Author Contributions
Conceptualization, H.-Y.Z. and X.-H.Z.; methodology, Z.-J.C., M.G., X.-H.Z. and H.-Y.Z.; software, Z.-J.C., M.G. and X.-H.Z.; validation, T.-F.D. and X.-H.Z.; formal analysis, Z.-J.C., M.G. and X.-H.Z.; resources, H.-Y.Z. and X.-H.Z.; data curation, Z.-J.C., M.G. and X.-H.Z.; writing—original draft preparation, Z.-J.C., M.G., B.-M.L., Y.Q., X.-Y.T. and X.-H.Z.; writing—review and editing, X.-H.Z.; Y.Q. and H.-Y.Z.; visualization, Z.-J.C., M.G. and X.-H.Z.; supervision, H.-Y.Z., X.-H.Z. and Y.Q. All authors have read and agreed to the published version of the manuscript.
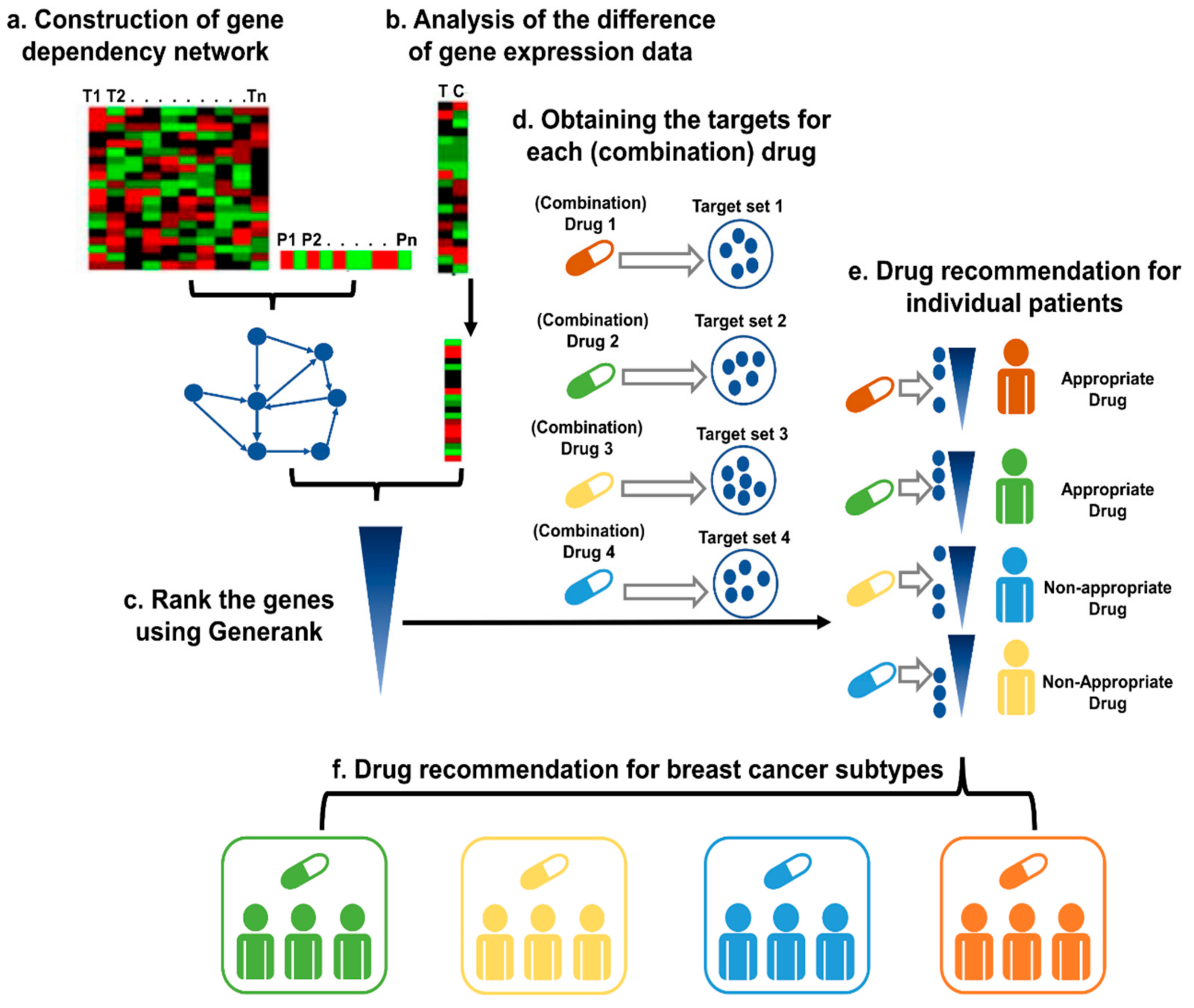
Figure 1.
The pipeline for precision drug discovery. (a) Construction of the gene dependency network based on the gene expression data and prognostic information of the cancer patients. (b) The difference in gene expression levels between cancer tissue and control tissue, as indicated by the fold-change. (c) Based on the gene dependency network and the fold-changes of the genes of the patient, GeneRank was applied to rank the genes. (d) Identification of the targets for the candidate (combination) drugs. (e) For each drug, the Kolmogorov–Smirnov test was applied to test whether the targets were enriched among the top-ranked genes with our method. A drug is recommended as a suitable therapy if the drug targets the important genes of the patient. (f) The candidate (combination) drugs were defined as an appropriate therapy for a specific subtype if the effective rate in the subtype was significantly higher than that in all patients, which was evaluated with a binomial test.
Figure 1.
The pipeline for precision drug discovery. (a) Construction of the gene dependency network based on the gene expression data and prognostic information of the cancer patients. (b) The difference in gene expression levels between cancer tissue and control tissue, as indicated by the fold-change. (c) Based on the gene dependency network and the fold-changes of the genes of the patient, GeneRank was applied to rank the genes. (d) Identification of the targets for the candidate (combination) drugs. (e) For each drug, the Kolmogorov–Smirnov test was applied to test whether the targets were enriched among the top-ranked genes with our method. A drug is recommended as a suitable therapy if the drug targets the important genes of the patient. (f) The candidate (combination) drugs were defined as an appropriate therapy for a specific subtype if the effective rate in the subtype was significantly higher than that in all patients, which was evaluated with a binomial test.
