
Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Solutions and Chemicals
2.2. GSC Cell Culture
2.3. Immunoblotting
2.4. Immunostaining
2.5. Intracellular Ca2+ Measurements
2.6. Cell Proliferation Assays
2.7. Sphere Forming and Self-Renewal Assays
2.8. Limit Dilution Assays
2.9. SOX2 Expression Assay
2.10. Statistics
3. Results
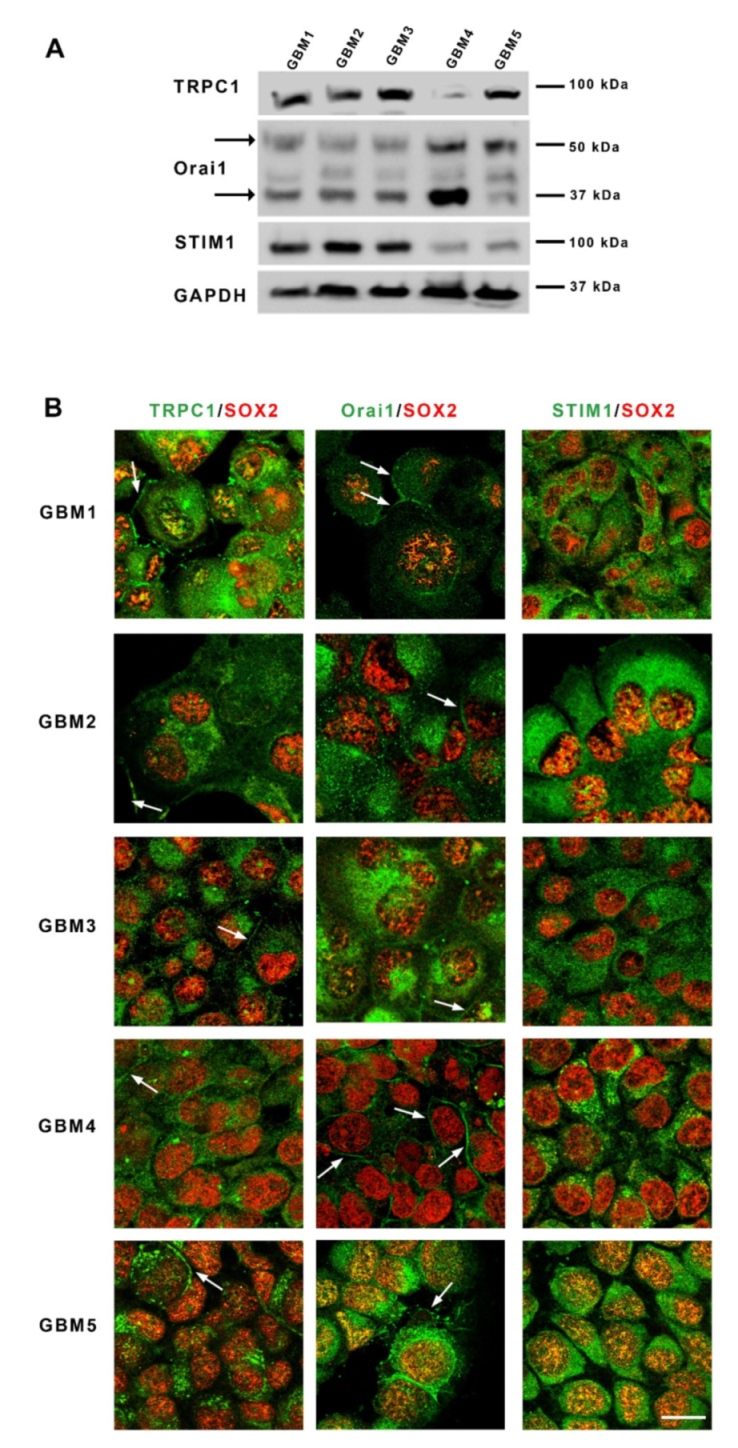
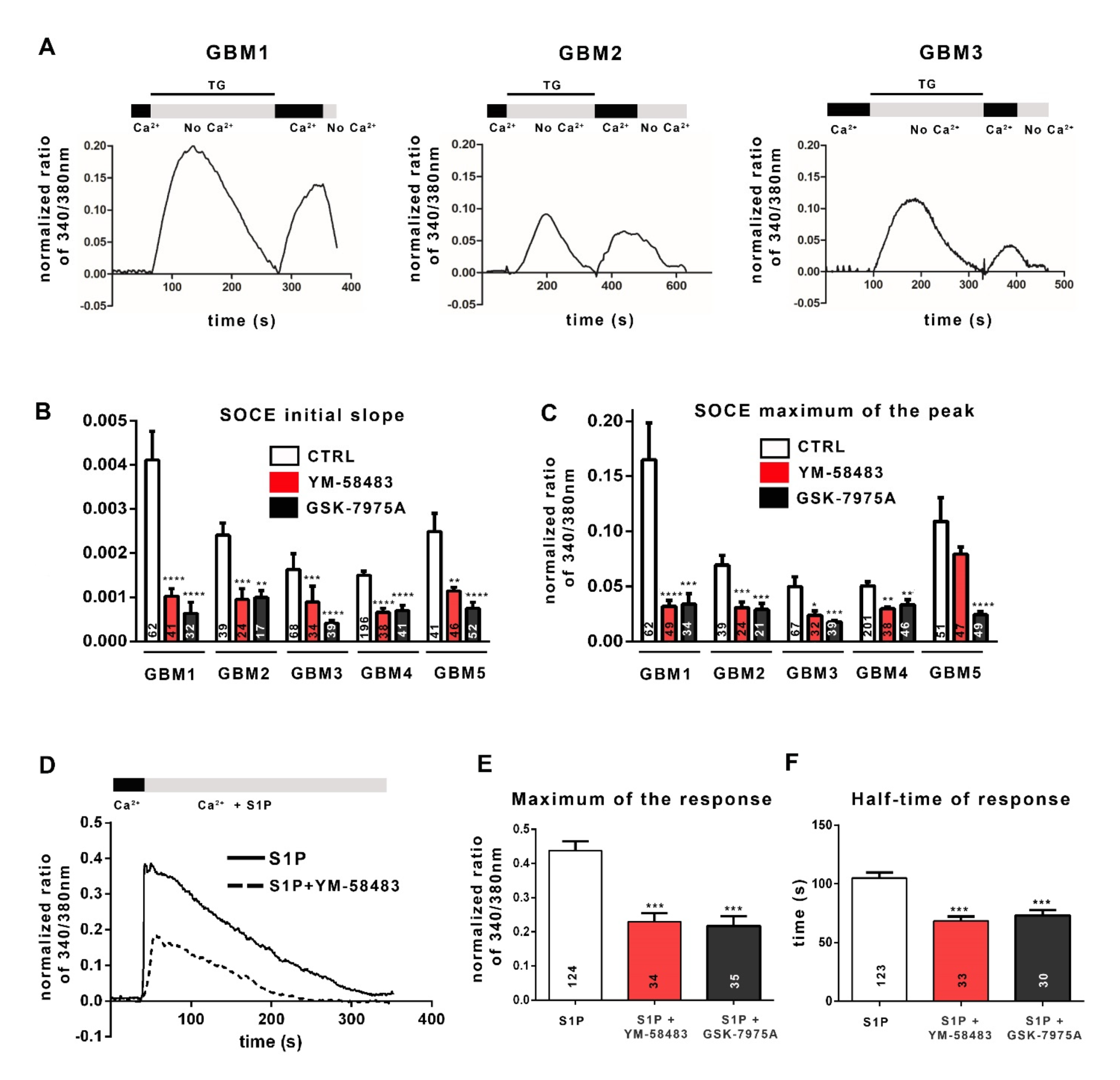
3.1. Patient-Derived GSC Express Functional SOC
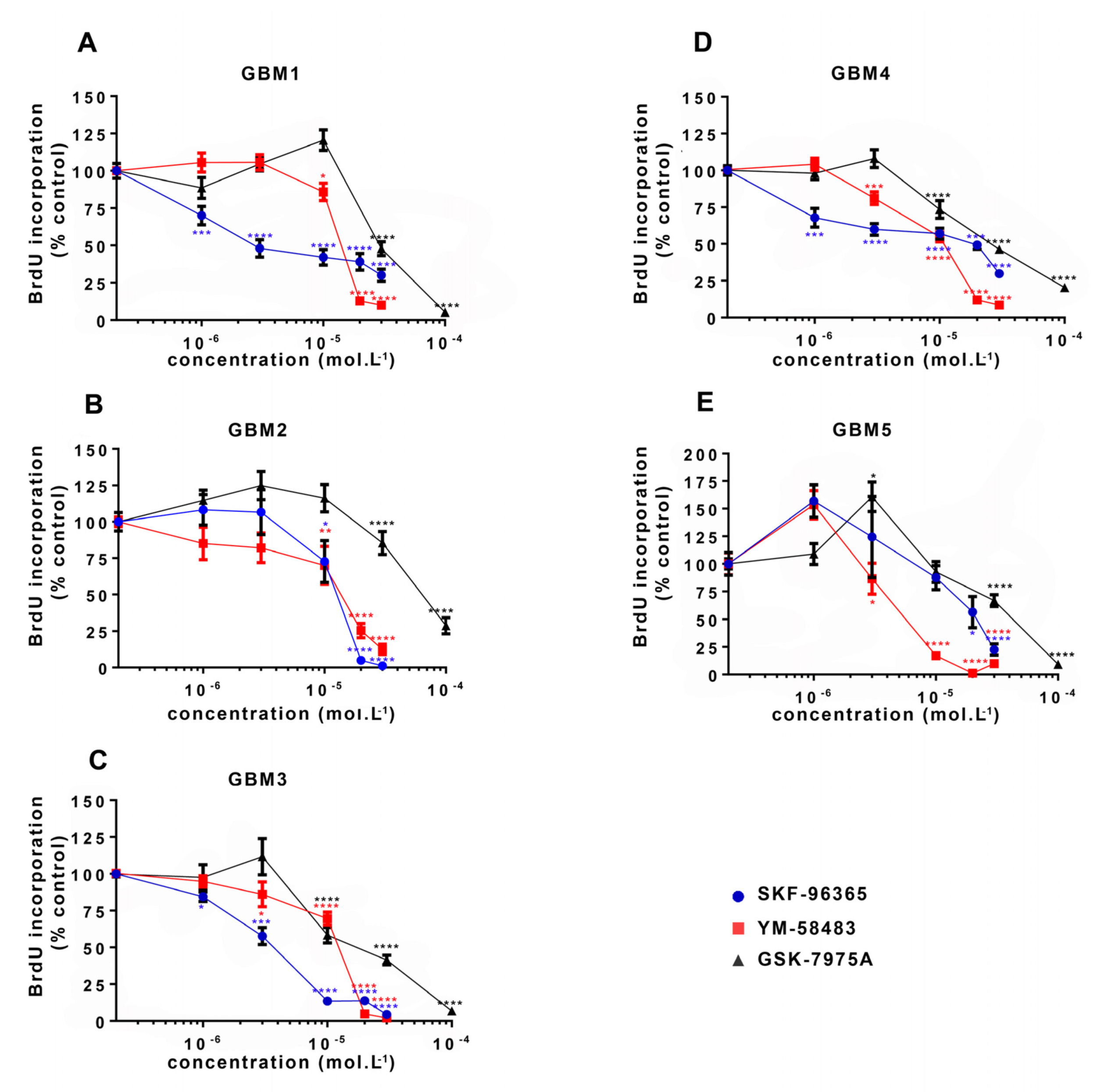
3.2. SOCE Inhibition Reduces GSC Proliferation
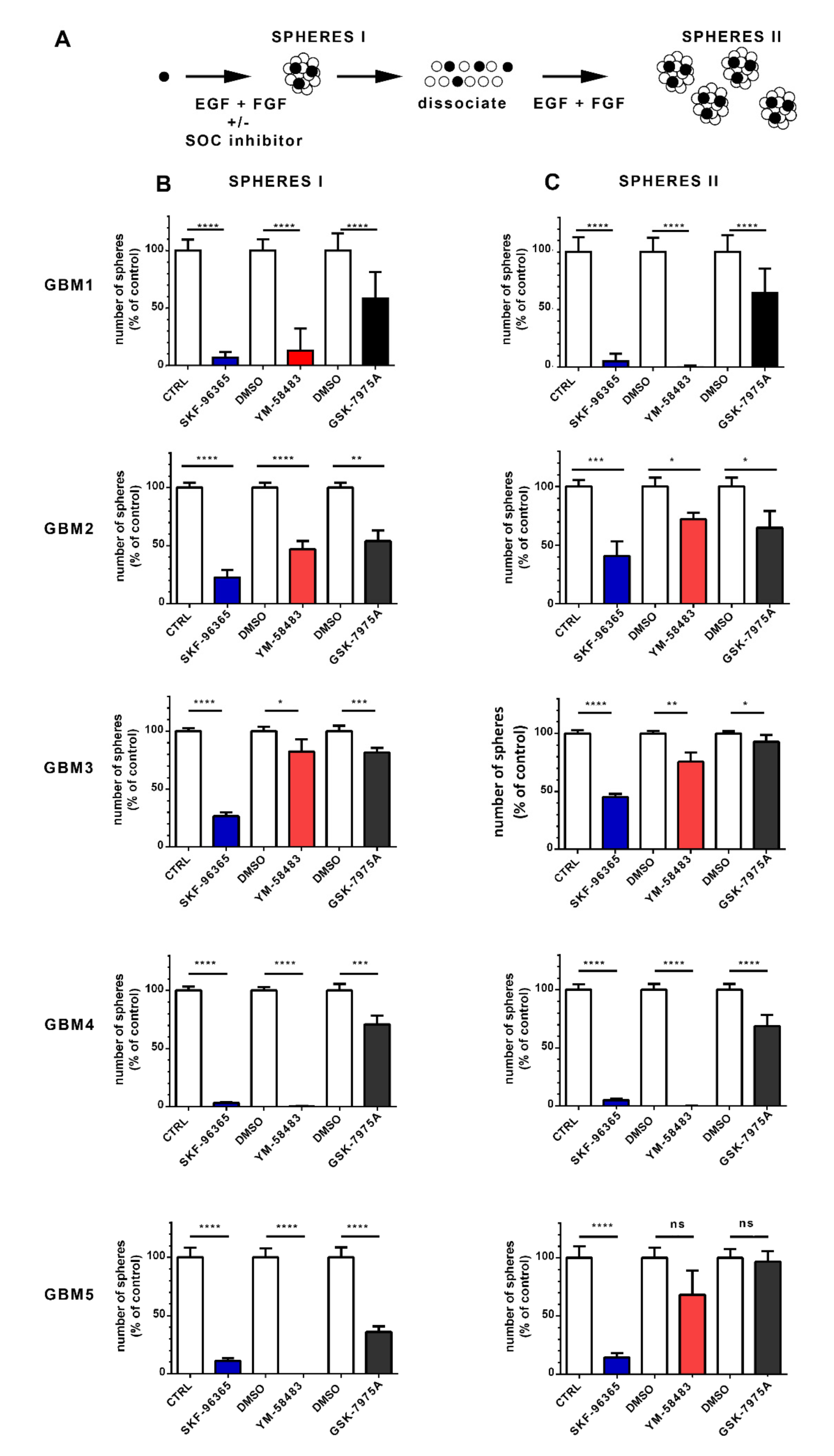
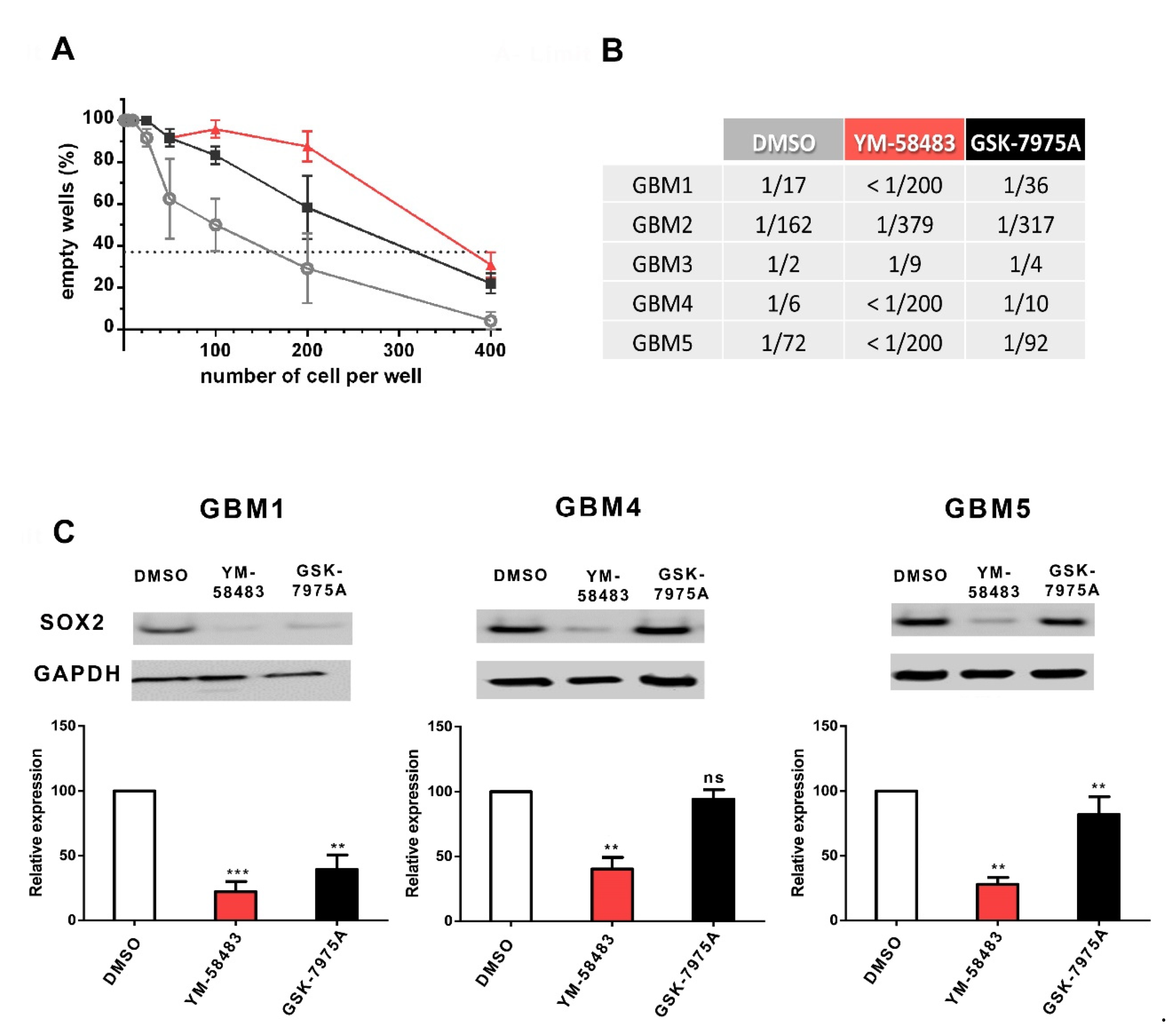
3.3. SOCE Inhibition Reduces the Stem Cell Population in GSC Cultures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Teo, W.-Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.-Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.-G.; Li, X.; et al. Relevance of a TCGA-derived glioblastoma subtype gene-classifier among patient populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Wee, S.; Niklasson, M.; Marinescu, V.D.; Segerman, A.; Schmidt, L.; Hermansson, A.; Dirks, P.; Forsberg-Nilsson, K.; Westermark, B.; Uhrbom, L.; et al. Selective calcium sensitivity in immature glioma cancer stem cells. PLoS ONE 2014, 9, e115698. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium: Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 Heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M.; Demiryurek, A.T.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumor Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef]
