
Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer

 ,
,  , ,
, ,  ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Culture and Treatments
2.3. Transient Transfection with siRNA
2.4. Preparation of Cell Lysates
2.5. SDS–PAGE and Immunoblot Analysis
2.6. Antibodies
2.7. Isolation of RNA, cDNA Synthesis, and Quantitative Real-Time PCR (qPCR)
2.8. Measurement of ROS Formation
2.9. Alkaline Comet Assay
2.10. Cell Cycle Analysis
2.11. Cell Death Analysis
2.12. Cell Viability Assay and Determination of IC50 Values
2.13. Analysis of Cell Morphology
2.14. Cultivation of Murine Tumor Organoids and Assessment of Viability
2.15. Immunofluorescence Microscopy of CRC Cells and Tumor Organoids
2.16. Statistics
3. Results
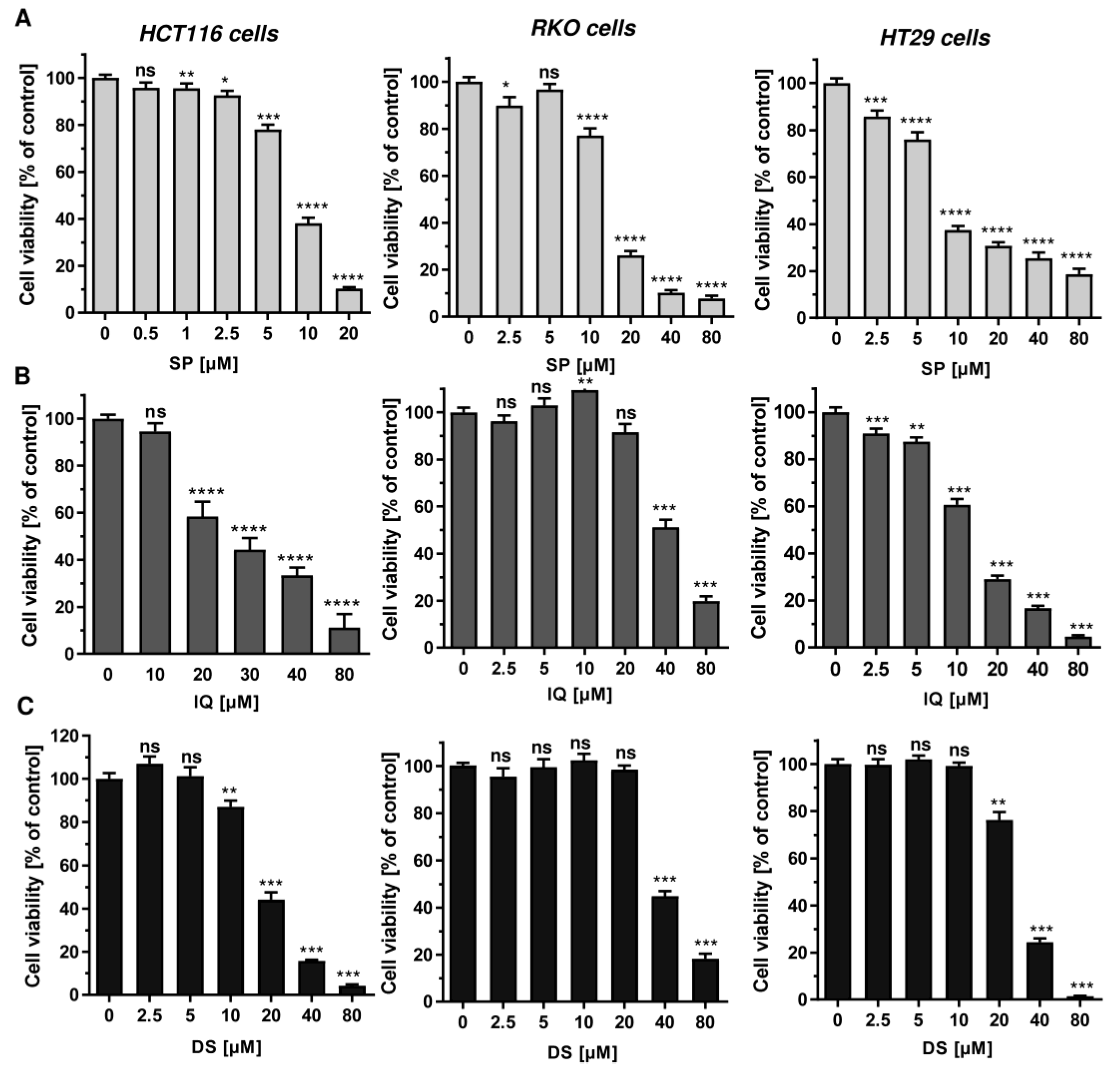
3.1. Cytotoxicity Screening of Merosesquiterpenes in Human CRC Cell Lines
3.2. Activation of the DNA Damage Response and Impact on Cell Cycle Progression
3.3. Cell Death Induction by Merosesquiterpenequinones and Impact of p53
3.4. Influence of Mutant p53 on Cell Death Induction
3.5. Therapeutic Efficacy of Merosesquiterpenquinone in Murine Tumor Organoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Maximo, P.; Ferreira, L.M.; Branco, P.; Lima, P.; Lourenco, A. The Role of Spongia sp. in the Discovery of Marine Lead Compounds. Mar. Drugs 2016, 14, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.H.; Chueh, S.C.; Kung, F.L.; Pan, S.L.; Shen, Y.C.; Guh, J.H. Ilimaquinone, a marine sponge metabolite, displays anticancer activity via GADD153-mediated pathway. Eur. J. Pharmacol. 2007, 556, 45–54. [Google Scholar] [CrossRef]
- Lin, C.W.; Bai, L.Y.; Su, J.H.; Chiu, C.F.; Lin, W.Y.; Huang, W.T.; Shih, M.C.; Huang, Y.T.; Hu, J.L.; Weng, J.R. Ilimaquinone Induces Apoptosis and Autophagy in Human Oral Squamous Cell Carcinoma Cells. Biomedicines 2020, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Do, M.T.; Na, M.; Kim, H.G.; Khanal, T.; Choi, J.H.; Jin, S.W.; Oh, S.H.; Hwang, I.H.; Chung, Y.C.; Kim, H.S.; et al. Ilimaquinone induces death receptor expression and sensitizes human colon cancer cells to TRAIL-induced apoptosis through activation of ROS-ERK/p38 MAPK-CHOP signaling pathways. Food Chem. Toxicol. 2014, 71, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kwak, C.H.; Jin, L.; Han, J.H.; Han, C.W.; Kim, E.; Cho, M.; Chung, T.W.; Bae, S.J.; Jang, S.B.; Ha, K.T. Ilimaquinone Induces the Apoptotic Cell Death of Cancer Cells by Reducing Pyruvate Dehydrogenase Kinase 1 Activity. Int. J. Mol. Sci. 2020, 21, 6021. