
Therapeutic Cancer Vaccination with Immunopeptidomics-Discovered Antigens Confers Protective Antitumor Efficacy

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Purification of MHC Class-I Complexes
2.3. Purification and Concentration of MHC-I Peptides
2.4. LC-MS Analysis of MHC-I Peptides
2.5. Proteomics Database Search
2.6. In Silico Analysis of the MHC-I Peptides
2.7. Mice and Animal Experiment
2.8. Immunogenicity Analysis
2.9. Flow Cytometric Analysis of Tumor-Infiltrating Lymphocytes
2.10. Statistical Analysis
3. Results
3.1. Direct Identification of Tumor-Associated Antigens in Mouse Triple-Negative Breast Tumor
3.2. Discovery of MHC-I-Presented Endogenous Retroviral Antigen
3.3. Control of Tumor Growth by MHC-I Ligand Immunization
3.4. Therapeutic Vaccination with PeptiCRAd Cancer Vaccine Targeted to Tumor-Associated Antigens Controls Tumor Growth
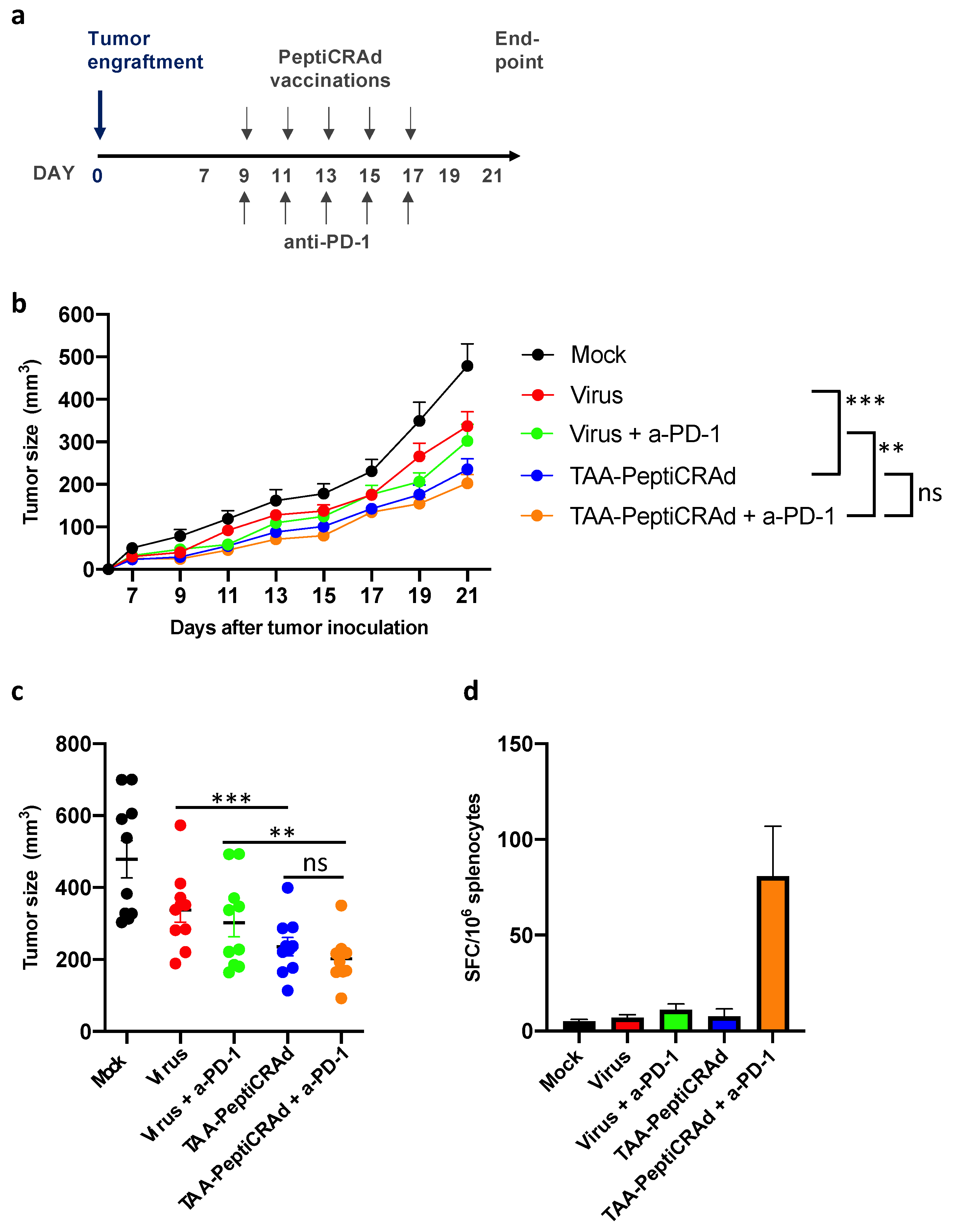
3.5. Therapeutic Vaccination with PeptiCRAd Cancer Vaccine Targeted to Endogenous Retroviral Antigen Shows Antitumor Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehmann, B.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lachapelle, J.; Leung, S.; Gao, D.; Foulkes, W.D.; O Nielsen, T. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012, 14, R48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.A.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef]
- Emens, L.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 379, 2108–2121. [Google Scholar]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Granados, D.P.; Laumont, C.M.; Thibault, P.; Perreault, C. The nature of self for T cells—A systems-level perspective. Curr. Opin. Immunol. 2015, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Haen, S.; Rammensee, H.-G. The repertoire of human tumor-associated epitopes—Identification and selection of antigens and their application in clinical trials. Curr. Opin. Immunol. 2013, 25, 277–283. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Solovyov, A.; Vabret, N.; Arora, K.S.; Snyder, A.; Funt, S.A.; Bajorin, D.F.; Rosenberg, J.E.; Bhardwaj, N.; Ting, D.T.; Greenbaum, B.D. Global cancer transcriptome quantifies repeat element polarization between immunotherapy responsive and T cell suppressive classes. Cell Rep. 2018, 23, 512–521. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.K.; Ørskov, A.D.; Bjerregaard, A.-M.; Unnikrishnan, A.; Holmberg-Thydén, S.; Borch, A.; Jensen, K.V.; Anande, G.; Bentzen, A.K.; Marquard, A.M.; et al. Human endogenous retroviruses form a reservoir of T cell targets in hematological cancers. Nat. Commun. 2020, 11, 5660. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M. Mass spectrometry based immunopeptidomics for the discovery of cancer neoantigens. Methods Mol. Biol. 2018, 1719, 209–221. [Google Scholar]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Alvarez, B.; Nielsen, M. GibbsCluster: Unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017, 45, W458–W463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapin, N.P.J.