- Scrideli, C.A.; Carlotti, C.G.; Okamoto, O.K.; Andrade, V.S.; Cortez, M.A.A.; Motta, F.J.N.; Lucio-Eterovic, A.K.; Neder, L.; Rosemberg, S.; Oba-Shinjo, S.M.; et al. Gene expression profile analysis of primary glioblastomas and non-neoplastic brain tissue: Identification of potential target genes by oligonucleotide microarray and real-time quantitative PCR. J. Neurooncol. 2008, 88, 281–291. [Google Scholar] [CrossRef]
- Bomben, V.C.; Turner, K.L.; Barclay, T.-T.C.; Sontheimer, H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J. Cell. Physiol. 2011, 226, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wei, Y.; Zhang, L.; Yee, P.P.; Johnson, M.; Zhang, X.; Gulley, M.; Atkinson, J.M.; Trebak, M.; Wang, H.-G.; et al. Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the hippo pathway transcriptional coactivators YAP/TAZ. Oncogene 2018, 38, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Coronas, V.; Terrié, E.; Déliot, N.; Arnault, P.; Constantin, B. Calcium channels in adult brain neural stem cells and in glioblastoma stem cells. Front. Cell. Neurosci. 2020, 14, 600018. [Google Scholar] [CrossRef]
- Domenichini, F.; Terrié, E.; Arnault, P.; Harnois, T.; Magaud, C.; Bois, P.; Constantin, B.; Coronas, V. Store-operated calcium entries control neural stem cell self-renewal in the adult brain subventricular zone. Stem Cells 2018, 36, 761–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.-C.; Teusan, R.; et al. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clin. Cancer Res. 2017, 23, 6292–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scicchitano, B.M.; Sorrentino, S.; Proietti, G.; Lama, G.; Dobrowolny, G.; Catizone, A.; Binda, E.; Larocca, L.M.; Sica, G. Levetiracetam enhances the temozolomide effect on glioblastoma stem cell proliferation and apoptosis. Cancer Cell Int. 2018, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Floyd, D.H.; Kefas, B.; Seleverstov, O.; Mykhaylyk, O.; Dominguez, C.; Comeau, L.; Plank, C.; Purow, B. Alpha-secretase inhibition reduces human glioblastoma stem cell growth in vitro and in vivo by inhibiting notch. Neuro Oncol. 