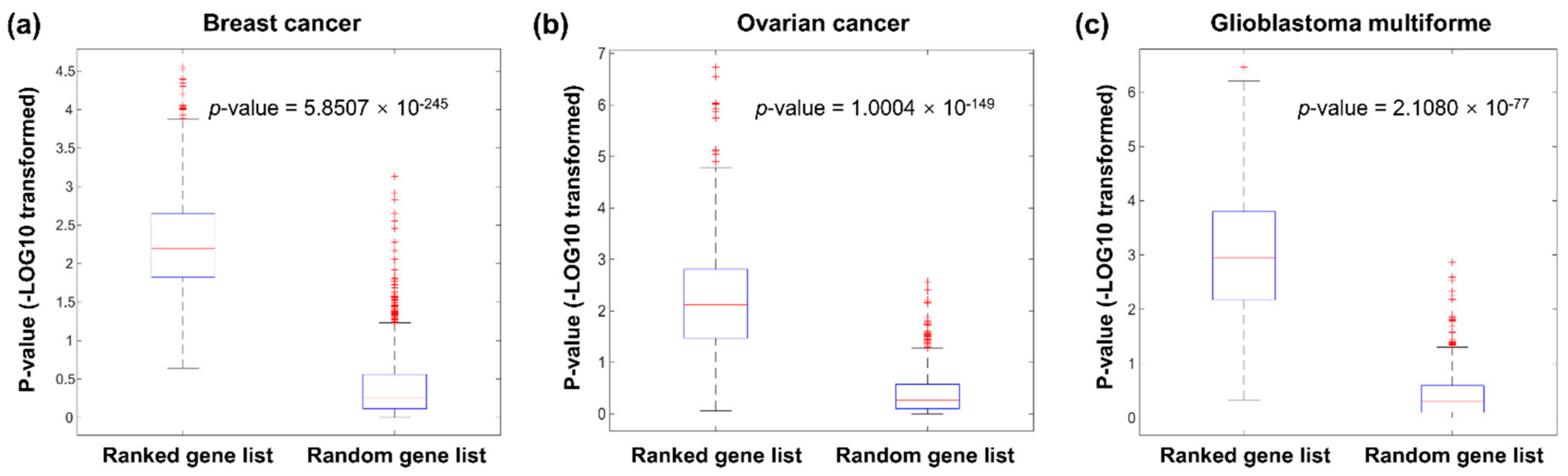
Figure 2.
Analysis of the enrichment of known cancer genes among the ranked gene lists of all assessed BC (a), OV (b), and GBM (c) patients.
Figure 2.
Analysis of the enrichment of known cancer genes among the ranked gene lists of all assessed BC (a), OV (b), and GBM (c) patients.
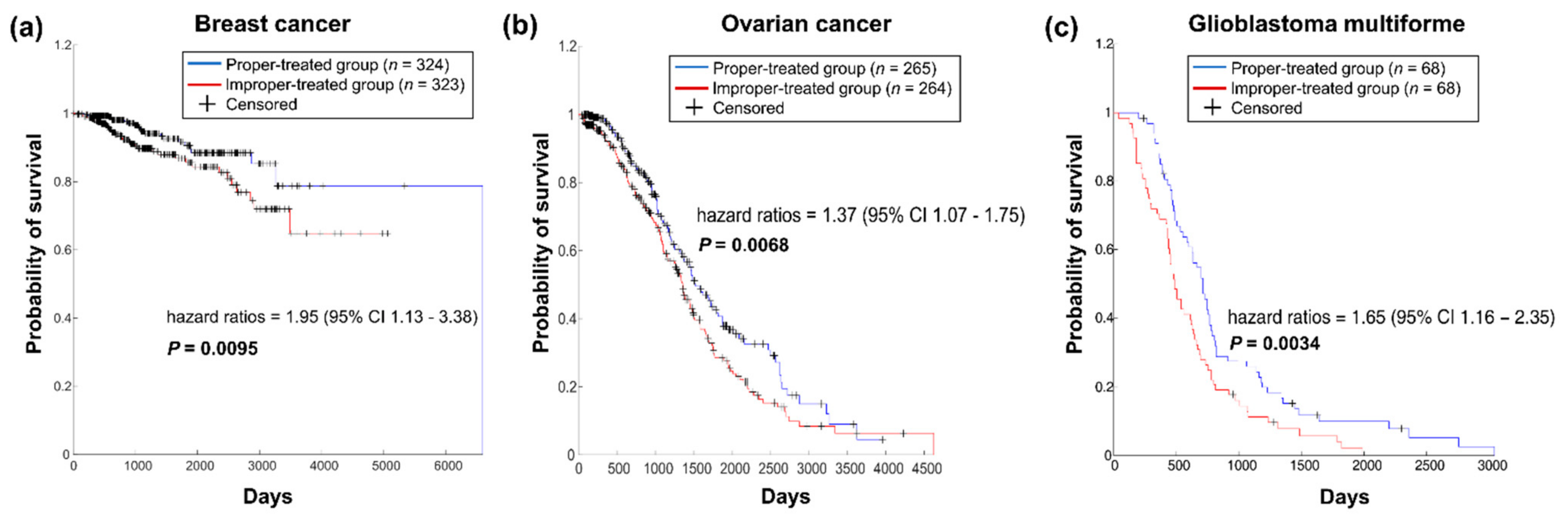
Figure 3.
Survival analysis of the two patient groups divided by administration of the appropriate therapy. (a). Survival analysis of BC patients. (b). Survival analysis of OV patients. (c). Survival analysis of GBM cancer patients.
Figure 3.
