
Genotoxic Treatment Enhances Immune Response in a Genetic Model of Lung Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
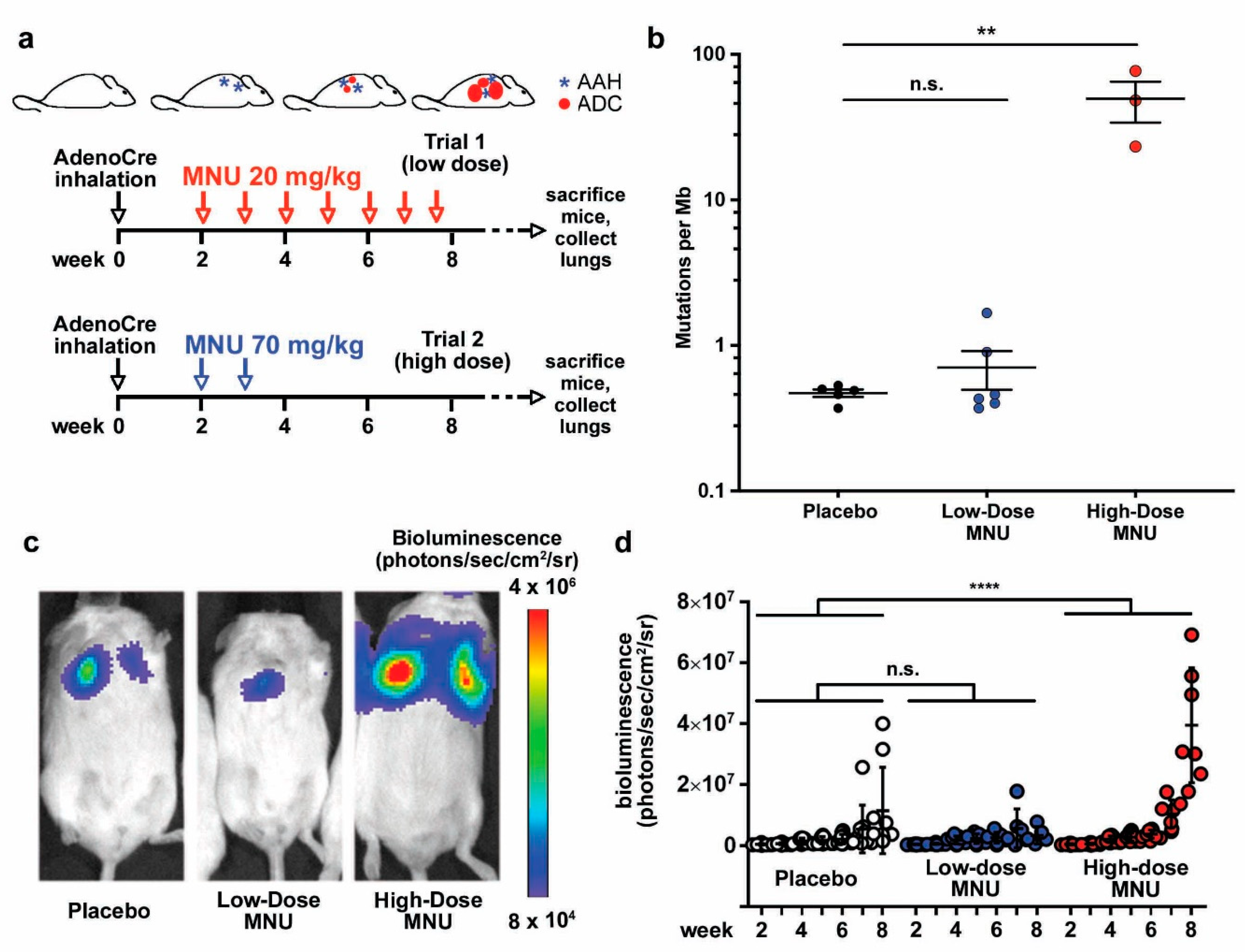
2.1. MNU Treatment Increases the Mutation Burden in a Genetically Engineered Murine Model of Lung Cancer
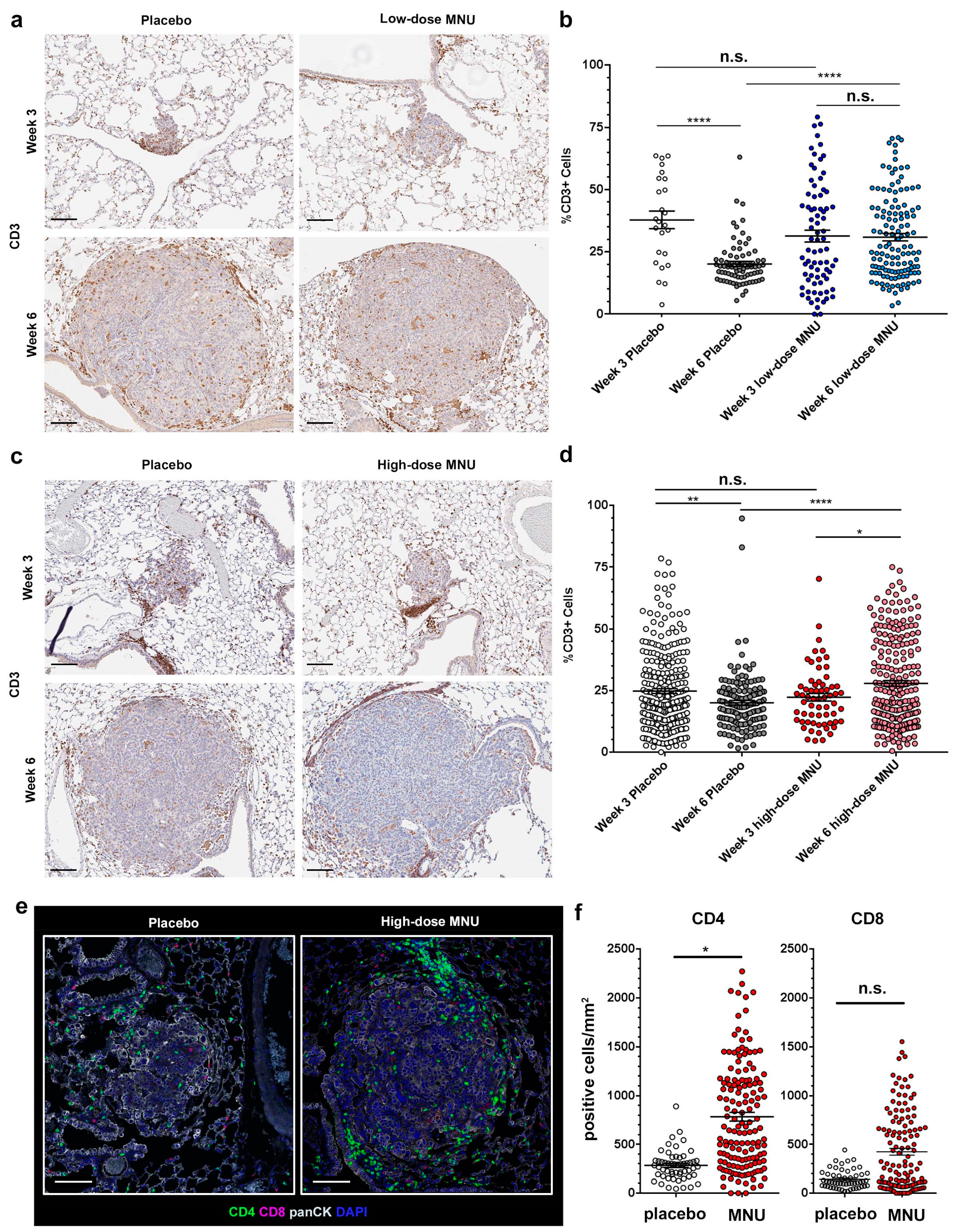
2.2. MNU Causes Sustained Tumor Lymphocyte Infiltration in KP Tumors
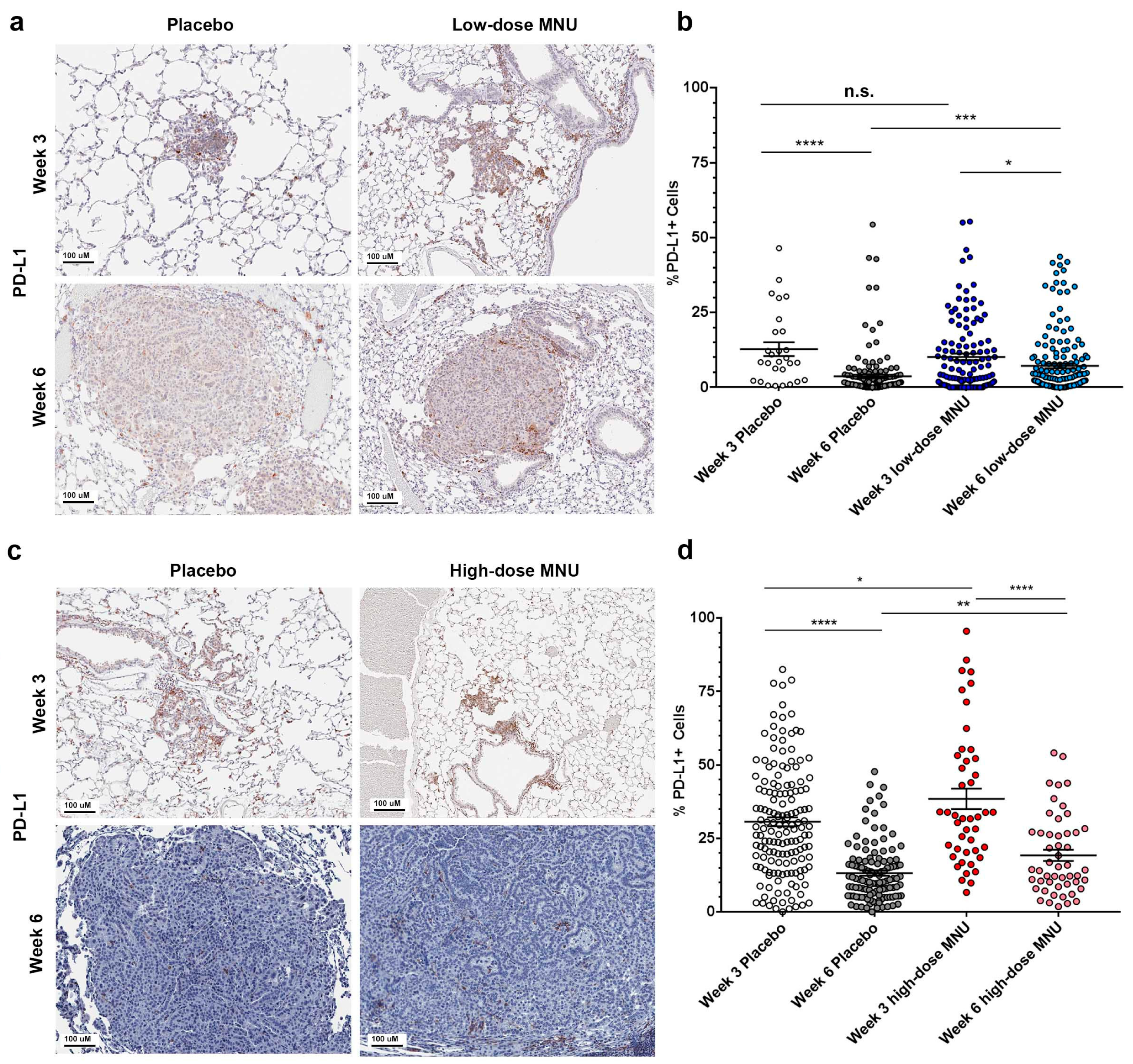
2.3. MNU Causes Sustained Immune Checkpoint Activation in KP Tumors
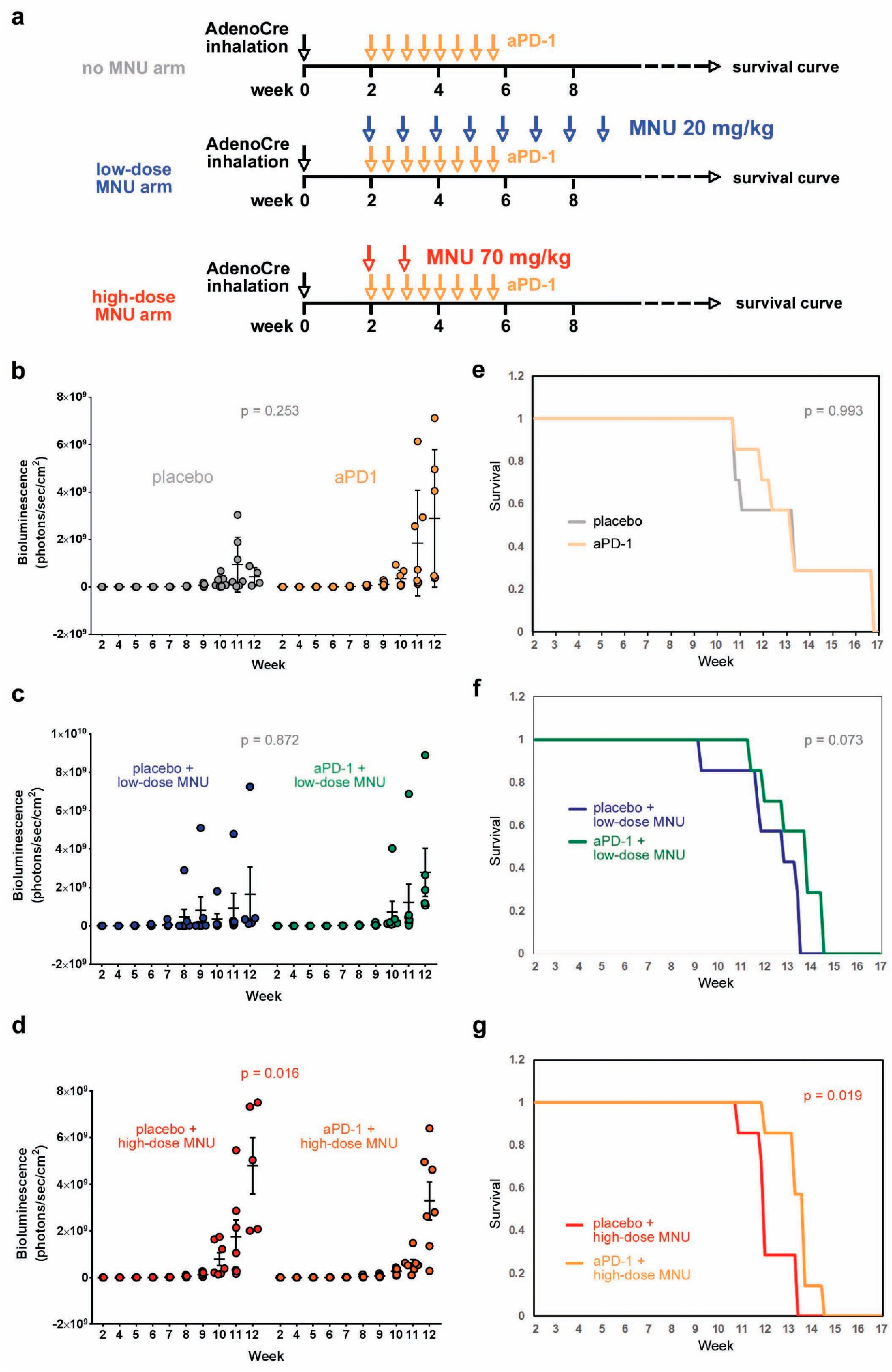
2.4. High-Dose MNU Treatment Improves the Response of KP Tumors to Checkpoint Inhibitors
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brahmer, J.R.; Govindan, R.; Anders, R.A.; Antonia, S.J.; Sagorsky, S.; Davies, M.J.; Dubinett, S.M.; Ferris, A.; Gandhi, L.; Garon, E.B.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (nsclc). J. Immunother. Cancer 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, G.B.; Farid, S.; Liu, S.V. Immunotherapy in lung cancer. J Surg Oncol 2021, 123, 718–729. [Google Scholar] [CrossRef]
- Mielgo-Rubio, X.; Uribelarrea, E.A.; Cortes, L.Q.; Moyano, M.S. Immunotherapy in non-small cell lung cancer: Update and new insights. J. Clin. Transl. Res. 2021, 7, 1–21. [Google Scholar]