2012, 14, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Tropepe, V.; Sibilia, M.; Ciruna, B.G.; Rossant, J.; Wagner, E.F.; van der Kooy, D. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev. Biol. 1999, 208, 166–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and functional characterization of orai proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [Green Version]
- Dörr, K.; Kilch, T.; Kappel, S.; Alansary, D.; Schwär, G.; Niemeyer, B.A.; Peinelt, C. Cell type-specific glycosylation of orai1 modulates store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra25. [Google Scholar] [CrossRef]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef]
- Lepannetier, S.; Zanou, N.; Yerna, X.; Emeriau, N.; Dufour, I.; Masquelier, J.; Muccioli, G.; Tajeddine, N.; Gailly, P. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 2016, 60, 373–383. [Google Scholar] [CrossRef]
- Marfia, G.; Campanella, R.; Navone, S.E.; Di Vito, C.; Riccitelli, E.; Hadi, L.A.; Bornati, A.; de Rezende, G.; Giussani, P.; Tringali, C.; et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells: S1P Prompts growth and stemness of GSCs. Glia 2014, 62, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Nakada, M.; Sugimoto, N.; Harada, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Yachie, A.; Takuwa, Y.; et al. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int J. Cancer 2010, 126, 2341–2352. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Aulestia, F.J.; Néant, I.