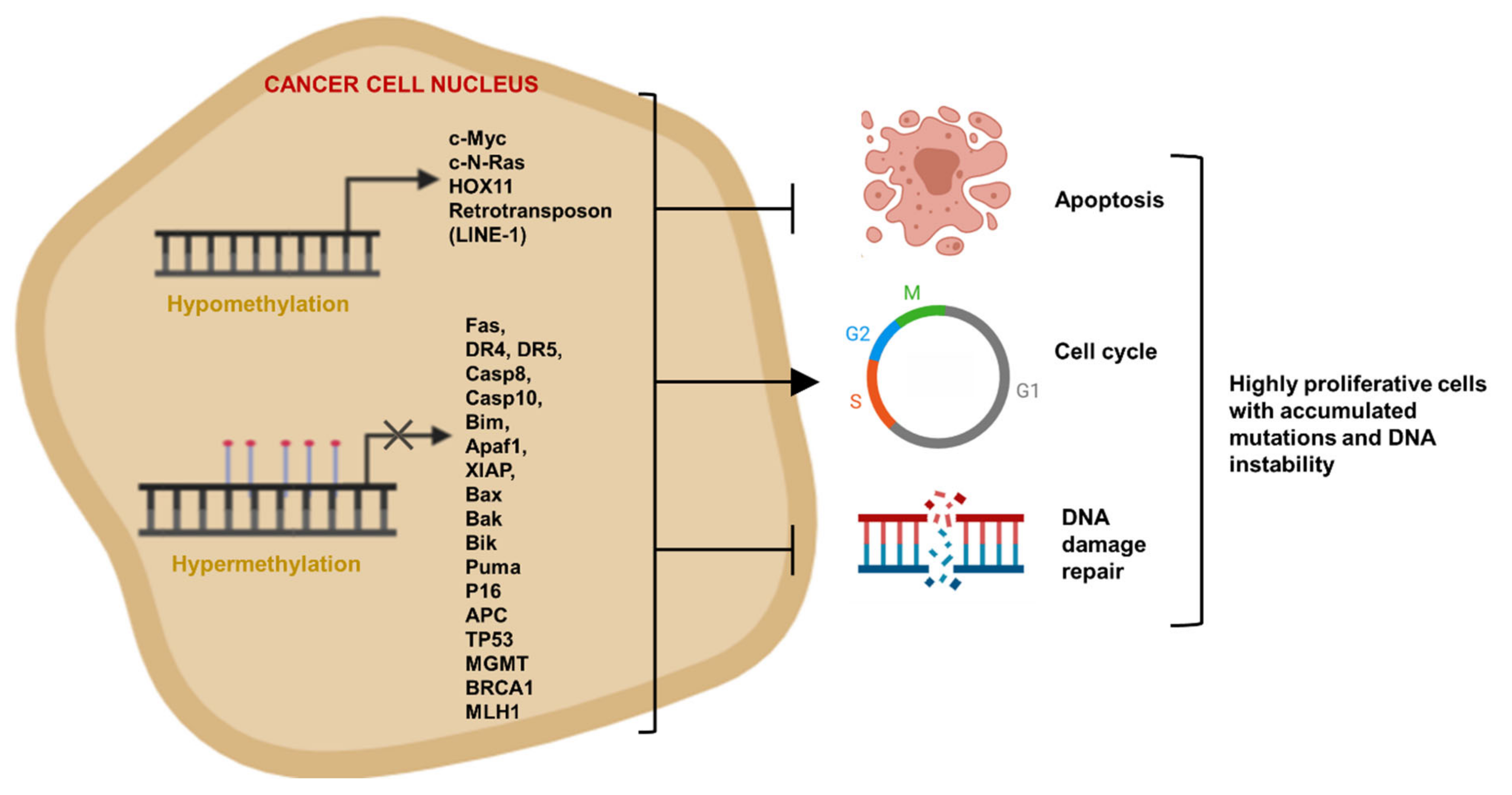
4. Epigenetics Mechanisms of Cancer Apoptosis Evasion
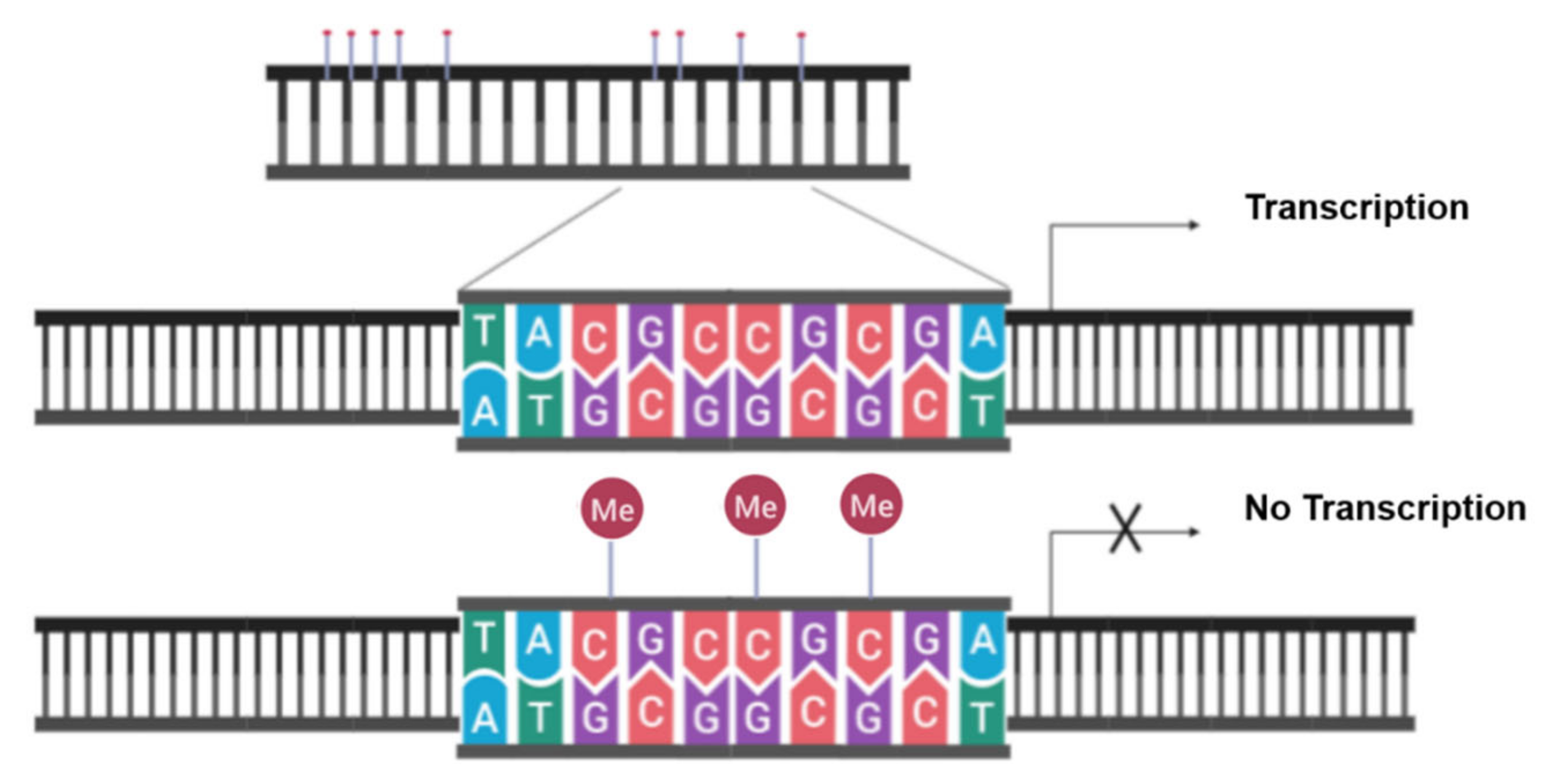
4.1. Evading Apoptosis by Aberrant DNA Methylation
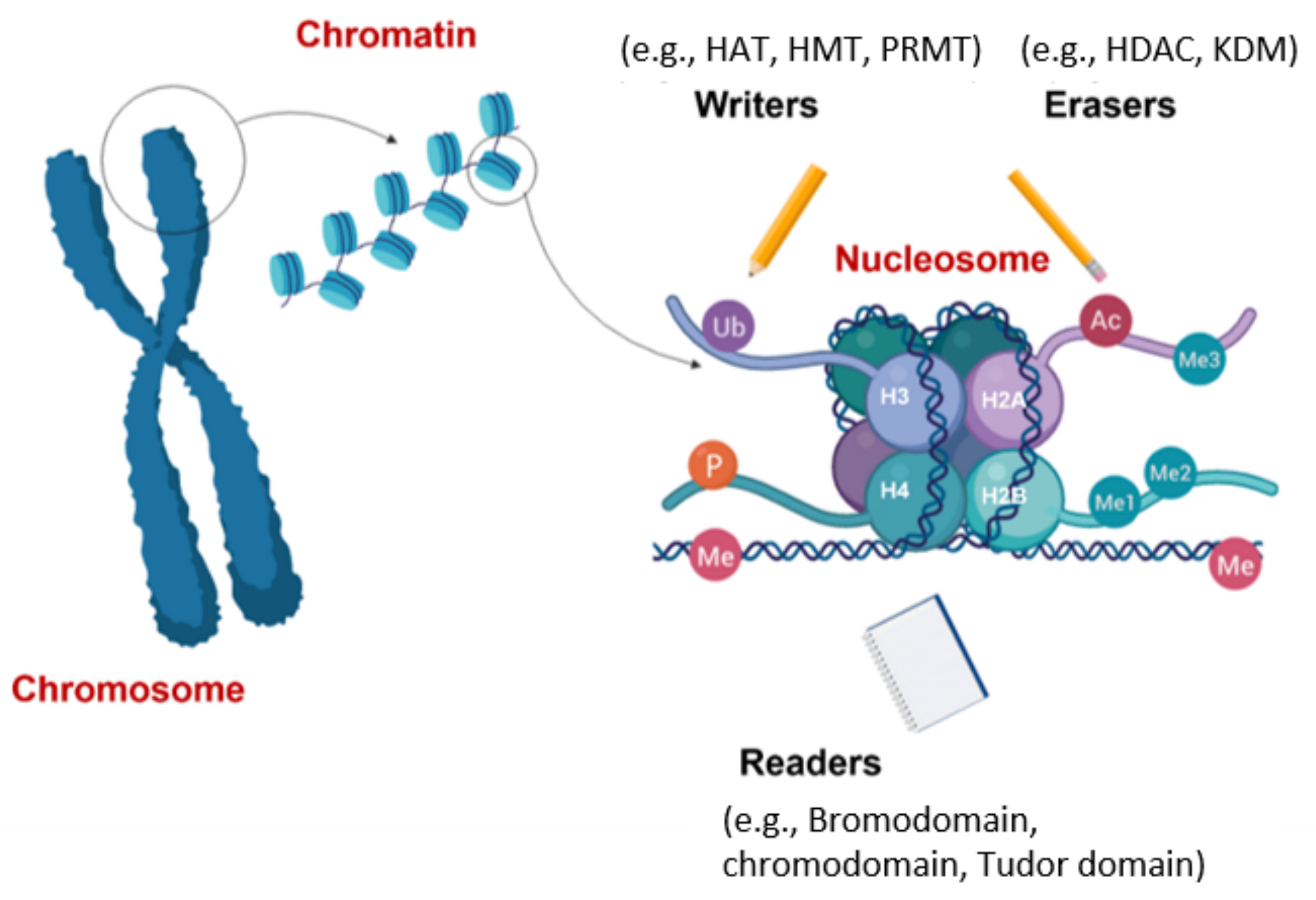
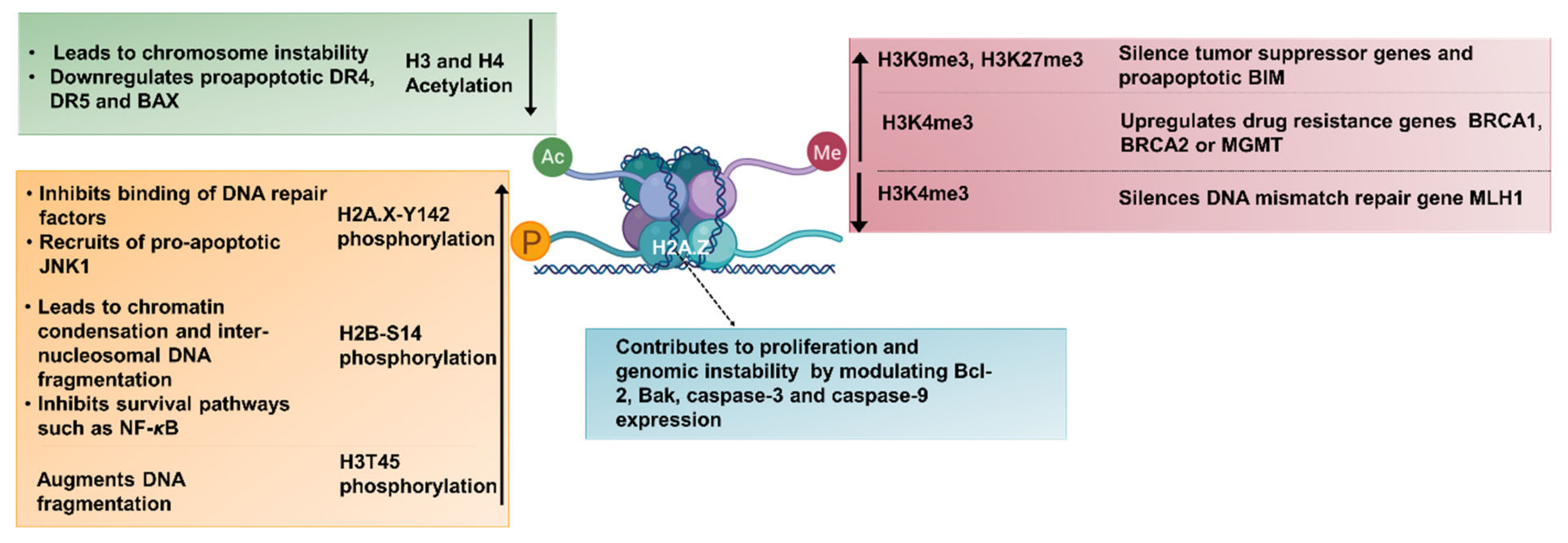
4.2. Evasion of Apoptosis by Aberrant Histone Modifications
4.3. Evading Apoptosis by Epigenetic Regulation of miRNAs
5. Reprogramming the Cancer Epigenome by Epigenetic Drugs to Trigger Tumor Cell Death
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Green, D.R. Means to an End: Apoptosis and Other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2010; ISBN 9780879698874. [Google Scholar]
- Wyllie, A.H.; Kerr, J.F.R.; Currie, A.R. Cell Death: The Significance of Apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer Review Douglas. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Fesik, S.W. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci. 2004, 13, 1979–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saelens, X.; Festjens, N.; Walle, L.V.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [Green Version]
- Henry-Mowatt, J.; Dive, C.; Martinou, J.C.; James, D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 2004, 23, 2850–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAf-1 · cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [Green Version]
- Breckenridge, D.G.; Xue, D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr. Opin. Cell Biol. 2004, 16, 647–652. [Google Scholar] [CrossRef]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Adams, J.M. Life in the balance: How BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 2005, 17, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, K.S.; Vousden, K.H. Complicating the complexity of p53. Carcinogenesis 2005, 26, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef] [Green Version]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar] [CrossRef]
- Mérino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell. Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov. 2008, 7, 1001–1012. [Google Scholar] [CrossRef]
- Wang, X.; Chen, W.; Zeng, W.; Bai, L.; Tesfaigzi, Y.; Belinsky, S.A.; Lin, Y. Akt-mediated eminent expression of c-FLIP and Mcl-1 confers acquired resistance to TRAIL-induced cytotoxicity to lung cancer cells. Mol. Cancer Ther. 2008, 7, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, H.; Xu, C.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 Type I/Type II model. Semin. Immunol. 2003, 15, 185–193. [Google Scholar] [CrossRef]
- Ozören, N.; El-Deiry, W.S. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 2002, 4, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- De Almagro, M.C.; Vucic, D. Necroptosis: Pathway diversity and characteristics. Semin. Cell Dev. Biol. 2015, 39, 56–62. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Chang, W.; Lin, J.; Dong, J.; Li, D. Pyroptosis: An inflammatory cell death implicates in atherosclerosis. Med. Hypotheses 2013, 81, 484–486. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braeuer, S.J.; Büneker, C.; Mohr, A.; Zwacka, R.M. Constitutively activated nuclear factor-kappaB, but not induced NF-kappaB, leads to TRAIL resistance by up-regulation of X-linked inhibitor of apoptosis protein in human cancer cells. Mol. Cancer Res. 2006, 4, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Roué, G.; Pérez-Galán, P.; López-Guerra, M.; Villamor, N.; Campo, E.; Colomer, D. Selective inhibition of IkappaB kinase sensitizes mantle cell lymphoma B cells to TRAIL by decreasing cellular FLIP level. J. Immunol. 2007, 178, 1923–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Allen, J.E.; Prabhu, V.V.; Talekar, M.K.; Finnberg, N.K.; El-Deiry, W.S. Targeting TRAIL in the treatment of cancer: New developments. Expert Opin. Ther. Targets 2015, 19, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.C.; Dietmann, S.; Irie, N.; Leitch, H.G.; Floros, V.I.; Bradshaw, C.R.; Hackett, J.A.; Chinnery, P.F.; Surani, M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell 2015, 161, 1453–1467. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Yan, W.; Duan, E. Epigenetic inheritance of acquired traits through sperm RNAs and sperm RNA modifications. Nat. Rev. Genet. 2016, 17, 733–743. [Google Scholar] [CrossRef]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S. The Nucleosome: From Genomic Organization to Genomic Regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; He, X.; Kingston, R.E.; Narlikar, G.J. Distinct strategies to make nucleosomal DNA accessible. Mol. Cell 2003, 11, 1311–1322. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Tsukiyama, T. Chromatin remodeling in vivo: Evidence for a nucleosome sliding mechanism. Mol. Cell 2003, 12, 1333–1340. [Google Scholar] [CrossRef]
- Kassabov, S.R.; Zhang, B.; Persinger, J.; Bartholomew, B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol. Cell 2003, 11, 391–403. [Google Scholar] [CrossRef]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.H.; Sen, S.; Wu, C. ATP-Driven Exchange of Histone H2AZ Variant Catalyzed by SWR1 Chromatin Remodeling Complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- Suto, R.K.; Clarkson, M.J.; Tremethick, D.J.; Luger, K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000, 7, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Roberti, A.; Dobay, M.P.; Bisig, B.; Vallois, D.; Boéchat, C.; Lanitis, E.; Bouchindhomme, B.; Parrens, M.C.; Bossard, C.; Quintanilla-Martinez, L.; et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Mar, B.G.; Bullinger, L.B.; McLean, K.M.; Grauman, P.V.; Harris, M.H.; Stevenson, K.; Neuberg, D.S.; Sinha, A.U.; Sallan, S.E.; Silverman, L.B.; et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun. 2014, 5, 3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007. [Google Scholar] [CrossRef] [PubMed]
- Theisen, E.R.; Pishas, K.I.; Saund, R.S.; Lessnick, S.L. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget 2016, 7, 17616–17630. [Google Scholar] [CrossRef] [Green Version]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.T.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef] [Green Version]
- Theisen, E.R.; Gajiwala, S.; Bearss, J.; Sorna, V.; Sharma, S.; Janat-Amsbury, M. Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. BMC Cancer 2014, 14, 752. [Google Scholar] [CrossRef] [Green Version]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the Histone Demethylase JMJD1A by Hypoxia-Inducible Factor 1α Enhances Hypoxic Gene Expression and Tumor Growth. Mol. Cell. Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadoss, S.; Guo, G.; Wang, C.Y. Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene 2017, 36, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Ohguchi, H.; Hideshima, T.; Bhasin, M.K.; Gorgun, G.T.; Santo, L.; Cea, M.; Samur, M.K.; Mimura, N.; Suzuki, R.; Tai, Y.T.; et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat. Commun. 2016, 7, 10258. [Google Scholar] [CrossRef]