Survival analysis of the two patient groups divided by administration of the appropriate therapy. (a). Survival analysis of BC patients. (b). Survival analysis of OV patients. (c). Survival analysis of GBM cancer patients.
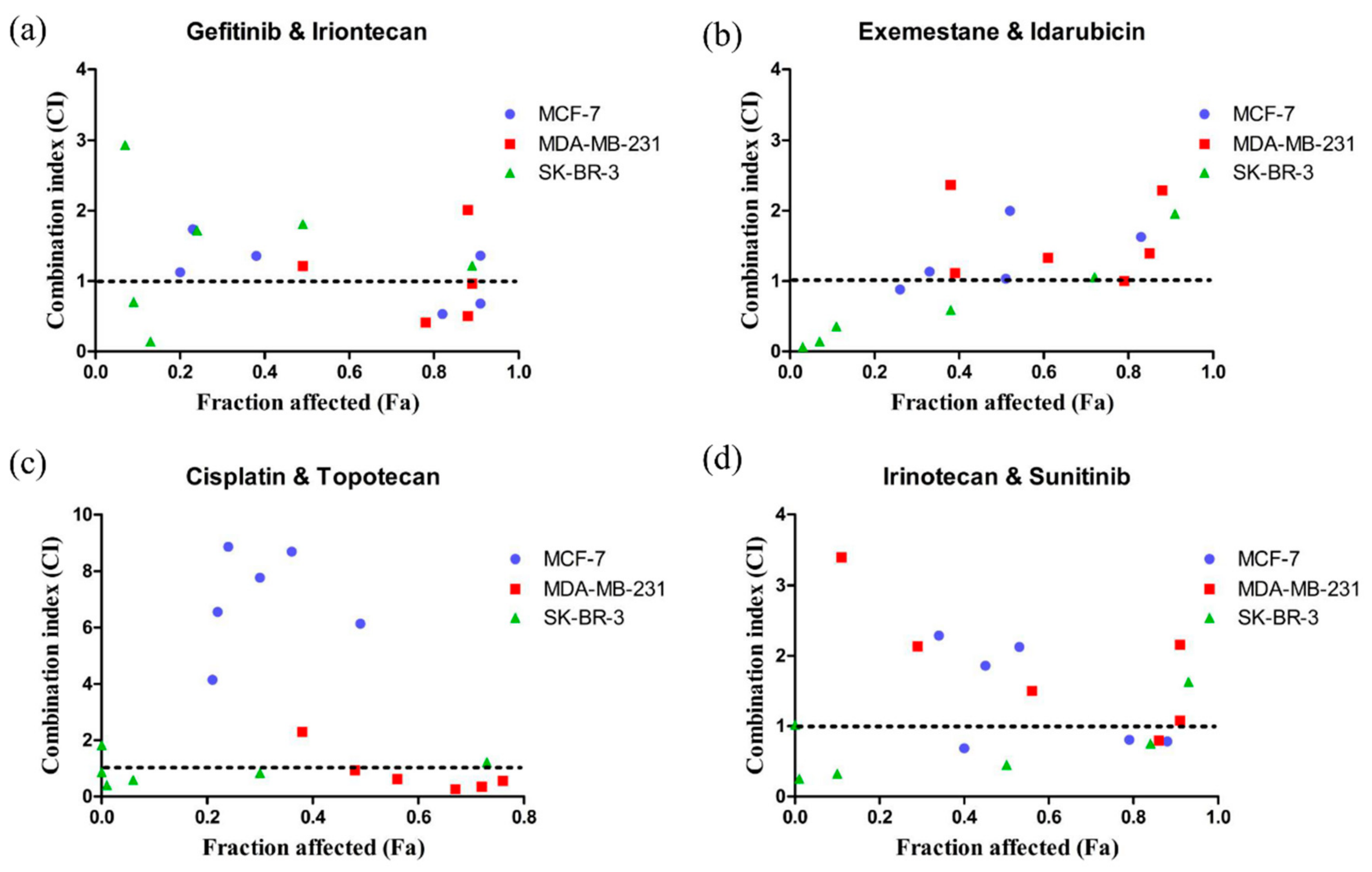
Figure 4.
Scatter plots of CI versus Fa. (a) gefitinib combined with irinotecan. (b) exemestane combined with idarubicin. (c) cisplatin combined with topotecan. (d) irinotecan combined with sunitinib. The synergistic effect of the combination of gefitinib and irinotecan on MCF-7, MDA-MB-231, and SK-BR-3 cells can be seen. The CI value and fraction affected (Fa) was calculated via CompuSyn software (ComboSyn, Inc., Paramus, NJ. 07652 USA). CI < 1 indicates a synergistic effect, CI = 1 indicates an additive effect, and CI > 1 indicates an antagonistic effect. Fa value represents the effect of the drug, that is, the tumour cell inhibition rate.
Figure 4.
Scatter plots of CI versus Fa. (a) gefitinib combined with irinotecan. (b) exemestane combined with idarubicin. (c) cisplatin combined with topotecan. (d) irinotecan combined with sunitinib. The synergistic effect of the combination of gefitinib and irinotecan on MCF-7, MDA-MB-231, and SK-BR-3 cells can be seen. The CI value and fraction affected (Fa) was calculated via CompuSyn software (ComboSyn, Inc., Paramus, NJ. 07652 USA). CI < 1 indicates a synergistic effect, CI = 1 indicates an additive effect, and CI > 1 indicates an antagonistic effect. Fa value represents the effect of the drug, that is, the tumour cell inhibition rate.