- Melosky, B.; Juergens, R.; McLeod, D.; Leighl, N.; Brade, A.; Card, P.B.; Chu, Q. Immune checkpoint-inhibitors and chemoradiation in stage iii unresectable non-small cell lung cancer. Lung Cancer 2019, 134, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.J.; Havel, L.S.; Crowley, M.J.; Ban, Y.; Lee, S.B.; Thalappillil, J.S.; Narula, N.; Bhinder, B.; Elemento, O.; Wong, S.T.; et al. Immune reprogramming via pd-1 inhibition enhances early-stage lung cancer survival. JCI Insight 2018, 3, e96836. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant pd-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Chang, X.; Brait, M.; Fertig, E.; Kagohara, L.T.; Bedi, A.; Marchionni, L.; Agrawal, N.; Ravi, R.; Jones, S.; et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat. Commun. 2015, 6, 8258. [Google Scholar] [CrossRef]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung cancer subtypes generate unique immune responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faget, J.; Groeneveld, S.; Boivin, G.; Sankar, M.; Zangger, N.; Garcia, M.; Guex, N.; Zlobec, I.; Steiner, L.; Piersigilli, A.; et al. Neutrophils and snail orchestrate the establishment of a pro-tumor microenvironment in lung cancer. Cell Rep. 2017, 21, 3190–3204. [Google Scholar] [CrossRef] [Green Version]