- Svotelis, A.; Bianco, S.; Madore, J.; Huppé, G.; Nordell-Markovits, A.; Mes-Masson, A.M.; Gévry, N. H3K27 demethylation by JMJD3 at a poised enhancer of anti-apoptotic gene BCL2 determines ERα ligand dependency. EMBO J. 2011, 30, 3947–3961. [Google Scholar] [CrossRef] [Green Version]
- Ene, C.I.; Edwards, L.; Riddick, G.; Baysan, M.; Woolard, K.; Kotliarova, S.; Lai, C.; Belova, G.; Cam, M.; Walling, J.; et al. Histone Demethylase Jumonji D3 (JMJD3) as a Tumor Suppressor by Regulating p53 Protein Nuclear Stabilization. PLoS ONE 2012, 7, e51407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, J.; Jelinek, J.; Lu, Y.; Cesaroni, M.; Madzo, J.; Neumann, F.; He, R.; Taby, R.; Vasanthakumar, A.; Macrae, T.; et al. TET2 mutations affect Non-CpG island DNA methylation at enhancers and transcription factor-binding sites in chronic myelomonocytic Leukemia. Cancer Res. 2015, 75, 2833–2843. [Google Scholar] [CrossRef] [Green Version]
- Teng, S.; Ma, C.; Yu, Y.; Yi, C. Hydroxyurea promotes TET1 expression and induces apoptosis in osteosarcoma cells. Biosci. Rep. 2019. [Google Scholar] [CrossRef] [Green Version]
- Sant, D.W.; Mustafi, S.; Gustafson, C.B.; Chen, J.; Slingerland, J.M.; Wang, G. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sánchez-Céspedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, M.; Pao, M.M.; Jeong, S.; Gal-Yam, E.N.; Egger, G.; Weisenberger, D.J.; Jones, P.A. Footprinting of mammalian promoters: Use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef] [Green Version]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Hajji, N.; Joseph, B. Epigenetic regulation of cell life and death decisions and deregulation in cancer. Essays Biochem. 2010, 48, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Roll, J.D.; Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer 2008, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012. [Google Scholar] [CrossRef] [PubMed]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Elmallah, M.I.Y.; Micheau, O. Epigenetic regulation of TRAIL signaling: Implication for cancer therapy. Cancers 2019, 11, 850. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wood, G.S. Reduction of Fas/CD95 promoter methylation, upregulation of fas protein, and enhancement of sensitivity to apoptosis in cutaneous T-Cell lymphoma. Arch. Dermatol. 2011, 147, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Petak, I.; Danam, R.P.; Tillman, D.M.; Vernes, R.; Howell, S.R.; Berczi, L.; Kopper, L.; Brent, T.P.; Houghton, J.A. Hypermethylation of the gene promoter and enhancer region can regulate Fas expression and sensitivity in colon carcinoma. Cell Death Differ. 2003, 10, 211–217. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; Van Bezouw, S.; Voûte, P.A.; Herman, J.G.; Pieters, R.; Versteeg, R. Clustering of hypermethylated genes in neuroblastoma. Genes Chromosom. Cancer 2003, 38, 226–233. [Google Scholar] [CrossRef]
- Bae, S.I.; Cheriyath, V.; Jacobs, B.S.; Reu, F.J.; Borden, E.C. Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene 2007, 27, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horak, P. Contribution of Epigenetic Silencing of Tumor Necrosis Factor-Related Apoptosis Inducing Ligand Receptor 1 (DR4) to TRAIL Resistance and Ovarian Cancer. Mol. Cancer Res. 2005, 3, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Lee, J.H.; Cho, S.B.; Yoon, K.W.; Park, S.Y.; Lee, W.S.; Park, C.H.; Joo, Y.E.; Kim, H.S.; Choi, S.K.; et al. Epigenetic methylation and expression of caspase 8 and survivin in hepatocellular carcinoma. Pathol. Int. 2010, 60, 203–211. [Google Scholar] [CrossRef]
- Malekzadeh, K.; Sobti, R.C.; Nikbakht, M.; Shekari, M.; Hosseini, S.A.; Tamandani, D.K.; Singh, S.K. Methylation patterns of Rb1 and Casp-8 promoters and their impact on their expression in bladder cancer. Cancer Invest. 2009, 27, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Shivapurkar, N.; Toyooka, S.; Eby, M.T.; Huang, C.X.; Sathyanarayana, U.G.; Cunningham, H.T.; Reddy, J.L.; Brambilla, E.; Takahashi, T.; Minna, J.D.; et al. Differential inactivation of caspase-8 in lung cancers. Cancer Biol. Ther. 2002, 1, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis. 2010, 1, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, K.; Toyooka, S.; Shivapurkar, N.; Maitra, A.; Reddy, J.L.; Matta, H.; Miyajima, K.; Timmons, C.F.; Tomlinson, G.E.; Mastrangelo, D.; et al. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002, 62, 5897–5901. [Google Scholar] [PubMed]
- San José-Eneriz, E.; Agirre, X.; Jiménez-Velasco, A.; Cordeu, L.; Martín, V.; Arqueros, V.; Gárate, L.; Fresquet, V.; Cervantes, F.; Martínez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer 2009, 45, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y. Methylation Silencing of the Apaf-1 Gene in Acute Leukemia. Mol. Cancer Res. 2005, 3, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Soengas, M.S.; Capodieci, P.; Polsky, D.; Mora, J.; Esteller, M.; Opitz-Araya, X.; McCombie, R.; Herman, J.G.; Gerald, W.L.; Lazebnik, Y.A.; et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001. [Google Scholar] [CrossRef]
- Wang, H.L.; Bai, H.; Li, Y.; Sun, J.; Wang, X.Q. Rationales for expression and altered expression of apoptotic protease activating factor-1 gene in gastric cancer. World J. Gastroenterol. 2007, 13, 5060–5064. [Google Scholar] [CrossRef] [Green Version]
- Christoph, F.; Kempkensteffen, C.; Weikert, S.; Köllermann, J.; Krause, H.; Miller, K.; Schostak, M.; Schrader, M. Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br. J. Cancer 2006, 95, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Kang, M.J.; Kim, H.R.; Chi, S.G. Hypermethylation of XIAP-associated Factor 1, a Putative Tumor Suppressor Gene from the 17p13.2 Locus, in Human Gastric Adenocarcinomas. Cancer Res. 2003, 63, 7068–7075. [Google Scholar]
- Shui, P.T.; Liston, P.; Jian, T.C.; Lin, M.C.M.; Xiao, H.J.; Yang, Y.; Gu, Q.; Shi, H.J.; Ching, T.L.; Hsiang, F.K.; et al. Restoration of XAF1 expression induces apoptosis and inhibits tumor growth in gastric cancer. Int. J. Cancer 2009, 125, 688–697. [Google Scholar] [CrossRef]