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Chung, K.J.; Hwang, I.H.; Gwak, J.; Park, S.; Ju, B.G.; Yun, E.; Kim, D.E.; Chung, Y.H.; Na, M.; et al. Activation of p53 with ilimaquinone and ethylsmenoquinone, marine sponge metabolites, induces apoptosis and autophagy in colon cancer cells. Mar. Drugs 2015, 13, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Van Stuijvenberg, J.; Proksch, P.; Fritz, G. Targeting the DNA damage response (DDR) by natural compounds. Bioorg. Med. Chem. 2020, 28, 115279. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Aoki, S.; Sowa, Y.; Sakai, T.; Kobayashi, M. Smenospongine, a sesquiterpene aminoquinone from a marine sponge, induces G1 arrest or apoptosis in different leukemia cells. Mar. Drugs 2008, 6, 480–488. [Google Scholar] [CrossRef]
- Tang, J.; Wu, W.; Yang, F.; Liu, L.; Yang, Z.; Liu, L.; Tang, W.; Sun, F.; Lin, H. Marine sponge-derived smenospongine preferentially eliminates breast cancer stem-like cells via p38/AMPKalpha pathways. Cancer Med. 2018, 7, 3965–3976. [Google Scholar] [CrossRef] [Green Version]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Seiwert, N.; Heylmann, D.; Hasselwander, S.; Fahrer, J. Mechanism of colorectal carcinogenesis triggered by heme iron from red meat. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188334. [Google Scholar] [CrossRef]
- Vuik, F.E.; Nieuwenburg, S.A.; Bardou, M.; Lansdorp-Vogelaar, I.; Dinis-Ribeiro, M.; Bento, M.J.; Zadnik, V.; Pellise, M.; Esteban, L.; Kaminski, M.F.; et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut 2019, 68, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörsam, B.; Fahrer, J. The disulfide compound alpha-lipoic acid and its derivatives: A novel class of anticancer agents targeting mitochondria. Cancer Lett. 2016, 371, 12–19. [Google Scholar] [CrossRef]
- Neitzel, C.; Demuth, P.; Wittmann, S.; Fahrer, J. Targeting Altered Energy Metabolism in Colorectal Cancer: Oncogenic Reprogramming, the Central Role of the TCA Cycle and Therapeutic Opportunities. Cancers 2020, 12, 1731. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Mauri, G.; Arena, S.; Siena, S.; Bardelli, A.; Sartore-Bianchi, A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 2020, 31, 1135–1147. [Google Scholar] [CrossRef]
- Balansa, W.; Mettal, U.; Wuisan, Z.G.; Plubrukarn, A.; Ijong, F.G.; Liu, Y.; Schaberle, T.F. A New Sesquiterpenoid Aminoquinone from an Indonesian Marine Sponge. Mar. Drugs 2019, 17, 158. [Google Scholar] [CrossRef] [Green Version]
- Jiso, A.; Kittiwisut, S.; Chantakul, R.; Yuenyongsawad, S.; Putchakarn, S.; Schaberle, T.F.; Temkitthaworn, P.; Ingkaninan, K.; Chaithirayanon, K.; Plubrukarn, A. Quintaquinone, a Merosesquiterpene from the Yellow Sponge Verongula cf. rigida Esper. J. Nat. Prod. 2020, 83, 532–536. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.; Warren, R.; Wells, R.; Blount, J. New quinones from a dictyoceratid sponge. Aust. J. Chem. 1978, 31, 2685–2697. [Google Scholar] [CrossRef]