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Moreau, M.; Leclerc, C. Quiescence status of glioblastoma stem-like cells involves remodelling of Ca2+ signalling and mitochondrial shape. Sci. Rep. 2018, 8, 9731. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a therapeutic strategy in glioblastoma. Front. Oncol 2016, 6, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.T.; Ellison, S.; Pandele, A.; Wood, S.; Nathan, E.; Forte, G.; Parker, H.; Zindy, E.; Elvin, M.; Dickson, A.; et al. Actinomycin D downregulates Sox2 and improves survival in preclinical models of recurrent glioblastoma. Neuro Oncol. 2020, 22, 1289–1301. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, K.H.; Kim, D.G.; Cho, H.J.; Kim, Y.; Rheey, J.; Shin, K.; Seo, Y.J.; Choi, Y.-S.; Lee, J.-I.; et al. FoxM1 promotes stemness and radio-resistance of glioblastoma by regulating the master stem cell regulator Sox2. PLoS ONE 2015, 10, e0137703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Joo, K.M.; Jin, J.; Kim, E.; Kim, K.H.; Kim, Y.; Kang, B.G.; Kang, Y.-J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef] [Green Version]
- Bacco, F.D.; Casanova, E.; Medico, E.; Pellegatta, S.; Orzan, F.; Albano, R.; Luraghi, P.; Reato, G.; D’Ambrosio, A.; Porrati, P.; et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012, 72, 4537–4550. [Google Scholar] [CrossRef] [Green Version]
- Hopson, K.P.; Truelove, J.; Chun, J.; Wang, Y.; Waeber, C. S1P activates store-operated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2011, 300, C919–C926. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Song, M.; Guo, R.; Wang, H.; Gao, P.; Shi, W.; Huang, L. Knockdown of stromal interaction molecule 1 attenuates hepatocyte growth factor-induced endothelial progenitor cell proliferation. Exp. Biol. Med. 2010, 235, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Jacques, T.S.; Swales, A.; Brzozowski, M.J.; Henriquez, N.V.; Linehan, J.M.; Mirzadeh, Z.; Malley, C.O.; Naumann, H.; Alvarez-Buylla, A.; Brandner, S. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010, 29, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Matarredona, E.R.; Pastor, A.M. Neural stem cells of the subventricular zone as the origin of human glioblastoma stem cells. Therapeutic implications. Front. Oncol. 2019, 9, 779. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.-H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 2009, 15, 110–116. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Z.; Wang, R.; Ma, W.; Yang, Y.; Wei, J.; Wei, Y. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J. Exp. Clin. Cancer Res. 2013, 32, 1. [Google Scholar] [CrossRef] [Green Version]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Reforgiato, M.; Poletto, V.; Lodola, F.; Bottino, C.; Guido, D.; Rappa, A.; Pareek, S.; et al. Enhanced expression of stim, orai, and TRPC transcripts and proteins in endothelial progenitor cells isolated from patients with primary myelofibrosis. PLoS ONE 2014, 9, e91099. [Google Scholar] [CrossRef]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal a breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef] [PubMed]
- Gronski, M.A.; Kinchen, J.M.; Juncadella, I.J.; Franc, N.C.; Ravichandran, K.S. An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory response. Cell Death Differ. 2009, 16, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Chen, X.Y.; Dong, H.; Xu, F. Cyclopiazonic acid-induced Ca2+ store depletion initiates endothelium-dependent hyperpolarization-mediated vasorelaxation of mesenteric arteries in healthy and colitis mice. Front. Physiol. 2021, 12, 639857. [Google Scholar] [CrossRef]