- Kempkensteffen, C.; Hinz, S.; Schrader, M.; Christoph, F.; Magheli, A.; Krause, H.; Schostak, M.; Miller, K.; Weikert, S. Gene expression and promoter methylation of the XIAP-associated Factor 1 in renal cell carcinomas: Correlations with pathology and outcome. Cancer Lett. 2007, 254, 227–235. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Q.; Cho, A.H.; Rondeau, G.; Welsh, J.; Adamson, E.; Mercola, D.; McClelland, M. Survey of Differentially Methylated Promoters in Prostate Cancer Cell Lines. Neoplasia 2005, 7, 748–IN7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompeia, C.; Hodge, D.R.; Plass, C.; Wu, Y.Z.; Marquez, V.E.; Kelley, J.A.; Farrar, W.L. Microarray analysis of epigenetic silencing of gene expression in the KAS-6/1 multiple myeloma cell line. Cancer Res. 2004, 64, 3465–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, S.P.; Jeffers, J.R.; Yang, C.; Nilsson, J.A.; Hall, M.A.; Rehg, J.E.; Yue, W.; Yu, J.; Zhang, L.; Onciu, M.; et al. Selection against PUMA Gene Expression in Myc-Driven B-Cell Lymphomagenesis. Mol. Cell. Biol. 2008, 28, 5391–5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Xu, J.; Cao, X.X.; Long, Z.W.; Liu, X.P.; Furuya, T.; Xu, J.W.; Liu, X.L.; De Xu, Z.; Sasaki, K.; Li, Q.Q. BCL2L10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J. Pathol. 2011, 223, 400–409. [Google Scholar] [CrossRef]
- Fabiani, E.; Leone, G.; Giachelia, M.; D’Alo’, F.; Greco, M.; Criscuolo, M.; Guidi, F.; Rutella, S.; Hohaus, S.; Voso, M.T. Analysis of genome-wide methylation and gene expression induced by 5-aza-2′-deoxycytidine identifies BCL2L10 as a frequent methylation target in acute myeloid leukemia. Leuk. Lymphoma 2010, 51, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Zhong, S.; Fields, C.R.; Kim, J.H.; Robertson, K.D. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006, 66, 7490–7501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, I.; Stephan, C.; Gillissen, B.; Siebert, R.; Janz, M.; Radetzki, S.; Jung, K.; Loening, S.; Dörken, B.; Daniel, P.T. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006, 13, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.M.; Sullivan, L.; Lane, C.; O’Connor, L.; Barrett, C.; Hollywood, D.; Lynch, T.; Lawler, M.; Perry, A.S. In silico analysis and DHPLC screening strategy identifies novel apoptotic gene targets of aberrant promoter hypermethylation in prostate cancer. Prostate 2011, 71, 1–17. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Dasoula, A.; Kounnis, V.; Benetatos, L.; Nigro, C.L.; Lattanzio, L.; Papoudou-Bai, A.; Dranitsaris, G.; Briasoulis, E.; Crook, T. Bcl2-interacting killer CpG methylation in multiple myeloma: A potential predictor of relapsed/refractory disease with therapeutic implications. Leuk. Lymphoma 2012, 53, 1709–1713. [Google Scholar] [CrossRef]
- Sugita, H.; Iida, S.; Inokuchi, M.; Kato, K.; Ishiguro, M.; Ishikawa, T.; Takagi, Y.; Enjoji, M.; Yamada, H.; Uetake, H.; et al. Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer. Oncol. Rep. 2011, 25, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Nakafusa, Y.; Nakamura, J.; Hashiguchi, K.; Sumi, K.; Noshiro, H.; Miyazaki, K. CpG island methylation of BNIP3 predicts resistance against S-1/CPT-11 combined therapy in colorectal cancer patients. Oncol. Rep. 2010, 23, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Pike, B.L.; Greiner, T.C.; Wang, X.; Weisenburger, D.D.; Hsu, Y.H.; Renaud, G.; Wolfsberg, T.G.; Kim, M.; Weisenberger, D.J.; Siegmund, K.D.; et al. DNA methylation profiles in diffuse large B-cell lymphoma and their relationship to gene expression status. Leukemia 2008, 22, 1035–1043. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Obata, T.; Toyota, M.; Satoh, A.; Sasaki, Y.; Ogi, K.; Akino, K.; Suzuki, H.; Murai, M.; Kikuchi, T.; Mita, H.; et al. Identification of HRK as a Target of Epigenetic Inactivation in Colorectal and Gastric Cancer. Clin. Cancer Res. 2003, 9, 6410–6418. [Google Scholar] [PubMed]
- Nakamura, M.; Ishida, E.; Shimada, K.; Nakase, H.; Sakaki, T.; Konishi, N. Frequent HRK inactivation associated with low apoptotic index in secondary glioblastomas. Acta Neuropathol. 2005, 110, 402–410. [Google Scholar] [CrossRef]
- Nakamura, M.; Ishida, E.; Shimada, K.; Nakase, H.; Sakaki, T.; Konishi, N. Defective expression of HRK is associated with promoter methylation in primary central nervous system lymphomas. Oncology 2006, 70, 212–221. [Google Scholar] [CrossRef]
- Higuchi, T.; Nakamura, M.; Shimada, K.; Ishida, E.; Hirao, K.; Konishi, N. HRK inactivation associated with promoter methylation and LOH in prostate cancer. Prostate 2008, 68, 105–113. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-Repair Gene MGMT and the Clinical Response of Gliomas to Alkylating Agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Mittag, F.; Kuester, D.; Vieth, M.; Peters, B.; Stolte, B.; Roessner, A.; Schneider-Stock, R. DAPK promotor methylation is an early event in colorectal carcinogenesis. Cancer Lett. 2006, 240, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, S.; Toyooka, K.O.; Miyajima, K.; Reddy, J.L.; Toyota, M.; Sathyanarayana, U.G.; Padar, A.; Tockman, M.S.; Lam, S.; Shivapurkar, N.; et al. Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin. Cancer Res. 2003, 9, 3034–3041. [Google Scholar] [CrossRef]
- Kissil, J.L.; Feinstein, E.; Cohen, O.; Jones, P.A.; Tsai, Y.C.; Knowles, M.A.; Eydmann, M.E.; Kimchi, A. DAP-kinase loss of expression in various carcinoma and B-cell lymphoma cell lines: Possible implications for role as tumor suppressor gene. Oncogene 1997, 15, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Biel, M.A.; Zhou, W.; Weitzman, S.A.; Baylin, S.B.; Gabrielson, E. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene 1998, 16, 2159–2164. [Google Scholar] [CrossRef] [Green Version]
- Waha, A.; Waha, A.; Koch, A.; Meyer-Puttlitz, B.; Weggen, S.; Sörensen, N.; Tonn, J.C.; Albrecht, S.; Goodyer, C.G.; Berthold, F.; et al. Epigenetic Silencing of the HIC-1 Gene in Human Medulloblastomas. J. Neuropathol. Exp. Neurol. 2003, 62, 1192–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agirre, X.; Novo, F.J.; Calasanz, M.J.; Larráyoz, M.J.; Lahortiga, I.; Valgañón, M.; García-Delgado, M.; Vizmanos, J.L. TP53 Is Frequently Altered by Methylation, Mutation, and/or Deletion in Acute Lymphoblastic Leukaemia. Mol. Carcinog. 2003, 38, 201–208. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Kiss, N.B.; Villablanca, A.; Haglund, F.; Nordenström, J.; Höög, A.; Larsson, C. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS ONE 2010, 5, e9472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Liu, H.; Chen, Y.; Liu, W.; Yu, J.; Wu, G. Methylation associated inactivation of RASSF1A and its synergistic effect with activated K-Ras in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 160. [Google Scholar] [CrossRef] [Green Version]