- Luibrand, R.T.; Erdman, T.R.; Vollmer, J.J.; Scheuer, P.J.; Finer, J.; Clardy, J. Ilimaquinone, a sesquiterpenoid quinone from a marine sponge. Tetrahydron 1979, 35, 609–612. [Google Scholar] [CrossRef]
- Carte, B.; Rose, C.B.; Faulkner, D.J. 5-Epi-Ilimaquinone, a metabolite of the sponge Fenestraspongia sp. J. Org. Chem. 1985, 50, 2785–2787. [Google Scholar] [CrossRef]
- Kondracki, M.-L.; Guyot, M. Smenospongine: A cytotoxic and antimicrobial aminoquinone isolated from Smenospongia sp. Tetrahedron Lett. 1987, 28, 5815–5818. [Google Scholar] [CrossRef]
- Kondracki, M.-L.; Guyot, M. Biologically active quinone and hydroquinone sesquiterpenoids from the sponge smenospongia sp. Tetrahedron 1989, 45, 1995–2004. [Google Scholar] [CrossRef]
- Kushlan, D.M.; Faulkner, D.J.; Parkanyi, L.; Clardy, J. Metabolites of the Palauan sponge dactylospongia sp. Tetrahydron 1989, 45, 3307–3312. [Google Scholar] [CrossRef]
- Venkateswarlu, Y.; Faulkner, D.J.; Steiner, J.L.R.; Corcoran, E.; Clardy, J. Smenochromenes, unusual macrocyclic sesquiterpene hydroquinone derivatives from a Seychelles sponge of the genus Smenospongia. J. Org. Chem. 1991, 56, 6271–6274. [Google Scholar] [CrossRef]
- Rodríguez, J.; Quiñoá, E.; Riguera, R.; Peters, B.M.; Abrell, L.M.; Crews, P. The structures and stereochemistry of cytotoxic sesquiterpene quinones from Dactylospongia elegans. Tetrahydron 1992, 48, 6667–6680. [Google Scholar] [CrossRef]
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787. [Google Scholar] [CrossRef]
- Göder, A.; Nagel, G.; Kraus, A.; Dörsam, B.; Seiwert, N.; Kaina, B.; Fahrer, J. Lipoic acid inhibits the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) and triggers its depletion in colorectal cancer cells with concomitant autophagy induction. Carcinogenesis 2015, 36, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef]
- Fahrer, J.; Schweitzer, B.; Fiedler, K.; Langer, T.; Gierschik, P.; Barth, H. C2-streptavidin mediates the delivery of biotin-conjugated tumor suppressor protein p53 into tumor cells. Bioconjug. Chem. 2013, 24, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, C.; Seiwert, N.; Göder, A.; Diehl, E.; Weber, C.; Nagel, G.; Stroh, S.; Rasenberger, B.; Christmann, M.; Fahrer, J. Lipoic Acid Synergizes with Antineoplastic Drugs in Colorectal Cancer by Targeting p53 for Proteasomal Degradation. Cells 2019, 8, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörsam, B.; Wu, C.F.; Efferth, T.; Kaina, B.; Fahrer, J. The eucalyptus oil ingredient 1,8-cineol induces oxidative DNA damage. Arch. Toxicol. 2015, 89, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Dörsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061–E4070. [Google Scholar] [CrossRef] [Green Version]