- Calderón-Sánchez, E.M.; Ávila-Medina, J.; Callejo-García, P.; Fernández-Velasco, M.; Ordóñez, A.; Smani, T. Role of orai1 and L-type CaV1.2 channels in endothelin-1 mediated coronary contraction under ischemia and reperfusion. Cell Calcium 2020, 86, 102157. [Google Scholar] [CrossRef] [PubMed]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.-M.; Rosado, J.A.; Ordóñez-Fernández, A.; Smani, T. SARAF and orai1 contribute to endothelial cell activation and angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef]
- Lee, S.H.; Rigas, N.K.; Lee, C.-R.; Bang, A.; Srikanth, S.; Gwack, Y.; Kang, M.K.; Kim, R.H.; Park, N.-H.; Shin, K.-H. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016, 7, 43239–43255. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, H.; Zheng, L.; Zhou, Y.; Wu, L.; Xu, Y.; Zhang, X.; Yan, G.; Sheng, H.; Xin, R.; et al. FGF19/SOCE/NFATc2 signaling circuit facilitates the self-renewal of liver cancer stem cells. Theranostics 2021, 11, 5045–5060. [Google Scholar] [CrossRef] [PubMed]
- Koguchi, M.; Nakahara, Y.; Ito, H.; Wakamiya, T.; Yoshioka, F.; Ogata, A.; Inoue, K.; Masuoka, J.; Izumi, H.; Abe, T. BMP4 induces asymmetric cell division in human glioma stem-like cells. Oncol. Lett. 2020, 19, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, M.S.; Clarke, R.; Merrill, B.J. Intracellular Ca2+ homeostasis and nuclear export mediate exit from naive pluripotency. Cell Stem Cell 2019, 25, 210.e6–224.e6. [Google Scholar] [CrossRef] [PubMed]