- Niklinska, W.; Naumnik, W.; Sulewska, A.; Kozłowski, M.; Pankiewicz, W.; Milewski, R. Prognostic significance of DAPK and RASSF1A promoter hypermethylation in Non-Small Cell Lung Cancer (NSCLC). Folia Histochem. Cytobiol. 2009, 47, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, S.; Haruta, M.; Sugawara, W.; Sasaki, F.; Ohira, M.; Matsunaga, T.; Yamaoka, H.; Horie, H.; Ohnuma, N.; Nakagawara, A.; et al. The methylation status of RASSF1A promoter predicts responsiveness to chemotherapy and eventual cure in hepatoblastoma patients. Int. J. Cancer 2008, 123, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988, 78, 151–155. [Google Scholar] [CrossRef]
- Rideout, W.M.; Coetzee, G.A.; Olumi, A.F.; Jones, P.A. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990, 249, 1288–1290. [Google Scholar] [CrossRef]
- Denissenko, M.F.; Chen, J.X.; Tang, M.-s.; Pfeifer, G.P. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc. Natl. Acad. Sci. USA 1997, 94, 3893–3898. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Fernandez, L.; Banyasz, A.; Esposito, L.; Markovitsi, D.; Improta, R. UV-induced damage to DNA: Effect of cytosine methylation on pyrimidine dimerization. Signal. Transduct. Target. Ther. 2017. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Gama-sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Bedford, M.T.; van Helden, P.D. Hypomethylation of dna in pathological conditions of the human prostate. Cancer Res. 1987. [Google Scholar]
- Wahlfors, J.; Hiltunen, H.; Heinonen, K.; Hmlinen, E.; Alhonen, L.; Jnne, J. Genomic hypomethylation in human chronic lymphocytic leukemia. Blood 1992, 80, 2074–2080. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Hsieh, S.Y.; Sheen, I.S.; Lee, W.C.; Chen, T.C.; Shyu, W.C.; Liaw, Y.F. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001, 61, 4238–4243. [Google Scholar] [PubMed]
- Kim, Y.-I.; Giuliano, A.; Hatch, K.D.; Schneider, A.; Nour, M.A.; Dallal, G.E.; Selhub, J.; Mason, J.B. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994, 74, 893–899. [Google Scholar] [CrossRef]
- Gaudet, F. Induction of Tumors in Mice by Genomic Hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Dante, R.; Dante-Paire, J.; Rigal, D.; Roizes, G. Methylation patterns of long interspersed repeated DNA and alphoid repetitive DNA from human cell lines and tumors. Anticancer Res. 1992, 12, 559–563. [Google Scholar] [PubMed]
- Jürgens, B.; Schmitz-Dräger, B.J.; Schulz, W.A. Hypomethylation of L1 LINE sequences prevailing in human urothelial carcinoma. Cancer Res. 1996, 56, 5698–5703. [Google Scholar] [PubMed]
- Takai, D. Hypomethylation of LINE1 Retrotransposon in Human Hepatocellular Carcinomas, but Not in Surrounding Liver Cirrhosis. Jpn. J. Clin. Oncol. 2002, 30, 306–309. [Google Scholar] [CrossRef] [Green Version]
- Santourlidis, S.; Florl, A.; Ackermann, R.; Wirtz, H.C.; Schulz, W.A. High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate 1999. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, V.; Ribieras, S.; Song-Wang, X.G.; Lasne, Y.; Frappart, L.; Rio, M.C.; Dante, R. Involvement of DNA methylation in the control of the expression of an estrogen-induced breast-cancer-associated protein (pS2) in human breast cancers. J. Cell. Biochem. 1997, 65, 95–106. [Google Scholar] [CrossRef]
- Watt, P.M.; Kumar, R.; Kees, U.R. Promoter demethylation accompanies reactivation of the HOX11 proto-oncogene in leukemia. Genes Chromosom. Cancer 2000, 29, 371–377. [Google Scholar] [CrossRef]
- Sharrard, R.M.; Royds, J.A.; Rogers, S.; Shorthouse, A.J. Patterns of methylation of the c-myc gene in human colorectal cancer progression. Br. J. Cancer 1992, 65, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Fang, J.; Qiu, D.; Zhang, T.; Yang, J.; Chen, S.; Xiao, S. Correlation between DNA methylation and pathological changes in human hepatocellular carcinoma. Hepatogastroenterology 1998, 45, 1753–1759. [Google Scholar]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [Green Version]
- Okitsu, C.Y.; Hsieh, C.-L. DNA Methylation Dictates Histone H3K4 Methylation. Mol. Cell. Biol. 2007, 27, 2746–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Gerson, A.; Fang, J.; Li, E.; Zhang, Y.; Shinkai, Y.; Cedar, H.; Bergman, Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol. 2006, 8, 188–194. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Th’ng, J.P. Histone modifications and apoptosis: Cause or consequence? Biochem. Cell Biol. 2011, 79, 305–311. [Google Scholar] [CrossRef]
- Lu, Y.; Chu, A.; Wajapeyee, N.; Turker, M.S.; Glazer, P.M. Abstract 2887: Epigenetic silencing of the DNA repair genes, BRCA1 and MLH1, induced by hypoxic stress in a pathway dependent on the histone demethylase, LSD1. Cancer Res. 2015, 75, 2887. [Google Scholar] [CrossRef]
- Lu, J.; Feng, Y.; Wang, X.; Xu, L.; Pan, H.; Zhu, S.; Liang, Q.; Huang, B. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. FEBS J. 2009, 276, 4197–4206. [Google Scholar] [CrossRef]
- Yang, X.; Karuturi, R.K.M.; Sun, F.; Aau, M.; Yu, K.; Shao, R.; Miller, L.D.; Tan, P.B.O.; Yu, Q. CDKN1C (p57KIP2) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS ONE 2009, 4, e5011. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stucki, M. Histone H2A.X Tyr142 phosphorylation: A novel sWItCH for apoptosis? DNA Repair 2009, 8, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, W.L.; Ajiro, K.; Samejima, K.; Kloc, M.; Cheung, P.; Mizzen, C.A.; Beeser, A.; Etkin, L.D.; Chernoff, J.; Earnshaw, W.C.; et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003, 113, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Ajiro, K.; Scoltock, A.B.; Smith, L.K.; Ashasima, M.; Cidlowski, J.A. Reciprocal epigenetic modification of histone H2B occurs in chromatin during apoptosis in vitro and in vivo. Cell Death Differ. 2010, 17, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.H.; Chan, H.; Ho, C.Y.; Lai, S.K.; Chan, K.S.; Koh, C.G.; Li, H.Y. Apoptotic histone modification inhibits nuclear transport by regulating RCC1. Nat. Cell Biol. 2009, 11, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Hurd, P.J.; Bannister, A.J.; Halls, K.; Dawson, M.A.; Vermeulen, M.; Olsen, J.V.; Ismail, H.; Somers, J.; Mann, M.; Owen-Hughes, T.; et al. Phosporylation of histone H3 Thr-45 is linked to apoptosis. J. Biol. Chem. 2009, 284, 16575–16583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Punj, V.; Choi, J.; Heo, K.; Kim, J.-M.; Laird, P.W.; An, W. Gene dysregulation by histone variant H2A.Z in bladder cancer. Epigenetics Chromatin 2013, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Tong, R.; Liu, H.; Wu, J.; Chen, D.; Xue, Z.; Ding, C.; Zhou, L.; Xie, H.; Wu, J.; et al. H2A.Z regulates tumorigenesis, metastasis and sensitivity to cisplatin in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2018, 52, 1235–1245. [Google Scholar] [CrossRef]
