
Phosphorylation of OGFOD1 by Cell Cycle-Dependent Kinase 7/9 Enhances the Transcriptional Activity of RNA Polymerase II in Breast Cancer Cells


 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. CRISPR/Cas9-Mediated Gene Knockout
2.4. Proximity-Dependent Labeling
2.5. Silver Staining
2.6. Protein Purification
2.7. Immunoprecipitation Assay
2.8. In Vitro Binding Assay
2.9. In Vitro Phosphorylation Assay
2.10. mRNA Sequencing and Analysis
2.11. Chromatin IP Assay
2.12. Cell Proliferation, Invasion, and Migration Assay
2.13. Lentivirus-Based shRNA Production and Expression
2.14. Animal Studies
2.15. Statistical Analysis
3. Results
3.1. OGFOD1 Has an Important Role for Rapid Proliferation of Breast Cancer Cell Lines
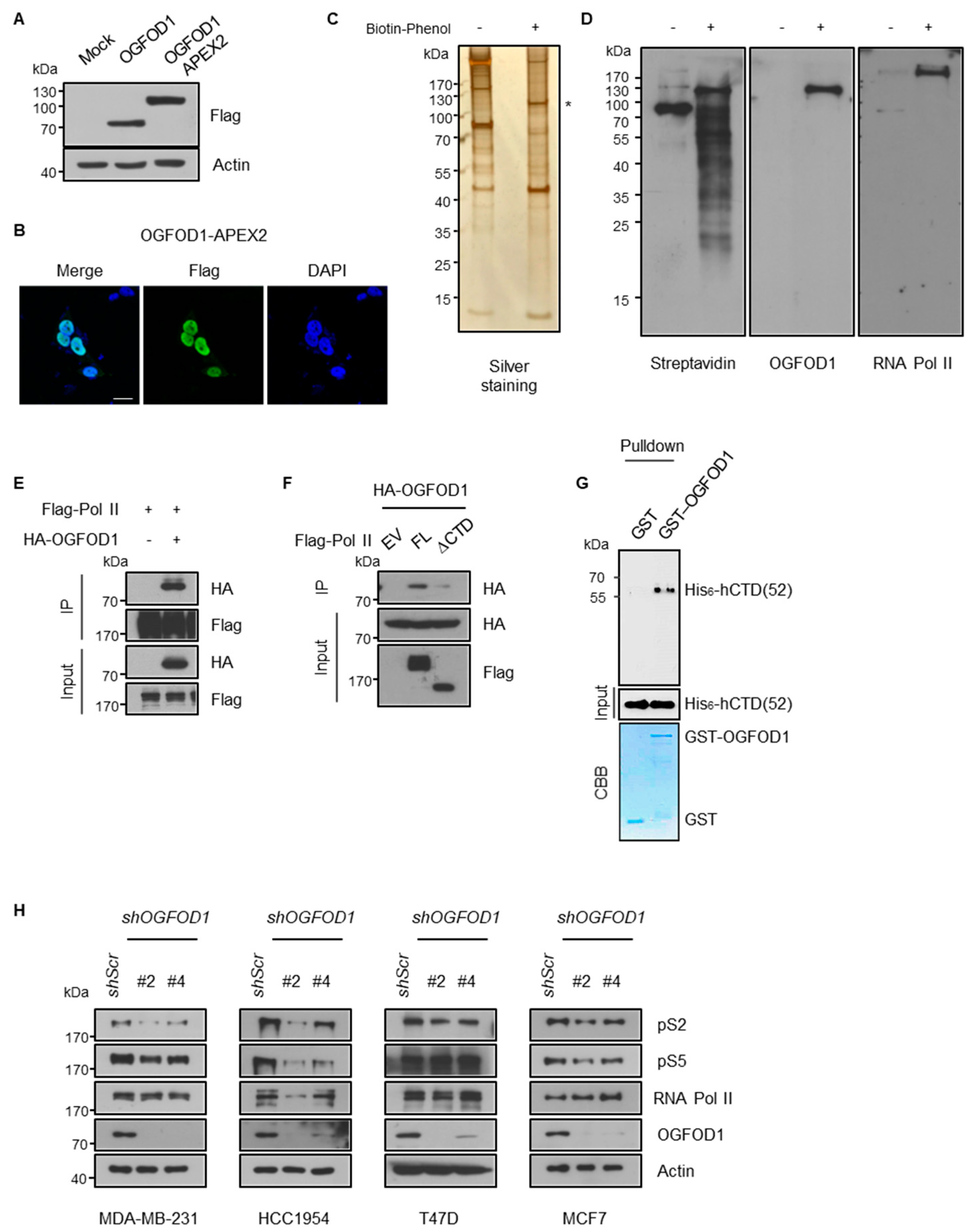
3.2. OGFOD1 Binds to the C-Terminal Domain of RNA Polymerase II and Attenuates Phosphorylation States
3.3. OGFOD1 Alters Phosphorylation States of RNA Polymerase II
3.4. Nuclear Localization of OGFOD1 Is Critical for Regulating RNA Polymerase II Activity
3.5. OGFOD1 KO Reduces Metastatic Gene Expressions in MDA-MB-231
3.6. CDK7 and CDK9 Phosphorylate Serine 256 of OGFOD1
3.7. CDK7/9 Enhance OGFOD1 Function
3.8. Non-Phosphorylated Mutation of Serine 256 Hampers Oncogenic Ability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wehner, K.A.; Schutz, S.; Sarnow, P. OGFOD1, a novel modulator of eukaryotic translation initiation factor 2alpha phosphorylation and the cellular response to stress. Mol. Cell Biol. 2010, 30, 2006–2016. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Adachi, N.; Koyama, H.; Matsushita, M. OGFOD1, a member of the 2-oxoglutarate and iron dependent dioxygenase family, functions in ischemic signaling. FEBS Lett. 2010, 584, 3340–3347. [Google Scholar] [CrossRef] [Green Version]
- Singleton, R.S.; Liu-Yi, P.; Formenti, F.; Ge, W.; Sekirnik, R.; Fischer, R.; Adam, J.; Pollard, P.J.; Wolf, A.; Thalhammer, A.; et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc. Natl. Acad. Sci. USA 2014, 111, 4031–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loenarz, C.; Sekirnik, R.; Thalhammer, A.; Ge, W.; Spivakovsky, E.; Mackeen, M.M.; McDonough, M.A.; Cockman, M.E.; Kessler, B.M.; Ratcliffe, P.J.; et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. USA 2014, 111, 4019–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, M.J.; Acevedo, J.M.; Loenarz, C.; Galagovsky, D.; Liu-Yi, P.; Perez-Pepe, M.; Thalhammer, A.; Sekirnik, R.; Ge, W.; Melani, M.; et al. Sudestada1, a Drosophila ribosomal prolyl-hydroxylase required for mRNA translation, cell homeostasis, and organ growth. Proc. Natl. Acad. Sci. USA 2014, 111, 4025–4030. [Google Scholar] [CrossRef] [Green Version]
- Stoehr, A.; Kennedy, L.; Yang, Y.; Patel, S.; Lin, Y.; Linask, K.L.; Fergusson, M.; Zhu, J.; Gucek, M.; Zou, J.; et al. The ribosomal prolyl-hydroxylase OGFOD1 decreases during cardiac differentiation and modulates translation and splicing. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Marina, O.; Hainz, U.; Biernacki, M.A.; Zhang, W.; Cai, A.; Duke-Cohan, J.S.; Liu, F.; Brusic, V.; Neuberg, D.; Kutok, J.L.; et al. Serologic markers of effective tumor immunity against chronic lymphocytic leukemia include nonmutated B-cell antigens. Cancer Res. 2010, 70, 1344–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, S.M.; Lee, J.H.; Chun, S.; Kang, B.H.; Kwak, S.; Roe, J.S.; Kim, T.W.; Kim, H.; Kim, W.H.; et al. OGFOD1 is required for breast cancer cell proliferation and is associated with poor prognosis in breast cancer. Oncotarget 2015, 6, 19528–19541. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.; Wang, Q.; Wang, S.; Zhu, G.; Tang, Q.; Liu, J. OGFOD1 negatively regulated by miR-1224-5p promotes proliferation in human papillomavirus-infected laryngeal papillomas. Mol. Genet. Genom. 2020, 295, 675–684. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Kim, M.; Borenstein-Auerbach, N.; Venkateswaran, N.; Hao, Y.H.; Ray, R.; Brabletz, T.; Scaglioni, P.P.; Shay, J.W.; Conacci-Sorrell, M. The aryl hydrocarbon receptor regulates nucleolar activity and protein synthesis in MYC-expressing cells. Genes Dev. 2018, 32, 1303–1308. [Google Scholar] [CrossRef] [Green Version]
- Kwak, H.; Lis, J.T. Control of transcriptional elongation. Annu. Rev. Genet. 2013, 47, 483–508. [Google Scholar] [CrossRef] [Green Version]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Zawel, L.; Fisher, L.; Egly, J.M.; Reinberg, D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature 1992, 358, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augert, A.; MacPherson, D. Treating transcriptional addiction in small cell lung cancer. Cancer Cell 2014, 26, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef] [Green Version]
- Valenciaga, A.; Saji, M.; Yu, L.; Zhang, X.; Bumrah, C.; Yilmaz, A.S.; Knippler, C.M.; Miles, W.; Giordano, T.J.; Cote, G.J.; et al. Transcriptional targeting of oncogene addiction in medullary thyroid cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Rosonina, E.; Blencowe, B.J. Analysis of the requirement for RNA polymerase II CTD heptapeptide repeats in pre-mRNA splicing and 3’-end cleavage. RNA 2004, 10, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.D.; Tu, L.C.; Mir, A.; Rodriguez, T.; Ding, Y.; Leszyk, J.; Dekker, J.; Shaffer, S.A.; Zhu, L.J.; Wolfe, S.A.; et al. C-BERST: Defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods 2018, 15, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Udeshi, N.D.; Lam, S.S.; Loh, K.H.; Cox, K.J.; Pedram, K.; Carr, S.A.; Ting, A.Y. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.P.; Greenleaf, A.L. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006, 20, 2922–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Larochelle, S.; Batliner, J.; Gamble, M.J.; Barboza, N.M.; Kraybill, B.C.; Blethrow, J.D.; Shokat, K.M.; Fisher, R.P. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat. Struct. Mol. Biol. 2006, 13, 55–62. [Google Scholar] [CrossRef]
- Kim, J.B.; Sharp, P.A. Positive transcription elongation factor B phosphorylates hSPT5 and RNA polymerase II carboxyl-terminal domain independently of cyclin-dependent kinase-activating kinase. J. Biol. Chem. 2001, 276, 12317–12323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardy, S.R.; Farnham, P.J. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem. 2001, 276, 48562–48571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluapuri, A.; Hofstetter, J.; Dudvarski Stankovic, N.; Endres, T.; Bhandare, P.; Vos, S.M.; Adhikari, B.; Schwarz, J.D.; Narain, A.; Vogt, M.; et al. MYC Recruits SPT5 to RNA Polymerase II to Promote Processive Transcription Elongation. Mol. Cell 2019, 74, 674–687. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Zheng, Y.; Yan, Y.; Bargonetti, J.; Foster, D.A. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 2006, 25, 7305–7310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; McGowan, P.M.; Harbeck, N.; Thomssen, C.; Schmitt, M. uPA and PAI-1 as biomarkers in breast cancer: Validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014, 16, 428. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Lester, R.D.; Montel, V.; Eastman, B.; Takimoto, S.; Gonias, S.L. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J. Biol. Chem. 2009, 284, 22825–22833. [Google Scholar] [CrossRef] [Green Version]