- Gengatharan, A.; Malvaut, S.; Marymonchyk, A.; Ghareghani, M.; Snapyan, M.; Fischer-Sternjak, J.; Ninkovic, J.; Götz, M.; Saghatelyan, A. Adult Neural stem cell activation in mice is regulated by the day/night cycle and intracellular calcium dynamics. Cell 2021, 184, 709.e13–722.e13. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.-H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2 + entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, B.; Xie, Q.; Ye, D.; Zhang, D.; Zhu, Y.; Chen, H.; Zhu, B. STIM1 mediates hypoxia-driven hepatocarcinogenesis via interaction with HIF-1. Cell Rep. 2015, 12, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Cabanas, H.; Harnois, T.; Magaud, C.; Cousin, L.; Constantin, B.; Bourmeyster, N.; Déliot, N. Deregulation of calcium homeostasis in Bcr-Abl-dependent chronic myeloid leukemia. Oncotarget 2018, 9, 26309–26327. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Lee, S.; Jung, H.J. A curcumin derivative hydrazinobenzoylcurcumin suppresses stem-like features of glioblastoma cells by targeting Ca2+ /calmodulin-dependent protein kinase II. J. Cell Biochem. 2019, 120, 6741–6752. [Google Scholar] [CrossRef]
- Song, Y.; Jiang, Y.; Tao, D.; Wang, Z.; Wang, R.; Wang, M.; Han, S. NFAT2-HDAC1 signaling contributes to the malignant phenotype of glioblastoma. Neuro-Oncology 2020, 22, 46–57. [Google Scholar] [CrossRef]
- Jiang, Y.; Song, Y.; Wang, R.; Hu, T.; Zhang, D.; Wang, Z.; Tie, X.; Wang, M.; Han, S. NFAT1-mediated regulation of NDEL1 promotes growth and invasion of glioma stem-like cells. Cancer Res. 2019, 79, 2593–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrié, E.; Coronas, V.; Constantin, B. Role of the calcium toolkit in cancer stem cells. Cell Calcium 2019, 80, 141–151. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.; Buchanan, P. Calcium channels and cancer stem cells. Cell Calcium 2019, 81, 21–28. [Google Scholar] [CrossRef]