- Aguilera, D.G.; Das, C.M.; Sinnappah-Kang, N.D.; Joyce, C.; Taylor, P.H.; Wen, S.; Hasselblatt, M.; Paulus, W.; Fuller, G.; Wolff, J.E.; et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J. Neurooncol. 2009, 93, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc min mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Ianari, A.; Gallo, R.; Palma, M.; Alesse, E.; Gulino, A. Specific role for p300/CREB-binding protein-associated factor activity in E2F1 stabilization in response to DNA damage. J. Biol. Chem. 2004, 279, 30830–30835. [Google Scholar] [CrossRef] [Green Version]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, A.; Piccin, D.; Tagliati, F.; Luchin, A.; Zatelli, M.C.; Uberti, E.C.D. miR-15a and miR-16-1 down-regulation in pituitary adenomas. J. Cell. Physiol. 2005, 204, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Liu, C.; Vasan, K.; Sulda, M.; Puduvalli, V.K.; Wierda, W.G.; Keating, M.J. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood 2012, 119, 1162–1172. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Hao, X.; Meng, Y.; Zhang, M.; DeSano, J.; Fan, D.; Xu, L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M. MiRNAs and cancer: An epigenetics view. Mol. Aspects Med. 2013, 34, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Körner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.A.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef] [Green Version]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.H.; Huang, J.; Wu, C.R.; Huang, L.Y.; Cui, J.; Xing, Z.Z.; Zhao, C.Y. Downregulation of miR-29b targets DNMT3b to suppress cellular apoptosis and enhance proliferation in pancreatic cancer. Mol. Med. Rep. 2017, 17, 2113–2120. [Google Scholar] [CrossRef] [Green Version]
- Ratert, N.; Meyer, H.A.; Jung, M.; Mollenkopf, H.J.; Wagner, I.; Miller, K.; Kilic, E.; Erbersdobler, A.; Weikert, S.; Jung, K. Reference miRNAs for miRNAome analysis of urothelial carcinomas. PLoS ONE 2012, 7, e39309. [Google Scholar] [CrossRef] [Green Version]
- Flavin, R.; Smyth, P.; Barrett, C.; Russell, S.; Wen, H.; Wei, J.; Laios, A.; O’Toole, S.; Ring, M.; Denning, K.; et al. MiR-29b expression is associated with disease-free survival in patients with ovarian serous carcinoma. Int. J. Gynecol. Cancer 2009, 19, 641–647. [Google Scholar] [CrossRef]
- Cortez, M.A.; Nicoloso, M.S.; Shimizu, M.; Rossi, S.; Gopisetty, G.; Molina, J.R.; Carlotti, C.; Tirapelli, D.; Neder, L.; Brassesco, M.S.; et al. miR-29b and miR-125a regulate podoplanin and suppress invasion in glioblastoma. Genes Chromosom. Cancer 2010, 49, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef]
- Saito, Y.; Suzuki, H.; Tsugawa, H.; Nakagawa, I.; Matsuzaki, J.; Kanai, Y.; Hibi, T. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 2009, 28, 2738–2744. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, A.; Fallah, S.; Ansari, M. MIR-153 as a tumor suppressor in glioblastoma multiforme is downregulated by DNA methylation. Clin. Lab. 2016, 59. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.; Batte, K.; Yu, L.; Wu, X.; Nuovo, G.J.; Marsh, C.B.; Otterson, G.A.; Nana-Sinkam, S.P. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem. Biophys. Res. Commun. 2009, 388, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, M.; Guo, M.; Xie, Y.; Cong, Y.S. miR-127 regulates cell proliferation and senescence by targeting BCL6. PLoS ONE 2013, 8, e80266. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrocca, F.; Visone, R.; Onelli, M.R.; Shah, M.H.; Nicoloso, M.S.; de Martino, I.; Iliopoulos, D.; Pilozzi, E.; Liu, C.G.; Negrini, M.; et al. E2F1-Regulated MicroRNAs Impair TGFβ-Dependent Cell-Cycle Arrest and Apoptosis in Gastric Cancer. Cancer Cell 2008, 13, 272–286. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lan, P.; Hou, Z.; Guan, Y.; Zhang, J.; Xu, W.; Tian, Z.; Zhang, C. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br. J. Cancer 2015, 112, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Z.; Zhang, J.X.; Zhang, A.L.; Shi, Z.D.; Han, L.; Jia, Z.F.; Yang, W.D.; Wang, G.X.; Jiang, T.; You, Y.P.; et al. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol. Cancer 2010, 9, 229. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Cai, L.; Wang, C.; Deng, X.; Yi, S.; Lei, Z.; Xiao, Q.; Xu, H.; Luo, H.; Sun, J. CASC2/miR-24/miR-221 modulates the TRAIL resistance of hepatocellular carcinoma cell through caspase-8/caspase-3. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Lu, C.; Zhou, G.P.; Zhang, W.; Xiao, H.; Wang, X.R. MicroRNA-221 silencing predisposed human bladder cancer cells to undergo apoptosis induced by TRAIL. Urol. Oncol. Semin. Orig. Investig. 2010, 28, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Garofalo, M.; Zanca, C.; Romano, G.; Iaboni, M.; Del Basso De Caro, M.; Martinez-Montero, J.C.; Incoronato, M.; Nuovo, G.; Croce, C.M.; et al. MiR-221/222 overexpession in human glioblastoma increases invasiveness by targeting the protein phosphate PTP. Oncogene 2012, 31, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.N.; Bai, J.X.; Zhou, Q.; Yan, B.; Qin, W.W.; Jia, L.T.; Meng, Y.L.; Jin, B.Q.; Yao, L.B.; Wang, T.; et al. TSA Suppresses miR-106b-93-25 Cluster Expression through Downregulation of MYC and Inhibits Proliferation and Induces Apoptosis in Human EMC. PLoS ONE 2012, 7, e45133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic MicroRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Navarro, A.; Diaz, T.; Martinez, A.; Gaya, A.; Pons, A.; Gel, B.; Codony, C.; Ferrer, G.; Martinez, C.; Montserrat, E.; et al. Regulation of JAK2 by miR-135a: Prognostic impact in classic Hodgkin lymphoma. Blood 2009, 114, 2945–2951. [Google Scholar] [CrossRef]
- Wu, H.; Huang, M.; Cao, P.; Wang, T.; Shu, Y.; Liu, P. MiR-135a targets JAK2 and inhibits gastric cancer cell proliferation. Cancer Biol. Ther. 2012, 13, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wen, Q. Downregulation of miR-135a predicts poor prognosis in acute myeloid leukemia and regulates leukemia progression via modulating HOXA10 expression. Mol. Med. Rep. 2018, 18, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Dong, X.; Hai, J.; Jiang, J.; Wang, W.; Yang, J.; Zhang, W.; Chen, C. MicroRNA-135a-3p is downregulated and serves as a tumour suppressor in ovarian cancer by targeting CCR2. Biomed. Pharmacother. 2018, 107, 712–720. [Google Scholar] [CrossRef]