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. Bet bromodomain inhibition cooperates with pd-1 blockade to facilitate antitumor response in kras-mutant non-small cell lung cancer. Cancer Immunol. Res. 2018, 6, 1234–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to pd-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westcott, P.M.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; Delrosario, R.; Jen, K.Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of kras-driven lung cancer. Nature 2015, 517, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Shioyama, Y.; Gondo, Y.; Nakao, K.; Katsuki, M. Different mutation frequencies and spectra among organs by n-methyl-n-nitrosourea in rpsl (stra) transgenic mice. Jpn. J. Cancer Res. 2000, 91, 482–491. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Candrian, U.; Maronpot, R.R.; Stoner, G.D.; Anderson, M.W. Activation of the ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain a mouse. Proc. Natl. Acad. Sci. USA 1989, 86, 3070–3074. [Google Scholar] [CrossRef] [Green Version]
- Salehi-Rad, R.; Li, R.; Tran, L.M.; Lim, R.J.; Abascal, J.; Momcilovic, M.; Park, S.J.; Ong, S.L.; Shabihkhani, M.; Huang, Z.L.; et al. Novel kras-mutant murine models of non-small cell lung cancer possessing co-occurring oncogenic mutations and increased tumor mutational burden. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Scafoglio, C.R.; Villegas, B.; Abdelhady, G.; Bailey, S.T.; Liu, J.; Shirali, A.S.; Wallace, W.D.; Magyar, C.E.; Grogan, T.R.; Elashoff, D.; et al. Sodium-glucose transporter 2 is a diagnostic and therapeutic target for early-stage lung adenocarcinoma. Sci. Transl. Med. 2018, 10, eaat5933. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.-Y.; Zeger, S.L. Longitudinal data analysis using generalized linear models. Biometrika 1986, 73, 13–22. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Bai, R.; Li, L.; Chen, X.; Chen, N.; Song, W.; Cui, J. Neoadjuvant and adjuvant immunotherapy: Opening new horizons for patients with early-stage non-small cell lung cancer. Front. Oncol. 2020, 10, 575472. [Google Scholar] [CrossRef] [PubMed]
- Biton, J.; Mansuet-Lupo, A.; Pecuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. Tp53, stk11, and egfr mutations predict tumor immune profile and the response to anti-pd-1 in lung adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. Stk11/lkb1 mutations and pd-1 inhibitor resistance in kras-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, D.; Mazzotta, M.; Scalera, S.; Terrenato, I.; Sperati, F.; D’Ambrosio, L.; Pallocca, M.; Corleone, G.; Krasniqi, E.; Pizzuti, L.; et al. Keap1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann. Oncol. 