- Gross, S.; Mallu, P.; Joshi, H.; Schultz, B.; Go, C.; Soboloff, J. Ca2+ as a therapeutic target in cancer. Adv. Cancer Res. 2020, 148, 233–317. [Google Scholar] [CrossRef] [PubMed]
- Tajada, S.; Villalobos, C. Calcium permeable channels in cancer hallmarks. Front. Pharm. 2020, 11, 968. [Google Scholar] [CrossRef]
- Omuro, A.; Beal, K.; McNeill, K.; Young, R.J.; Thomas, A.; Lin, X.; Terziev, R.; Kaley, T.J.; DeAngelis, L.M.; Daras, M.; et al. Multicenter phase IB trial of carboxyamidotriazole orotate and temozolomide for recurrent and newly diagnosed glioblastoma and other anaplastic gliomas. J. Clin. Oncol. 2018, 36, 1702–1709. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrié, E.; Déliot, N.; Benzidane, Y.; Harnois, T.; Cousin, L.; Bois, P.; Oliver, L.; Arnault, P.; Vallette, F.; Constantin, B.; et al. Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma. Cancers 2021, 13, 3428. https://doi.org/10.3390/cancers13143428
Terrié E, Déliot N, Benzidane Y, Harnois T, Cousin L, Bois P, Oliver L, Arnault P, Vallette F, Constantin B, et al. Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma. Cancers. 2021; 13(14):3428. https://doi.org/10.3390/cancers13143428
Chicago/Turabian StyleTerrié, Elodie, Nadine Déliot, Yassine Benzidane, Thomas Harnois, Laëtitia Cousin, Patrick Bois, Lisa Oliver, Patricia Arnault, François Vallette, Bruno Constantin, and et al. 2021. "Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma" Cancers 13, no. 14: 3428. https://doi.org/10.3390/cancers13143428
APA StyleTerrié, E., Déliot, N., Benzidane, Y., Harnois, T., Cousin, L., Bois, P., Oliver, L., Arnault, P., Vallette, F., Constantin, B., & Coronas, V. (2021). Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma. Cancers, 13(14), 3428. https://doi.org/10.3390/cancers13143428