- Nakano, H.; Miyazawa, T.; Kinoshita, K.; Yamada, Y.; Yoshida, T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-XLin colorectal cancer cells. Int. J. Cancer 2010, 127, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Nakamura, Y. EGR2 induces apoptosis in various cancer cell lines by direct transactivation of BNIP3L and BAK. Oncogene 2003, 22, 2172–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Jin, H.; Yang, Z.; Luo, G.; Lu, Y.; Li, K.; Ren, G.; Su, T.; Pan, Y.; Feng, B.; et al. MiR-150 promotes gastric cancer proliferation by negatively regulating the pro-apoptotic gene EGR2. Biochem. Biophys. Res. Commun. 2010, 392, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E.J.; Pinchuk, I.V.; Suarez, G.; Sierra, J.C.; Reyes, V.E. Helicobacter pylori CagA-Dependent Macrophage Migration Inhibitory Factor Produced by Gastric Epithelial Cells Binds to CD74 and Stimulates Procarcinogenic Events. J. Immunol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Gong, J.-M.; Zhou, L.-L.; Sheng, J.-H. MiR-451 as a new tumor marker for gastric cancer. Oncotarget 2017, 8, 56542–56545. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, H.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Shah, K.M.; Young, L.S. Epstein-Barr virus and carcinogenesis: Beyond Burkitt’s lymphoma. Clin. Microbiol. Infect. 2009, 15, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Choy, E.Y.-W.; Siu, K.-L.; Kok, K.-H.; Lung, R.W.-M.; Tsang, C.M.; To, K.-F.; Kwong, D.L.-W.; Tsao, S.W.; Jin, D.-Y. An Epstein-Barr virus–encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Meco, M.T.; Abu-Baker, S. The Par-4/PTEN connection in tumor suppression. Cell Cycle 2009, 8, 2518–2522. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, Y.; Nakada, C.; Noguchi, T.; Tanigawa, M.; Nguyen, L.T.; Uchida, T.; Hijiya, N.; Matsuura, K.; Fujioka, T.; Seto, M.; et al. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3ζ. Cancer Res. 2010, 70, 2339–2349. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.M.; Tergaonkar, V. NFκB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 2009, 14, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′- deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Suciu, S.; Hagemeijer, A.; Rüter, B.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Decitabine improves progression-free survival in older high-risk MDS patients with multiple autosomal monosomies: Results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group. Ann. Hematol. 2016, 95, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.P.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous t-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, Y.L.; Chen, X.H.; et al. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.P.; Rouge, T.d.L.M.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A. Clinical response of carcinomas harboring the BRD4–NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol. Cancer Res. 2010, 8, 1610–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2010, 8, 1610–1618. [Google Scholar] [CrossRef] [Green Version]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [Green Version]
- Vester, B.; Wengel, J. LNA (Locked Nucleic Acid): High-affinity targeting of complementary RNA and DNA. Biochemistry 2004, 43, 13233–13241. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Monroig, P. del C.; Chen, L.; Zhang, S.; Calin, G.A. Small molecule compounds targeting miRNAs for cancer therapy. Adv. Drug Deliv. Rev. 2015, 81, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griveau, A.; Bejaud, J.; Anthiya, S.; Avril, S.; Autret, D.; Garcion, E. Silencing of miR-21 by locked nucleic acid-lipid nanocapsule complexes sensitize human glioblastoma cells to radiation-induced cell death. Int. J. Pharm. 2013, 454, 765–774. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Trang, P.; Medina, P.P.; Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Homer, R.; Brown, D.; Bader, A.G.; Weidhaas, J.B.; et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene 2010, 29, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daige, C.; Priddy, L.; Wiggins, J.; Nelligan-Davis, T.; Enzler, D.; Vadnagara, K.; Brown, D. MRX34, a liposomal miR-34 mimic and potential first-in-class microRNA therapeutic: Activity in animal models of liver cancer. J. Clin. Oncol. 2016, 34, e14076. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Humphreys, K.J.; Cobiac, L.; Le Leu, R.K.; Van der Hoek, M.B.; Michael, M.Z. Histone deacetylase inhibition in colorectal cancer cells reveals competing roles for members of the oncogenic miR-17-92 cluster. Mol. Carcinog. 2013, 52, 459–474. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re- expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar] [PubMed]
- Belinsky, S.A.; Klinge, D.M.; Stidley, C.A.; Issa, J.P.; Herman, J.G.; March, T.H.; Baylin, S.B. Inhibition of DNA Methylation and Histone Deacetylation Prevents Murine Lung Cancer. Cancer Res. 2003, 63, 7089–7093. [Google Scholar] [PubMed]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Mönkemeyer, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor effects of a combined 5-aza-2′deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res. 2009, 69, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdy, S.C.; Jiang, S.; Zhou, X.C.; Hou, X.; Jin, F.; Podratz, K.C.; Jiang, S.-W. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006, 5, 2767–2776. [Google Scholar] [CrossRef] [Green Version]
- Arnold, N.B.; Arkus, N.; Gunn, J.; Korc, M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin. Cancer Res. 2007, 13, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Rikiishi, H.; Shinohara, F.; Sato, T.; Sato, Y.; Suzuki, M.; Echigo, S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int. J. Oncol. 2007, 30, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of Histone Deacetylase Increases Cytotoxicity to Anticancer Drugs Targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar]
- Gomyo, Y.; Sasaki, J.I.; Branch, C.; Roth, J.A.; Mukhopadhyay, T. 5-Aza-2′-deoxycytidine upregulates caspase-9 expression cooperating with p53-induced apoptosis in human lung cancer cells. Oncogene 2004, 23, 6779–6787. [Google Scholar] [CrossRef] [Green Version]
- Shang, D.; Liu, Y.; Liu, Q.; Zhang, F.; Feng, L.; Lv, W.; Tian, Y. Synergy of 5-aza-2′-deoxycytidine (DAC) and paclitaxel in both androgen-dependent and -independent prostate cancer cell lines. Cancer Lett. 2009, 278, 82–87. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef]
- Arundel, C.M.; Glicksman, A.S.; Leith, J.T. Enhancement of Radiation Injury in Human Colon Tumor Cells by the Maturational Agent Sodium Butyrate (NaB). Radiat. Res. 2006. [Google Scholar] [CrossRef]