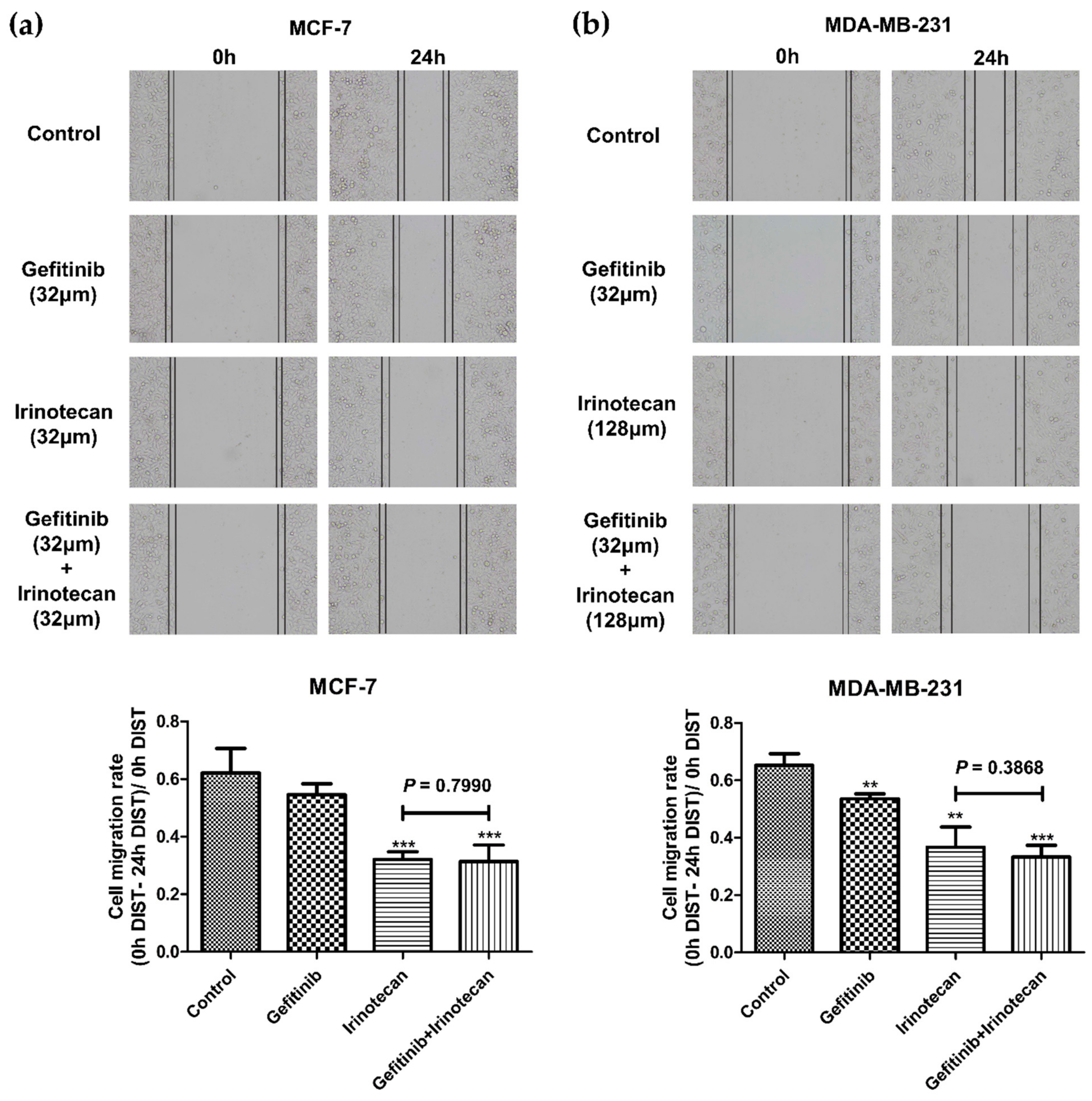
Figure 5.
Effects of gefitinib and irinotecan on the migration of BC cell lines. The cell migration rate was calculated as (0 h distance–24 h distance)/ 0 h distance. (a) In the MCF-7 cell line, the cell migration rates of the gefitinib, irinotecan, and gefitinib plus irinotecan groups were significantly lower than that of the control group at 24 h, while there was no significant difference between the gefitinib plus irinotecan group and the irinotecan group. (b) In the MDA-MB-231 cell line, the cell migration rates of the gefitinib, irinotecan, and gefitinib plus irinotecan groups were significantly lower than that of the control group at 24 h, and the migration rate of the gefitinib plus irinotecan group was significantly lower than that of the irinotecan group. * p < 0.05, ** p < 0.01, and *** p < 0.001, as compared with the control group.
Figure 5.
Effects of gefitinib and irinotecan on the migration of BC cell lines. The cell migration rate was calculated as (0 h distance–24 h distance)/ 0 h distance. (a) In the MCF-7 cell line, the cell migration rates of the gefitinib, irinotecan, and gefitinib plus irinotecan groups were significantly lower than that of the control group at 24 h, while there was no significant difference between the gefitinib plus irinotecan group and the irinotecan group. (b) In the MDA-MB-231 cell line, the cell migration rates of the gefitinib, irinotecan, and gefitinib plus irinotecan groups were significantly lower than that of the control group at 24 h, and the migration rate of the gefitinib plus irinotecan group was significantly lower than that of the irinotecan group. * p < 0.05, ** p < 0.01, and *** p < 0.001, as compared with the control group.