2020, 31, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Humeau, J.; Buque, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. Qupath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saggese, P.; Martinez, C.A.; Tran, L.M.; Lim, R.; Dumitras, C.; Grogan, T.; Elashoff, D.; Salehi-Rad, R.; Dubinett, S.M.; Liu, B.; et al. Genotoxic Treatment Enhances Immune Response in a Genetic Model of Lung Cancer. Cancers 2021, 13, 3595. https://doi.org/10.3390/cancers13143595
Saggese P, Martinez CA, Tran LM, Lim R, Dumitras C, Grogan T, Elashoff D, Salehi-Rad R, Dubinett SM, Liu B, et al. Genotoxic Treatment Enhances Immune Response in a Genetic Model of Lung Cancer. Cancers. 2021; 13(14):3595. https://doi.org/10.3390/cancers13143595
Chicago/Turabian StyleSaggese, Pasquale, Cesar A. Martinez, Linh M. Tran, Raymond Lim, Camelia Dumitras, Tristan Grogan, David Elashoff, Ramin Salehi-Rad, Steven M. Dubinett, Bin Liu, and et al. 2021. "Genotoxic Treatment Enhances Immune Response in a Genetic Model of Lung Cancer" Cancers 13, no. 14: 3595. https://doi.org/10.3390/cancers13143595
APA StyleSaggese, P., Martinez, C. A., Tran, L. M., Lim, R., Dumitras, C., Grogan, T., Elashoff, D., Salehi-Rad, R., Dubinett, S. M., Liu, B., & Scafoglio, C. (2021). Genotoxic Treatment Enhances Immune Response in a Genetic Model of Lung Cancer. Cancers, 13(14), 3595. https://doi.org/10.3390/cancers13143595