- Karagiannis, T.C.; El-Osta, A. Modulation of cellular radiation responses by histone deacetylase inhibitors. Oncogene 2006, 25, 3885–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, T.C.; El-Osta, A. The paradox of histone deacetylase inhibitor-mediated modulation of cellular responses to radiation. Cell Cycle 2005, 5, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangert, A.; Cristofanon, S.; Eckhardt, I.; Abhari, B.A.; Kolodziej, S.; Häcker, S.; Vellanki, S.H.K.; Lausen, J.; Debatin, K.M.; Fulda, S. Histone deacetylase inhibitors sensitize glioblastoma cells to TRAIL-induced apoptosis by c-myc-mediated downregulation of cFLIP. Oncogene 2012, 31, 4677–4688. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Liapis, V.; Bouralexis, S.; Welldon, K.; Hay, S.; Thai, L.M.; Labrinidis, A.; Tilley, W.D.; Findlay, D.M.; Evdokiou, A. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAIL. Int. J. Cancer 2006, 119, 944–954. [Google Scholar] [CrossRef]
- Lagneaux, L.; Gillet, N.; Stamatopoulos, B.; Delforge, A.; Dejeneffe, M.; Massy, M.; Meuleman, N.; Kentos, A.; Martiat, P.; Willems, L.; et al. Valproic acid induces apoptosis in chronic lymphocytic leukemia cells through activation of the death receptor pathway and potentiates TRAIL response. Exp. Hematol. 2007, 35, 1527–1537. [Google Scholar] [CrossRef]
- VanOosten, R.L.; Moore, J.M.; Ludwig, A.T.; Griffith, T.S. Depsipeptide (FR901228) enhances the cytotoxic activity of TRAIL by redistributing TRAIL receptor to membrane lipid rafts. Mol. Ther. 2005, 11, 542–552. [Google Scholar] [CrossRef]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate: A potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem. Pharmacol. 2003, 66, 1537–1545. [Google Scholar] [CrossRef]
- Guo, F.; Sigua, C.; Tao, J.; Bali, P.; George, P.; Li, Y.; Wittmann, S.; Moscinski, L.; Atadja, P.; Bhalla, K. Cotreatment with Histone Deacetylase Inhibitor LAQ824 Enhances Apo-2L/ Tumor Necrosis Factor-Related Apoptosis Inducing Ligand-Induced Death Inducing Signaling Complex Activity and Apoptosis of Human Acute Leukemia Cells. Cancer Res. 2004, 64, 2580–2589. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Küfer, M.U.; Meyer, E.; Van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Debatin, K.M. 5-Aza-2′-deoxycytidine and IFN-γ cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene 2006, 25, 5125–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotzer, M.; Eggert, A.; Zuzak, T.; Janss, A.; Marwaha, S.; Wiewrodt, B.; Ikegaki, N.; Brodeur, G.; Phillips, P. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene 2000, 19, 4604–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminskyyy, V.O.; Surovay, O.V.; Vaculova, A.; Zhivotovsky, B. Combined inhibition of DNA methyltransferase and histone deacetylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis 2011, 32, 1450–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, S.; Higuchi, H.; Saito, Y.; Nakamoto, N.; Takaishi, H.; Tada, S.; Saito, H.; Gores, G.J.; Hibi, T. DNMT1 and DNMT3b silencing sensitizes human hepatoma cells to TRAIL-mediated apoptosis via up-regulation of TRAIL-R2/DR5 and caspase-8. Cancer Sci. 2010, 101, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Florean, C.; Schnekenburger, M.; Lee, J.-Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.-M.; Grandjenette, C.; Kim, J.-G.; Yoon, A.-Y.; et al. Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef]
- Weigt, D.; Hopf, C.; Médard, G. Studying epigenetic complexes and their inhibitors with the proteomics toolbox. Clin. Epigenetics 2016, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.H.; Plass, C.; Chen, C.S. Effects of histone deacetylase inhibitors on modulating H3K4 methylation marks—A novel cross-talk mechanism between histone-modifying enzymes. Mol. Cell. Pharmacol. 2011, 3, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Roy, S.; Namjoshi, S.; Malarkey, C.S.; Jones, D.N.M.; Kutateladze, T.G.; Churchill, M.E.A.; Tyler, J.K. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc. Natl. Acad. Sci. USA 2014, 111, E1072–E1081. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006. [Google Scholar] [CrossRef]
- Day, J.J. New approaches to manipulating the epigenome. Dialogues Clin. Neurosci. 2014, 16, 345–357. [Google Scholar]
- Geel, T.M.; Ruiters, M.H.J.; Cool, R.H.; Halby, L.; Voshart, D.C.; Andrade Ruiz, L.; Niezen-Koning, K.E.; Arimondo, P.B.; Rots, M.G. The past and presence of gene targeting: From chemicals and DNA via proteins to RNA. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170077. [Google Scholar] [CrossRef] [PubMed]
- Tye, K.M.; Deisseroth, K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat. Rev. Neurosci. 2012. [Google Scholar] [CrossRef]
- Cano-Rodriguez, D.; Gjaltema, R.A.F.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.J.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef] [PubMed]
- Mlambo, T.; Nitsch, S.; Hildenbeutel, M.; Romito, M.; Müller, M.; Bossen, C.; Diederichs, S.; Cornu, T.I.; Cathomen, T.; Mussolino, C. Designer epigenome modifiers enable robust and sustained gene silencing in clinically relevant human cells. Nucleic Acids Res. 2018, 46, 4456–4468. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pro/Antiapoptotic Genes | Epigenetic Modification | Outcome | Cancer Type |
|---|---|---|---|
| FAS | DNA hypermethylation | Downregulation | T-cell lymphoma [84], colon carcinoma [85] |
| DR4/DR5 | DNA hypermethylation | Downregulation | Neuroblastoma [86], melanoma [87] and ovarian cancer [88], |
| H3 and H4 deacetylation | Downregulation | Medulloblastoma [170] | |
| CASPASE-8/10 | DNA hypermethylation | Downregulation | Hepatocellular carcinoma [89], bladder cancer [90], small-cell lung carcinoma [91], GBM [92], retinoblastoma, and neuroblastoma [93] |
| BIM | DNA hypermethylation | Downregulation | Renal cell carcinoma and chronic myeloid leukemia [94] |
| H3K27me3 repressive mark | Downregulation | Burkitt’s lymphoma [172] | |
| APAF-1 | DNA hypermethylation | Downregulation | Leukemia [95], melanoma [96], and gastric [97], bladder, and kidney cancer [98] |
| XAF1 | DNA hypermethylation | Downregulation | Gastric and bladder cancer [99,100,101] |
| BCL-2 | DNA hypermethylation | Downregulation | Prostate cancer [102] |
| miR-15/16 silencing by histone deacetylation | Upregulation | Pituitary adenoma [180] and B-cell chronic lymphocyte leukemia [181] | |
| miR-34 hypermethylation | Upregulation | Gastric cancer, chronic lymphocytic leukemia, pancreatic, breast, colon, and kidney cancer, and Burkitt’s lymphoma [184,185] | |
| BAX | DNA hypermethylation | Downregulation | Multiple myeloma cells [103] and Burkitt’s lymphoma [104] |
| H3 and H4 deacetylation | Downregulation | Colon cancer [171] | |
| BAK | DNA hypermethylation | Downregulation | Multiple myeloma cells [103] and Burkitt’s lymphoma [104] |