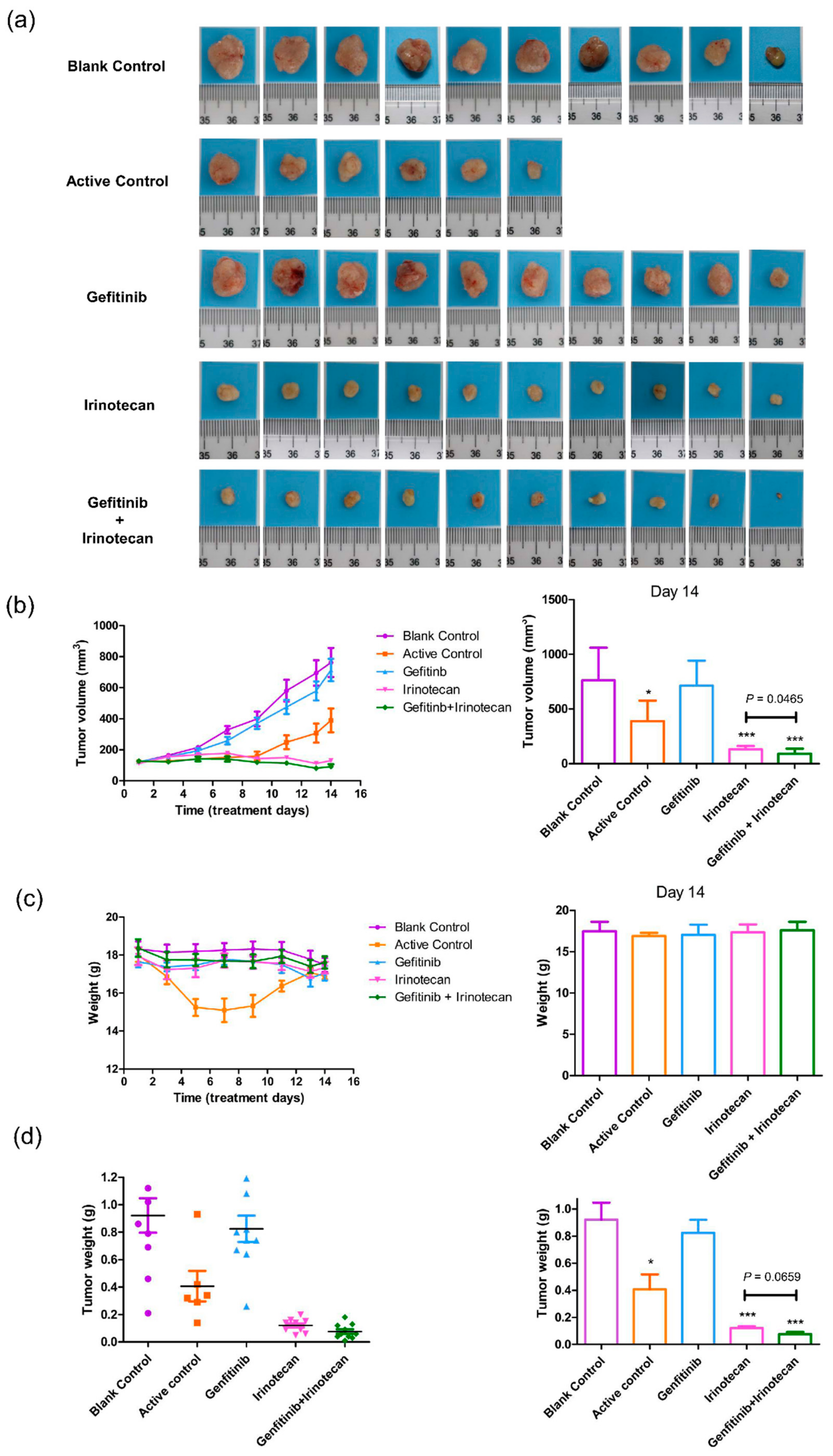
Figure 6.
Effects of gefitinib and irinotecan on cell-derived xenograft models constructed with MDA-MB-231 cell lines. (a) After 14 days of drug treatment, the tumours of the nude mice were removed and photographed on a 2 cm × 2 cm light blue cardboard platform (ten nude mice in each group; four nude mice in the active control group died; therefore, four pictures are missing from (a)). (b) Trends in tumour volume in each drug treatment group during the 14-day dosing cycle (measured with Vernier callipers). At the end of the 14th day, the tumour volumes of the active control (paclitaxel plus gemcitabine), irinotecan, and gefitinib plus irinotecan groups were significantly smaller than that of the blank control group, and the tumour volume of the gefitinib plus irinotecan group was significantly smaller than that of the irinotecan group. (c) The positive control group continued to decrease in weight five days after administration, indicating that the drug combination was highly toxic to nude mice according to this metric and resulting in intolerance. After ceasing the administration of gemcitabine on the sixth day, the weight of the surviving mice in this group gradually recovered. The other groups of mice remained stable during the 14-day treatment period. At the end of the 14th day, there was no significant difference in body weight between the groups. (d) On the 14th day, the subcutaneous tumours of all nude mice were removed for weighing. The tumour weights of the active control, irinotecan, and gefitinib plus irinotecan groups were significantly lower than those of the blank control group. The tumour weight of the gefitinib plus irinotecan group was lower than that of the irinotecan group, and the p value was 0.0659, which indicated marginal significance. * p < 0.05, and *** p < 0.001, as compared with the blank control group.
Figure 6.
Effects of gefitinib and irinotecan on cell-derived xenograft models constructed with MDA-MB-231 cell lines. (a) After 14 days of drug treatment, the tumours of the nude mice were removed and photographed on a 2 cm × 2 cm light blue cardboard platform (ten nude mice in each group; four nude mice in the active control group died; therefore, four pictures are missing from (a)). (b) Trends in tumour volume in each drug treatment group during the 14-day dosing cycle (measured with Vernier callipers). At the end of the 14th day, the tumour volumes of the active control (paclitaxel plus gemcitabine), irinotecan, and gefitinib plus irinotecan groups were significantly smaller than that of the blank control group, and the tumour volume of the gefitinib plus irinotecan group was significantly smaller than that of the irinotecan group. (c) The positive control group continued to decrease in weight five days after administration, indicating that the drug combination was highly toxic to nude mice according to this metric and resulting in intolerance. After ceasing the administration of gemcitabine on the sixth day, the weight of the surviving mice in this group gradually recovered. The other groups of mice remained stable during the 14-day treatment period. At the end of the 14th day, there was no significant difference in body weight between the groups. (d) On the 14th day, the subcutaneous tumours of all nude mice were removed for weighing. The tumour weights of the active control, irinotecan, and gefitinib plus irinotecan groups were significantly lower than those of the blank control group. The tumour weight of the gefitinib plus irinotecan group was lower than that of the irinotecan group, and the p value was 0.0659, which indicated marginal significance. * p < 0.05, and *** p < 0.001, as compared with the blank control group.