-P.; Hoof, I.; Lund, O.; Nielsen, M. MHC motif viewer. Immunogenetics 2008, 60, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Ylösmäki, E.; Ylösmäki, L.; Fusciello, M.; Martins, B.; Ahokas, P.; Cojoc, H.; Uoti, A.; Feola, S.; Kreutzman, A.; Ranki, T.; et al. Characterization of a novel OX40 ligand and CD40 ligand-expressing oncolytic adenovirus used in the PeptiCRAd cancer vaccine platform. Mol. Ther. Oncolytics 2021, 20, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. OncoImmunology 2016, 5, e1105429. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.J.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nat. Cell Biol. 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Schuster, H.; Shao, W.; Weiss, T.; Pedrioli, P.G.; Roth, P.; Weller, M.; Campbell, D.S.; Deutsch, E.W.; Moritz, R.L.; Planz, O.; et al. A tissue-based draft map of the murine MHC class I immunopeptidome. Sci. Data 2018, 5, 180157. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, H.J.; Consortia Oslo Breast Cancer Research Consortium (OSBREAC); Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez, A.; Aure, M.R.; Sennblad, B.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [Green Version]
- Chiaro, J.; Kasanen, H.H.E.; Whalley, T.; Capasso, C.; Gronholm, M.; Feola, S.; Peltonen, K.; Hamdan, F.; Hemberg, M.; Mäkelä, S.; et al. Viral Molecular Mimicry Influences the Antitumor Immune Response in Murine and Human Melanoma. Cancer Immunol. Res. 2021. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2008, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.-J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feola, S.; Capasso, C.; Fusciello, M.; Martins, B.; Tähtinen, S.; Medeot, M.; Carpi, S.; Frascaro, F.; Ylosmäki, E.; Peltonen, K.; et al. Oncolytic vaccines increase the response to PD-L1 blockade in immunogenic and poorly immunogenic tumors. OncoImmunology 2018, 7, e1457596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strausberg, R.L.; Feingold, E.A.; Grouse, L.H.; Derge, J.G.; Klausner, R.D.; Collins, F.S.; Wagner, L.; Shenmen, C.M.; Schuler, G.D.; Altschul, S.F.; et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [PubMed] [Green Version]
- Ternette, N.; Olde Nordkamp, M.J.M.; Muller, J.; Anderson, A.P.; Nicastri, A.; Hill, A.V.S.; Kessler, B.M.; Li, D. Immunopeptidomic profiling of HLA-A2-positive triple negative breast cancer identifies potential immunotherapy target antigens. Proteomics 2018, 18, e1700465. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.-Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Toh, U.; Iwakuma, N.; Takenaka, M.; Otsuka, H.; Furukawa, M.; Fujii, T.; Seki, N.; Kawahara, A.; Kage, M.; et al. Feasibility study of personalized peptide vaccination for metastatic recurrent triple-negative breast cancer patients. Breast Cancer Res. 2014, 16, R70. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Laverdure, J.-P.; Lanoix, J.; Durette, C.; Côté, C.; Bonneil, É.; Laumont, C.M.; Gendron, P.; Vincent, K.; Courcelles, M.; et al. Proteogenomics Uncovers a Vast Repertoire of Shared Tumor-Specific Antigens in Ovarian Cancer. Cancer Immunol. Res. 2020, 8, 544–555. [Google Scholar] [CrossRef]
- Liu, J.; Blake, S.J.; Yong, M.C.R.; Harjunpää, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [Green Version]