| PUMA | DNA hypermethylation | Downregulation | Multiple myeloma cells [103] and Burkitt’s lymphoma [104] |
| BAD | DNA hypermethylation | Downregulation | Multiple myeloma cells [103] |
| BCL-2L10 | DNA hypermethylation | Downregulation | Gastric cancer [105] and leukemia [106] |
| BIK | DNA hypermethylation | Downregulation | Glioma [107], RCC [108], prostate cancer [109], and myeloma [110] |
| BNIP3 | DNA hypermethylation | Downregulation | Gastric cancer [111], colorectal cancer [112], leukemia [113], and HCC [114] |
| HRK | DNA hypermethylation | Downregulation | Colorectal, gastric [115], GBM [116], PCNSL [117], and prostate cancer [118] |
| miRNA | miRNA Status in Cancer | Target Gene & Outcome | Cancer Type |
|---|---|---|---|
| miR-29b | Downregulated | HRK upregulation | Lung [187], prostate [188], bladder [189], and ovarian cancers [190] and GBM [191] |
| miR-193a-3p, miR-512-5p, miR-153, miR-133B | Hypermethylated | MLC1 upregulation | AML [192], gastric tumors [193], GBM [194], and lung cancer [195] |
| miR-127 | Hypermethylated | BCL-6 upregulation | Bladder, prostate, breast, and lung cancer and lymphoma [197] |
| miR-221, miR-222 | Upregulated | PUMA and CASPASE-3 downregulation | Bladder [203] and glioma [204] |
| miR-17-92, miR-106b-93-25 | Overexpressed | BIM downregulation | Lung, colon, lymphoma, medulloblastoma, and multiple myeloma [206,207] |
| miR-135a | Downregulated | BCL-xL upregulation | Hodgkin lymphoma, AML [210], and ovarian cancer [211] |
| miR-451 | Downregulated | BCL-2 upregulation | Gastric cancer [216] |
| Other Apoptosis Related Genes | Epigenetic Modification | Outcome |
|---|---|---|
| DAPK | DNA hypermethylation [121,122,123] | Downregulation |
| HIC1 | DNA hypermethylation [124,125] | Downregulation |
| p16INK4a | DNA hypermethylation [119] | Downregulation |
| APC | DNA hypermethylation [119] | Downregulation |
| TP53 | DNA hypermethylation [126] | Downregulation |
| RASSF1 | DNA hypermethylation [127,128,129,130] | Downregulation |
| MGMT | DNA hypermethylation [119,120] | Downregulation |
| Increasing H3K4me3 [35] | Downregulation | |
| BRCA1 | DNA Hypermethylation [119] | Downregulation |
| Increased H3K4me3 [35] | Downregulation | |
| MLH1 | DNA hypermethylation [119] | Downregulation |
| Decreased H3K4 methylation [158] | ||
| pS2 | DNA hypomethylation [147] | Upregulation |
| HOX11 | DNA hypomethylation [148] | Upregulation |
| c-MYC | DNA hypomethylation [149,150] | Upregulation |
| c-N-RAS | DNA hypomethylation [149,150] | Upregulation |
| MRE11 | H2A.X-Y142 phosphorylation [162,163] | Binding to γ-H2A.X sites is blocked |
| RAD50 | ||
| NBS1 | ||
| 53BP1 | ||
| BRCA1 | ||
| NF-κB | H2B-S14ph [166] | Inhibited by reduced nuclear trafficking |
| E2F1 | Protein acetylation [174] | Half-life and DNA binding affinity is increased |
| KU70 | Protein acetylation [175] | Interaction with BAX is disrupted |
| Targeted Epigenetic Modification | Class | Agent | FDA Approval Status | Targeted Disease |
|---|---|---|---|---|
| DNA Methylation | DNMT1 inhibitors | Azacitidine | Approved (2004) | MDS and AML |
| Decitabine | Approved (2006) | MDS and AML | ||
| Guadecitabine | Clinical trial | Liver, pancreatic, bile duct, or gallbladder cancer (NCT03257761), non-small-cell lung cancer (NCT03220477), kidney cancer (NCT03220477), AML (NCT02878785), and urothelial cancer (NCT03179943) | ||
| 4-Thio-2-deoxycytidine (TdCyd) | Clinical trial | Solid tumors (NCT02423057) | ||
| IDH1 and IDH2 inhibitors | AG-120 (Ivosidenib) | Clinical trial | AML (NCT03173248), myelodysplastic syndrome, chronic myelomonocytic leukemia (NCT03564821), and hematologic malignancies (NCT03471260) | |
| AG-221 (Enasidenib) | Clinical trial | AML (NCT03013998, NCT02577406, NCT03825796, NCT03728335, NCT03683433, NCT03881735) and MDS (NCT03383575) | ||
| AG-881 (Vorasidenib) | Clinical trial | Hematologic malignancies (NCT02492737) | ||
| IDH305 | Clinical trial | Low-grade gliomas (NCT02987010) | ||
| Histone Acetylation | HDAC inhibitors | Vorinostat (SAHA) | Approved (2006) | Cutaneous or peripheral T-cell lymphoma |
| Belinostat | Approved (2014) | Peripheral T-cell lymphoma | ||
| Romidepsin | Approved (2009) | Cutaneous or peripheral T-cell lymphoma | ||
| Trichostatin A | Clinical trial | Hematologic malignancies (NCT03838926) | ||
| Panobinostat | Approved (2015) | Multiple myeloma | ||
| Histone acetylation reader inhibitors | OTX015 | Clinical trial | Hematologic malignancies (NCT01713582) | |
| ABBV-075 (Mivebresib) | Clinical trial | Breast cancer, non-small-cell lung cancer, AML, multiple myeloma, prostate cancer, small-cell lung cancer, and non-Hodgkin’s lymphoma (NCT02391480) | ||
| BMS-986158 | Clinical trial | Advanced cancers (NCT02419417), refractory solid tumors, central nervous system tumors, or lymphoma (NCT03936465) | ||
| GSK2820151 | Clinical trial | Advanced or recurrent solid tumors (NCT02630251) | ||
| Histone Methylation | HMT inhibitors | Tazemetostat | Clinical trial | Advanced solid tumors, non-Hodgkin’s lymphoma, histiocytic disorders (NCT03213665), synovial sarcoma (NCT02601937), urothelial carcinoma (NCT03854474), and ovarian or endometrial cancer (NCT03348631) |
| CPI-1205 | Clinical trial | Castration-resistant prostate cancer (NCT03480646) | ||
| DS-3201 (Valemetostat) | Clinical trial | Lymphomas (NCT02732275), AML, and ALL (NCT03110354) | ||
| GSK2816126 | Clinical trial | Diffuse large B-cell lymphoma, transformed follicular lymphoma, other non-Hodgkin’s lymphomas, solid tumors, and multiple myeloma (NCT02082977) | ||
| Pinometostat | Clinical trial | AML (NCT03724084, NCT03701295) | ||
| HDM inhibitors | GSK2879552 | Clinical trial | Small-cell lung carcinoma (NCT02034123) and AML (NCT02177812) | |
| INCB059872 | Clinical trial | Ewing sarcoma (NCT03514407), solid tumors, and hematologic malignancy (NCT02712905) | ||
| miRNA Regulation | miRNA mimics | MesomiR-1 | Clinical trial | NSCLC and MPM (NCT02369198) |
| MRX34 | Clinical trial | Hepatocellular carcinoma, renal cell carcinoma, and melanoma (NCT01829971) | ||
| miRNA antagonist | MRG-106 | Clinical trial | T-cell lymphoma (NCT03713320) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozyerli-Goknar, E.; Bagci-Onder, T. Epigenetic Deregulation of Apoptosis in Cancers. Cancers 2021, 13, 3210. https://doi.org/10.3390/cancers13133210
Ozyerli-Goknar E, Bagci-Onder T. Epigenetic Deregulation of Apoptosis in Cancers. Cancers. 2021; 13(13):3210. https://doi.org/10.3390/cancers13133210
Chicago/Turabian StyleOzyerli-Goknar, Ezgi, and Tugba Bagci-Onder. 2021. "Epigenetic Deregulation of Apoptosis in Cancers" Cancers 13, no. 13: 3210. https://doi.org/10.3390/cancers13133210