- Xue, A.; Xue, M.; Jackson, C.; Smith, R.C. Suppression of urokinase plasminogen activator receptor inhibits proliferation and migration of pancreatic adenocarcinoma cells via regulation of ERK/p38 signaling. Int. J. Biochem. Cell Biol. 2009, 41, 1731–1738. [Google Scholar] [CrossRef]
- LeBeau, A.M.; Duriseti, S.; Murphy, S.T.; Pepin, F.; Hann, B.; Gray, J.W.; VanBrocklin, H.F.; Craik, C.S. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013, 73, 2070–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehner, C.; Hockla, A.; Miller, E.; Ran, S.; Radisky, D.C.; Radisky, E.S. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 2014, 5, 2736–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, M.; Mineva, N.D.; Polmear, M.; Conrad, C.; Srinivasan, S.; Loussouarn, D.; Barille-Nion, S.; Georgakoudi, I.; Dagg, A.; McDermott, E.W.; et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol. Med. 2014, 6, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhou, H.; Feng, P.; Zhou, X.; Wen, H.; Xie, X.; Shen, H.; Zhu, X. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J. Exp. Clin. Cancer Res. 2010, 29, 92. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, B.; Wang, T.; Xu, L.; He, C.; Wen, H.; Yan, J.; Su, H.; Zhu, X. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS ONE 2014, 9, e109980. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, H.; Fu, Y.; Zhu, Y.; Kong, L.; Chen, L.; Zhao, F.; Yu, L.; Chen, X. TLR4/MyD88 signaling determines the metastatic potential of breast cancer cells. Mol. Med. Rep. 2018, 18, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Rajput, S.; Volk-Draper, L.D.; Ran, S. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol. Cancer Ther. 2013, 12, 1676–1687. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, S.; Ye, Q.; Pan, J. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol. Cancer 2019, 18, 140. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-T.; Lee, I.-H.; Kim, J.-H.; Lee, S.; Kwak, S.; Suh, M.-Y.; Hwang, I.-Y.; Kang, B.-G.; Cha, S.-S.; Lee, B.-I.; et al. Phosphorylation of OGFOD1 by Cell Cycle-Dependent Kinase 7/9 Enhances the Transcriptional Activity of RNA Polymerase II in Breast Cancer Cells. Cancers 2021, 13, 3418. https://doi.org/10.3390/cancers13143418
Lee H-T, Lee I-H, Kim J-H, Lee S, Kwak S, Suh M-Y, Hwang I-Y, Kang B-G, Cha S-S, Lee B-I, et al. Phosphorylation of OGFOD1 by Cell Cycle-Dependent Kinase 7/9 Enhances the Transcriptional Activity of RNA Polymerase II in Breast Cancer Cells. Cancers. 2021; 13(14):3418. https://doi.org/10.3390/cancers13143418
Chicago/Turabian StyleLee, Han-Teo, Il-Hwan Lee, Jae-Hwan Kim, Sangho Lee, Sojung Kwak, Min-Young Suh, In-Young Hwang, Bu-Gyeong Kang, Sun-Shin Cha, Byung-Il Lee, and et al. 2021. "Phosphorylation of OGFOD1 by Cell Cycle-Dependent Kinase 7/9 Enhances the Transcriptional Activity of RNA Polymerase II in Breast Cancer Cells" Cancers 13, no. 14: 3418. https://doi.org/10.3390/cancers13143418