![Cancers 13 03586 g006]()
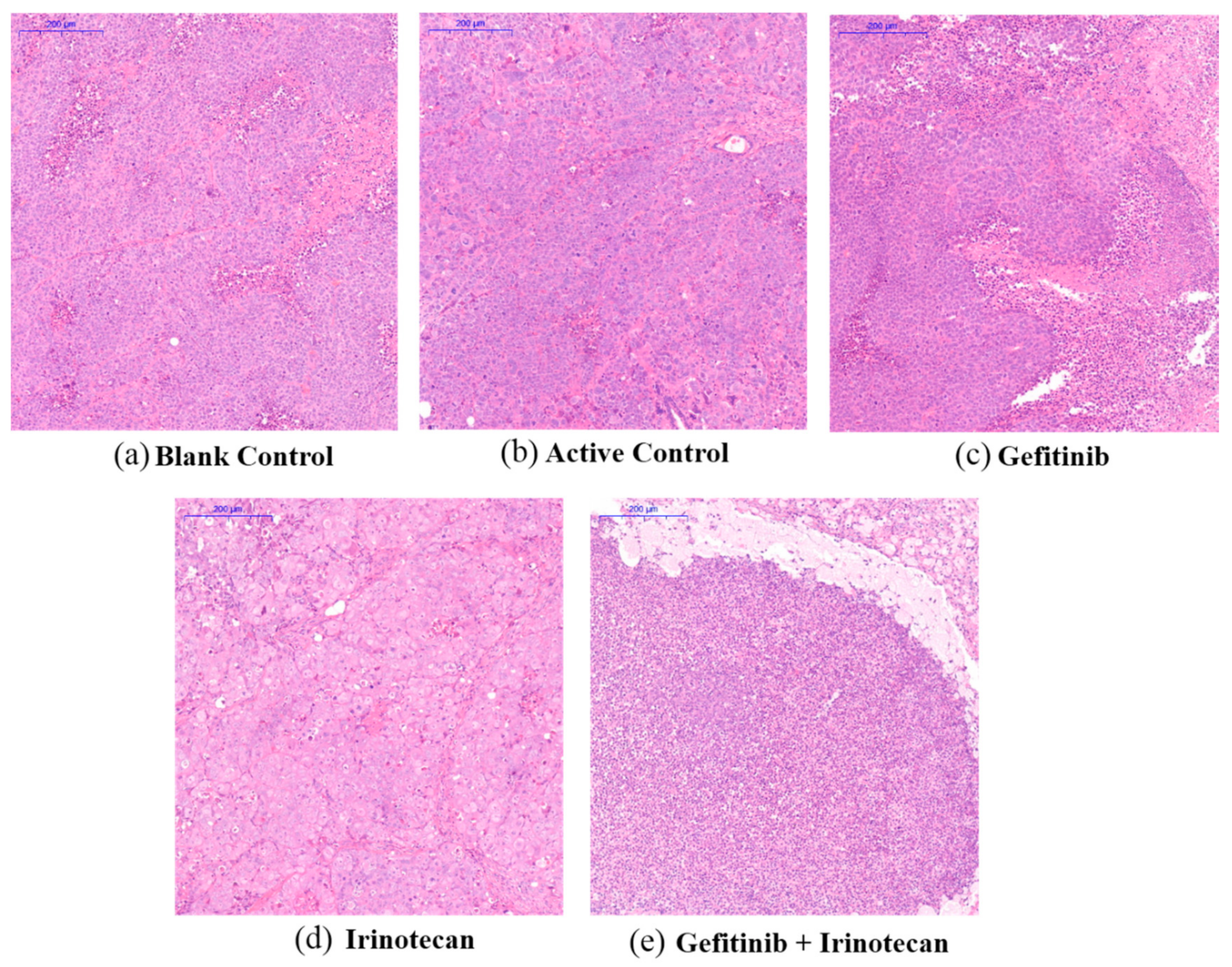
Figure 7.
Effect of drug treatment on the pathological changes of transplanted tumours in nude mice, as indicated by H&E staining. (a,b) The solvent blank control group and the active control group of paclitaxel plus gemcitabine, respectively. (c,d) The single-agent gefitinib group and the single-agent irinotecan group, respectively. (e) Gefitinib plus irinotecan combination group. The scale bar of the subfigures is 200 μm.
Figure 7.
Effect of drug treatment on the pathological changes of transplanted tumours in nude mice, as indicated by H&E staining. (a,b) The solvent blank control group and the active control group of paclitaxel plus gemcitabine, respectively. (c,d) The single-agent gefitinib group and the single-agent irinotecan group, respectively. (e) Gefitinib plus irinotecan combination group. The scale bar of the subfigures is 200 μm.
Table 1.
Group information of wound healing analysis.
Table 1.
Group information of wound healing analysis.
| Group | MCF-7 (Drug Treatment) | MDA-MB-231 (Drug Treatment) |
|---|
| Control | Solvent | Solvent |
| Gefitinib | Gefitinib (32 μM) | Gefitinib (32 μM) |
| Irinotecan | Irinotecan (32 μM) | Irinotecan (128 μM) |
| Gefitinib + Irinotecan | Gefitinib (32 μM) +Irinotecan (32 μM) | Gefitinib (32 μM) +Irinotecan (128 μM) |
Table 2.
Grouping of xenotransplantation experiments and drug treatment information.
Table 2.
Grouping of xenotransplantation experiments and drug treatment information.
| Groups | Number | Drug Treatment (Dosage of Administration; Administration Route; Delivery Days) |
|---|
| Blank Control | 1–10 | 5% glucose solution (the dose volume was approximately equal to group Gefitinib plus Irinotecan) |
| Active Control | 11–20 | Paclitaxel (20 mg/kg; ip a; 1,8); Gemcitabine (35 mg/kg, ip, 1,2,3,14) |
| Gefitinib | 21–30 | Gefitinib (30 mg/kg; ig b; 1,3,5,7,9,11,13) |
| Irinotecan | 31–40 | Irinotecan (8 mg/kg; ip; 1,3,5,7,9,11,13) |
| Gefitinib + Irinotecan | 41–50 | Gefitinib (30 mg/kg; ig; 1,3,5,7,9,11,13), Irinotecan (8 mg/kg; ip; 1,3,5,7,9,11,13) |
Table 3.