- Mimmler, M.; Peter, S.; Kraus, A.; Stroh, S.; Nikolova, T.; Seiwert, N.; Hasselwander, S.; Neitzel, C.; Haub, J.; Monien, B.H.; et al. DNA damage response curtails detrimental replication stress and chromosomal instability induced by the dietary carcinogen PhIP. Nucleic Acids Res. 2016, 44, 10259–10276. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, N.; Neitzel, C.; Stroh, S.; Frisan, T.; Audebert, M.; Toulany, M.; Kaina, B.; Fahrer, J. AKT2 suppresses pro-survival autophagy triggered by DNA double-strand breaks in colorectal cancer cells. Cell Death Dis. 2017, 8, e3019. [Google Scholar] [CrossRef] [Green Version]
- Dörsam, B.; Göder, A.; Seiwert, N.; Kaina, B.; Fahrer, J. Lipoic acid induces p53-independent cell death in colorectal cancer cells and potentiates the cytotoxicity of 5-fluorouracil. Arch. Toxicol. 2015, 89, 1829–1846. [Google Scholar] [CrossRef]
- Ripani, P.; Delp, J.; Bode, K.; Delgado, M.E.; Dietrich, L.; Betzler, V.M.; Yan, N.; von Scheven, G.; Mayer, T.U.; Leist, M.; et al. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain. Oncogene 2020, 39, 2345–2357. [Google Scholar] [CrossRef]
- Bode, K.J.; Mueller, S.; Schweinlin, M.; Metzger, M.; Brunner, T. A fast and simple fluorometric method to detect cell death in 3D intestinal organoids. Biotechniques 2019, 67, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Rodrigues, N.R.; Rowan, A.; Smith, M.E.; Kerr, I.B.; Bodmer, W.F.; Gannon, J.V.; Lane, D.P. p53 mutations in colorectal cancer. Proc. Natl. Acad. Sci. USA 1990, 87, 7555–7559. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Jiso, A.; Yurasakpong, L.; Janta, S.; Chaithirayanon, K.; Plubrukarn, A. Exerting DNA Damaging Effects of the Ilimaquinones through the Active Hydroquinone Species. Sci. Pharm. 2021, 89, 26. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Strzeszewska-Potyrała, A.; Staniak, K.; Czarnecka-Herok, J.; Rafiee, M.-R.; Herok, M.; Mosieniak, G.; Krijgsveld, J.; Sikora, E. Chromatin-Directed Proteomics Identifies ZNF84 as a p53-Independent Regulator of p21 in Genotoxic Stress Response. Cancers 2021, 13, 2115. [Google Scholar] [CrossRef]
- Liu, Y.; Bodmer, W.F. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Yun, E.; Hwang, I.H.; Yoon, S.; Kim, D.-E.; Kim, J.S.; Na, M.; Song, G.-Y.; Oh, S. Ilimaquinone and Ethylsmenoquinone, Marine Sponge Metabolites, Suppress the Proliferation of Multiple Myeloma Cells by Down-Regulating the Level of β-Catenin. Mar. Drugs 2014, 12, 3231–3244. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Noh, K.; Park, I.; Na, M.; Oh, S.; Shin, B.S.; Kang, W. Stereo-Selective Pharmacokinetics of Ilimaquinone Epimers Extracted from a Marine Sponge in Rats. Mar. Drugs 2019, 17, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qPCR Target | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| ACTB | TGGCATCCACGAAACTACC | GTGTTGGCGTACAGGTCTT |