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.K.; Nielsen, T.O. TIM-3 expression in breast cancer. OncoImmunology 2018, 7, e1502128. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Peptide | UniProt ID | Protein Description | Short Name | H-2Kd IC50 (nM) | Known MHC-I Lgand (IEDB) * | MHC-I Ligand in Normal Tissue ** |
|---|---|---|---|---|---|---|---|
| Peptide pool1 | |||||||
| #1 | RYLPAPTAL | Q9JL70 | Fanconi anemia group A protein homolog | Fanca | 13.44 | Yes | - |
| #2 | FYITSRTQF | F8WI90 | Tyrosine-protein kinase | Scr | 15.05 | Yes | Yes |
| #3 | SYFPEITHI | B1ASP2 | Tyrosine-protein kinase | Jak1 | 21.79 | Yes | - |
| #4 | FYLETQQQI | Q99MP8 | BRCA1-associated protein | Brap | 29.37 | - | - |
| #5 | NYVPGKFTV | E9PXX8 | Metastasis-associated in colon cancer 1 | Macc1 | 59.45 | - | - |
| Peptide pool2 | |||||||
| #6 | EYVHTKNFI | H7BXB1 | Casein kinase I isoform alpha | Csnk1a1 | 65.81 | - | - |
| #7 | NYQDTIGRL | A0A0A6YWC8 | Vimentin | Vim | 470.08 | Yes | Yes |
| #8 | KYLATLETL | B1ASP2 | Tyrosine-protein kinase | Jak1 | 13.72 | Yes | Yes |
| #9 | YFISSTTRI | A0A0A0MQ80 | Spermatogenesis-associated protein 5 | Spata5 | 29.64 | Yes | - |
| #10 | SYLKSELGL | A2AQD5 | Sperm-specific antigen 2 homolog | Ssfa2 | 121.09 | - | - |
| Peptide pool3 | |||||||
| #11 | SYHPALNAI | S4R1L5 | Baculoviral IAP repeat-containing protein 6 | Birc6 | 9.9 | Yes | - |
| #12 | SYYAVAHAV | A0A0R4J170 | Transcription activator BRG1 | Smarca4 | 11.16 | - | - |
| #13 | AYKAVLNYL | D3YXN3 | Testis-expressed protein 30 | Tex30 | 43.77 | Yes | - |
| #14 | EYVANLTEL | A0A0R4J170 | Transcription activator BRG1 | Smarca4 | 100.38 | Yes | - |
| #15 | KYSAQIEDL | B1AUF1 | Ski oncogene | Ski | 443.63 | - | - |
| Peptide pool4 | |||||||
| #16 | EYIHSKNFI | Q9JMK2 | Casein kinase I isoform epsilon | Csnk1e | 26.77 | Yes | - |
| #17 | KYQAVTATL | P19253 | 60S ribosomal protein L13a | Rpl13a | 14.51 | Yes | Yes |
| #18 | KYQEALDVI | Q8BWZ3 | N-alpha-acetyltransferase 25, NatB auxiliary subunit UV excision repair protein RAD23 homolog B, HR23B | Naa25 | 31.04 | Yes | - |
| #19 | SYENMVTEI | P54728 | mHR23B | Rad23b | 9.65 | Yes | - |
| #20 | SYKPIVEYI | Q8C1B7 | Septin 11 | Sept11 | 45.35 | Ye | - |
| #21 | TYVPIAQQV | A2APB8 | Targeting protein for Xklp2 | Tpx2 | 90.44 | Yes | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peltonen, K.; Feola, S.; Umer, H.M.; Chiaro, J.; Mermelekas, G.; Ylösmäki, E.; Pesonen, S.; Branca, R.M.M.; Lehtiö, J.; Cerullo, V. Therapeutic Cancer Vaccination with Immunopeptidomics-Discovered Antigens Confers Protective Antitumor Efficacy. Cancers 2021, 13, 3408. https://doi.org/10.3390/cancers13143408
Peltonen K, Feola S, Umer HM, Chiaro J, Mermelekas G, Ylösmäki E, Pesonen S, Branca RMM, Lehtiö J, Cerullo V. Therapeutic Cancer Vaccination with Immunopeptidomics-Discovered Antigens Confers Protective Antitumor Efficacy. Cancers. 2021; 13(14):3408. https://doi.org/10.3390/cancers13143408
Chicago/Turabian StylePeltonen, Karita, Sara Feola, Husen M. Umer, Jacopo Chiaro, Georgios Mermelekas, Erkko Ylösmäki, Sari Pesonen, Rui M. M. Branca, Janne Lehtiö, and Vincenzo Cerullo. 2021. "Therapeutic Cancer Vaccination with Immunopeptidomics-Discovered Antigens Confers Protective Antitumor Efficacy" Cancers 13, no. 14: 3408. https://doi.org/10.3390/cancers13143408