The enriched pathways of important genes in BC (FDR q-value < 0.01).
Table 3.
The enriched pathways of important genes in BC (FDR q-value < 0.01).
| Gene Set Name | p-Value | FDR q-Value |
|---|
| Calcium signalling pathway | 1.77 × 10−9 | 3.29 × 10−7 |
| Pathways in cancer | 2.72 × 10−8 | 2.53 × 10−6 |
| MAPK signalling pathway | 2.65 × 10−7 | 1.64 × 10−5 |
| Focal adhesion | 3.79 × 10−7 | 1.76 × 10−5 |
| Neuroactive ligand–receptor interaction | 5.95 × 10−6 | 2.21 × 10−4 |
| Wnt signalling pathway | 7.19 × 10−6 | 2.23 × 10−4 |
| Vascular smooth muscle contraction | 1.33 × 10−5 | 3.55 × 10−4 |
| Gap junction | 5.68 × 10−5 | 1.22 × 10−3 |
| Purine metabolism | 5.91 × 10−5 | 1.22 × 10−3 |
| Lysosome | 1.14 × 10−4 | 2.11 × 10−3 |
| Melanogenesis | 1.50 × 10−4 | 2.54 × 10−3 |
| Small cell lung cancer | 2.05 × 10−4 | 3.18 × 10−3 |
| Oocyte meiosis | 3.48 × 10−4 | 4.98 × 10−3 |
| Fructose and mannose metabolism | 4.38 × 10−4 | 5.81 × 10−3 |
| Glycosaminoglycan degradation | 6.06 × 10−4 | 6.83 × 10−3 |
| Basal cell carcinoma | 6.17 × 10−4 | 6.83 × 10−3 |
| Phosphatidylinositol signalling system | 6.24 × 10−4 | 6.83 × 10−3 |
| Hedgehog signalling pathway | 6.81 × 10−4 | 7.03 × 10−3 |
| Regulation of actin cytoskeleton | 9.83 × 10−4 | 9.63 × 10−3 |
Table 4.
Actual and predictive activity of BC drugs (consistent).
Table 4.
Actual and predictive activity of BC drugs (consistent).
| DCDB ID | Drug Combination | Predicted Subtypes | Clinical Trial Subtypes | Clinical Effectiveness |
|---|
| DC000083 | arzoxifene + LG100268 | TNBC/HER2+ | TNBC/HER2+ | efficacious |
| DC000132 | paclitaxel + trastuzumab | HER2+/Luminal B | HER2+/Luminal B | efficacious |
| DC000225 | gemcitabine + trastuzumab | HER2+ | HER2+/Luminal B | efficacious |
| DC000227 | epirubicin + trastuzumab | HER2+ | HER2+/Luminal B | efficacious |
| DC000231 | trastuzumab + vinorelbine | HER2+ | HER2+/Luminal B | efficacious |
| DC000233 | cyclophosphamide + trastuzumab | HER2+/Luminal B | HER2+/Luminal B | efficacious |
| DC000236 | carboplatin + trastuzumab | HER2+ | HER2+/Luminal B | efficacious |
| DC000649 | lapatinib + paclitaxel | HER2+/Luminal B | HER2+/Luminal B | efficacious |
| DC006857 | cediranib + olaparib | TNBC | TNBC/HER2+ | efficacious |
| DC001856 | anastrozole + gefitinib | TNBC/HER2+ | Luminal A/Luminal B | non-efficacious |
| DC002772 | fulvestrant + gefitinib | TNBC/HER2+ | Luminal A/Luminal B | non-efficacious |
Table 5.
Actual and predictive activity of BC drugs (inconsistent).
Table 5.
Actual and predictive activity of BC drugs (inconsistent).
| DCDB ID | Drug Combination | Predicted Subtypes | Clinical Trial Subtypes | Clinical Effectiveness |
|---|
| DC000089 | gefitinib + trastuzumab | HER2+ | HER2+ | non-efficacious |
| DC000220 | lapatinib + letrozole | TNBC/HER2+ | Luminal A/Luminal B | efficacious |
| DC000229 | doxorubicin + trastuzumab | Luminal A | HER2+ | efficacious |
Table 6.
Subtype prediction results for four drug combinations (p-values evaluated with binomial tests).
Table 6.
Subtype prediction results for four drug combinations (p-values evaluated with binomial tests).
| Drug Combination | Luminal A | Luminal B | HER2+ | TNBC |
|---|
| gefitinib + irinotecan | 0.9233 | 0.2787 | 0.1644 | 0.0401 |
| exemestane + idarubicin | 0.9998 | 0.0819 | 0.0241 | 7.9100 × 10−11 |
| cisplatin + topotecan | 1.0000 | 0.6619 | <1 × 10−16 | <1 × 10−16 |
| irinotecan + sunitinib | 0.9233 | 0.6619 | 0.5039 | 0.0006 |
Table 7.
Single drug IC50 value determination.
Table 7.