| BAX | CAGAAGGCACTAATCAAG | ATCAGATGTGGTCTATAATG |
| BCL2 | TTCAGAGACAGCCAGGAGAAA | AGTACCTGAACCGGCACCT |
| BCL-XL | AAGCGTAGACAAGGAGAT | TAGGTGGTCATTCAGGTAA |
| BID | GTGTGGATGATATGAAGGC | GAAGACAGGCTGGAAGATA |
| BIM | CCAAATGGCAAAGCAACCTTCTG | CTGTCAATGCATTCTCCACACC |
| cIAP1 | TTCCCAGGTCCCTCGTATCA | CCGGCGGGGAAAGTTGAATA |
| cIAP2 | TCACTCCCAGACTCTTTCCA | CCCCGTGTTCTACAAGTGTC |
| FASL | GGGATGTTTCAGCTCTTCCA | TAAATGGGCCACTTTCCTCA |
| FASR | TTATCTGATGTTGACTTGAGTAA | GGCTTCATTGACACCATT |
| GAPDH | CATGAGAAGTATGACAACAG | ATGAGTCCTTCCACGAT |
| MDM2 | ATCTTGATGCTGGTGTAA | AGGCTATAATCTTCTGAGTC |
| NOXA | TCTTCGGTCACTACACAAC | CCAACAGGAACACATTGAAT |
| p21 | ACCATGTCAGAACCGGCTGGG | TGGGCGGATTAGGGCTTC |
| PUMA | TAAGGATGGAAAGTGTAG | TTCAGTTTCTCATTGTTAC |
| Survivin | ATGACTTGTGTGTGATGA | GTTTGTGCTATTCTGTGAA |
| Compound | HCT116 | RKO | HT29 |
|---|---|---|---|
| smenospongine | 8 µM | 15 µM | 10 µM |
| smenospongorine | 39 µM | 58 µM | 37 µM |
| smenospongiarine | - | n.d. | n.d. |
| smenospongidine | 80 µM | n.d. | n.d. |
| ilimaquinone | 27 µM | 43 µM | 13 µM |
| 5-epi-ilimaquinone | 47 µM | n.d. | n.d. |
| quintaquinone | 80 µM | n.d. | n.d. |
| cyclospongiaquinone-1 | 79 µM | n.d. | n.d. |
| smenodiol | 31 µM | 66 µM | 42 µM |
| dactylospontriol | 19 µM | 40 µM | 29 µM |
| 3-farnesyl-2-hydroxy-5-methoxyquinone | - | n.d. | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiso, A.; Demuth, P.; Bachowsky, M.; Haas, M.; Seiwert, N.; Heylmann, D.; Rasenberger, B.; Christmann, M.; Dietrich, L.; Brunner, T.; et al. Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer. Cancers 2021, 13, 3282. https://doi.org/10.3390/cancers13133282
Jiso A, Demuth P, Bachowsky M, Haas M, Seiwert N, Heylmann D, Rasenberger B, Christmann M, Dietrich L, Brunner T, et al. Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer. Cancers. 2021; 13(13):3282. https://doi.org/10.3390/cancers13133282
Chicago/Turabian StyleJiso, Apisada, Philipp Demuth, Madeleine Bachowsky, Manuel Haas, Nina Seiwert, Daniel Heylmann, Birgit Rasenberger, Markus Christmann, Lea Dietrich, Thomas Brunner, and et al. 2021. "Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer" Cancers 13, no. 13: 3282. https://doi.org/10.3390/cancers13133282
APA StyleJiso, A., Demuth, P., Bachowsky, M., Haas, M., Seiwert, N., Heylmann, D., Rasenberger, B., Christmann, M., Dietrich, L., Brunner, T., Riyanti, Schäberle, T. F., Plubrukarn, A., & Fahrer, J. (2021). Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer. Cancers, 13(13), 3282. https://doi.org/10.3390/cancers13133282