Single drug IC50 value determination.
| Drug Name | MCF-7 | MDA-MB-231 | SK-BR-3 |
|---|
| IC50 (µM) | Concentration Range (µM) | IC50 (µM) | Concentration Range (µM) | IC50 (µM) | Concentration Range (µM) |
|---|
| Gefitinib | 35.19 | 4.69–600 | 55.20 | 4.69–600 | 5.55 | 0.00768–600 |
| Irinotecan | 34.76 | 0.002–150 | 201.27 | 0.1–300 | 26.36 | 0.00768–600 |
| Exemestane | 145.78 | 4.688–600 | 127.37 | 4.688–600 | 128.38 | 0.00768–600 |
| Idarubicin | 0.31 | 0.00256–200 | 0.26 | 0.00256–200 | 0.02 | 0.00768–600 |
| Cisplatin | 47.12 | 2.34–300 | 47.12 | 2.34–300 | 8.46 | 0.00768–600 |
| Topotecan | 0.48 | 0.1–300 | 103.57 | 0.1–300 | 0.74 | 0.00768–600 |
| Sunitinib | 4.05 | 0.1–300 | 8.37 | 0.1–300 | 5.94 | 0.00768–600 |
Table 8.
Fa and CI values for actual experimental points of gefitinib combined with irinotecan.
Table 8.
Fa and CI values for actual experimental points of gefitinib combined with irinotecan.
| MCF-7 (1:1) a | MDA-MB-231 (1:4) b | SK-BR-3 (0.03:0.16) c |
|---|
| Total Dose | Fa | CI Value | Total Dose | Fa | CI Value | Total Dose | Fa | CI Value |
|---|
| 256.00 | 0.91 | 1.36100 | 640.00 | 0.88 | 2.01029 | 93.00 | 0.89 | 1.22031 |
| 128.00 | 0.91 | 0.68050 | 320.00 | 0.89 | 0.96101 | 31.00 | 0.49 | 1.81269 |
| 64.00 | 0.82 | 0.53076 | 160.00 | 0.88 | 0.50257 | 10.34 | 0.24 | 1.71940 |
| 32.00 | 0.38 | 1.35817 | 80.00 | 0.78 | 0.41251 | 3.45 | 0.07 | 2.93259 |
| 16.00 | 0.23 | 1.73336 | 40.00 | 0.49 | 1.21187 | 1.15 | 0.09 | 0.70258 |
| 8.00 | 0.20 | 1.12579 | | | | 0.38 | 0.13 | 0.14314 |
Table 9.
Fa and CI values for actual experimental points of exemestane combined with idarubicin.
Table 9.
Fa and CI values for actual experimental points of exemestane combined with idarubicin.
| MCF-7 (4.69:0.01) a | MDA-MB-231 (4.69:0.01) b | SK-BR-3 (1.6:2.0E-4) c |
|---|
| Total Dose | Fa | CI Value | Total Dose | Fa | CI Value | Total Dose | Fa | CI Value |
|---|
| 300.60 | 0.83 | 1.62448 | 300.60 | 0.88 | 2.28575 | 390.06 | 0.91 | 1.95615 |
| 150.30 | 0.52 | 1.99761 | 150.30 | 0.85 | 1.39243 | 130.02 | 0.72 | 1.05564 |
| 75.15 | 0.51 | 1.03379 | 75.15 | 0.79 | 0.99996 | 43.34 | 0.38 | 0.59039 |
| 37.58 | 0.33 | 1.13269 | 37.58 | 0.61 | 1.32759 | 14.44 | 0.11 | 0.35679 |
| 18.79 | 0.26 | 0.87867 | 18.79 | 0.38 | 2.36631 | 4.81 | 0.07 | 0.14347 |
| 9.40 | 0.05 | 9.02758 | 9.40 | 0.39 | 1.11416 | 1.60 | 0.03 | 0.06721 |
Table 10.
Fa and CI values for actual experimental points of cisplatin combined with topotecan.
Table 10.
Fa and CI values for actual experimental points of cisplatin combined with topotecan.
| MCF-7 (3.13:0.03) a | MDA-MB-231 (0.41:1.23) b | SK-BR-3 (0.12:0.01) c |
|---|
| Total Dose | Fa | CI Value | Total Dose | Fa | CI Value | Total Dose | Fa | CI Value |
|---|
| 100.80 | 0.49 | 6.13662 | 400.00 | 0.76 | 0.55871 | 32.25 | 0.73 | 1.22184 |
| 50.40 | 0.36 | 8.68534 | 133.33 | 0.72 | 0.34791 | 10.75 | 0.30 | 0.83846 |
| 25.20 | 0.30 | 7.77286 | 44.44 | 0.67 | 0.26373 | 3.58 | 0.06 | 0.59108 |
| 12.60 | 0.24 | 8.85453 | 14.81 | 0.56 | 0.62049 | 1.19 | 0.01 | 0.40758 |
| 6.30 | 0.22 | 6.55762 | 4.93 | 0.48 | 0.93238 | 0.40 | 1.0E-4 | 0.86656 |
| 3.16 | 0.21 | 4.14541 | 1.64 | 0.38 | 2.29294 | 0.13 | 1.0E-6 | 1.82670 |
Table 11.
Fa and CI values for actual experimental points of irinotecan combined with sunitinib.
Table 11.
Fa and CI values for actual experimental points of irinotecan combined with sunitinib.
| MCF-7 (3.13:0.03) a | MDA-MB-231 (0.41:1.23) b | SK-BR-3 (0.12:0.01) c |
|---|
| Total Dose | Fa | CI Value | Total Dose | Fa | CI Value | Total Dose | Fa | CI Value |
|---|
| 114.80 | 0.88 | 0.78207 | 537.60 | 0.91 | 2.15513 | 96.00 | 0.93 | 1.62976 |
| 57.40 | 0.79 | 0.80721 | 268.80 | 0.91 | 1.07757 | 32.00 | 0.84 | 0.75227 |
| 28.70 | 0.53 | 2.12400 | 134.40 | 0.86 | 0.79456 | 10.67 | 0.50 | 0.44974 |
| 14.35 | 0.45 | 1.85993 | 67.20 | 0.56 | 1.49972 | 3.56 | 0.10 | 0.32681 |
| 7.18 | 0.34 | 2.28449 | 33.60 | 0.29 | 2.13264 | 1.18 | 0.01 | 0.25476 |
| 3.59 | 0.40 | 0.68404 | 16.80 | 0.11 | 3.39470 | 0.39 | 1 × 10−5 | 1.02124 |


 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}