
Functional Roles of Bromodomain Proteins in Cancer

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Bromodomains Are Histone Lysine Acetylation Reader Domains
3. Expression and Dependency Patterns of Bromodomain Genes across Cancer
4. An Interconnected Network of Functional Groups in Bromodomain Proteins
4.1. BET Family of Bromodomain Proteins
4.2. Chromatin Remodeling Factors
4.3. HAT Bromodomain Proteins
4.4. WD Repeat Proteins
4.5. AAA-ATPase Bromodomain Proteins
4.6. HMT Enzymes
4.7. TRIM Family of Bromodomain Proteins
4.8. Speckled Protein Family of Bromodomain Proteins
4.9. Zinc Finger MYND Bromodomain Proteins
5. Emerging Strategies to Target Bromodomain Proteins
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gregorc, V.; Lazzari, C.; Mandala, M.; Ippati, S.; Bulotta, A.; Cangi, M.G.; Khater, A.; Vigano, M.G.; Mirabile, A.; Pecciarini, L.; et al. Intratumoral Cellular Heterogeneity: Implications for Drug Resistance in Patients with Non-Small Cell Lung Cancer. Cancers 2021, 13, 2023. [Google Scholar] [CrossRef]
- Vignoli, A.; Risi, E.; McCartney, A.; Migliaccio, I.; Moretti, E.; Malorni, L.; Luchinat, C.; Biganzoli, L.; Tenori, L. Precision Oncology via NMR-Based Metabolomics: A Review on Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4687. [Google Scholar] [CrossRef] [PubMed]
- Ieni, A.; Vita, R.; Pizzimenti, C.; Benvenga, S.; Tuccari, G. Intratumoral Heterogeneity in Differentiated Thyroid Tumors: An Intriguing Reappraisal in the Era of Personalized Medicine. J. Pers. Med. 2021, 11, 333. [Google Scholar] [CrossRef] [PubMed]
- Castells-Roca, L.; Tejero, E.; Rodriguez-Santiago, B.; Surralles, J. CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer. Cancers 2021, 13, 1591. [Google Scholar] [CrossRef]
- Li, Q.; Sun, M.; Wang, M.; Feng, M.; Yang, F.; Li, L.; Zhao, J.; Chang, C.; Dong, H.; Xie, T.; et al. Dysregulation of Wnt/beta-catenin signaling by protein kinases in hepatocellular carcinoma and its therapeutic application. Cancer Sci. 2021, 112, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Gilman, E.A.; Pruthi, S.; Hofstatter, E.W.; Mussallem, D.M. Preventing Breast Cancer Through Identification and Pharmacologic Management of High-Risk Patients. Mayo Clin. Proc. 2021, 96, 1033–1040. [Google Scholar] [CrossRef]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar] [CrossRef]
- Soffer, R.L. Post-translational modification of proteins catalyzed by aminoacyl-tRNA-protein transferases. Mol. Cell Biochem. 1973, 2, 3–14. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Perez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenet. Off. J. DNA Methylation Soc. 2017, 12, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Tweedie-Cullen, R.Y.; Brunner, A.M.; Grossmann, J.; Mohanna, S.; Sichau, D.; Nanni, P.; Panse, C.; Mansuy, I.M. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS ONE 2012, 7, e36980. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, Q.; Li, S.; Plotnikov, A.N.; Walsh, M.J.; Zhou, M.M. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 2010, 466, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.; Li, W.; et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sabari, B.R.; Panchenko, T.; Wen, H.; Zhao, D.; Guan, H.; Wan, L.; Huang, H.; Tang, Z.; Zhao, Y.; et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 2016, 62, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef] [Green Version]
- Jeanmougin, F.; Wurtz, J.M.; Le Douarin, B.; Chambon, P.; Losson, R. The bromodomain revisited. Trends Biochem. Sci. 1997, 22, 151–153. [Google Scholar] [CrossRef]
- Owen, D.J.; Ornaghi, P.; Yang, J.C.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, F.A.; Taylor, A.M.; Crawford, T.D.; Tsui, V.; Cote, A.; Magnuson, S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J. Med. Chem. 2016, 59, 1271–1298. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Hewings, D.S.; Rooney, T.P.; Jennings, L.E.; Hay, D.A.; Schofield, C.J.; Brennan, P.E.; Knapp, S.; Conway, S.J. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J. Med. Chem. 2012, 55, 9393–9413. [Google Scholar] [CrossRef]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone modifications. Cell 2014, 159, 458.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauser, R.; Jeltsch, A. Application of modified histone peptide arrays in chromatin research. Arch. Biochem. Biophys. 2019, 661, 31–38. [Google Scholar] [CrossRef]
- Quinn, A.M.; Bedford, M.T.; Espejo, A.; Spannhoff, A.; Austin, C.P.; Oppermann, U.; Simeonov, A. A homogeneous method for investigation of methylation-dependent protein-protein interactions in epigenetics. Nucleic Acids Res. 2010, 38, e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, J.T.; McLaughlin, K.; Lubula, M.Y.; Gay, J.C.; Dest, A.; Gao, C.; Phillips, M.; Tonelli, M.; Cornilescu, G.; Marunde, M.R.; et al. Structural Insights into the Recognition of Mono- and Diacetylated Histones by the ATAD2B Bromodomain. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.J.; Fernandez-Montalvan, A.E.; Badock, V.; Ott, C.J.; Holton, S.J.; von Ahsen, O.; Toedling, J.; Vittori, S.; Bradner, J.E.; Gorjanacz, M. ATAD2 is an epigenetic reader of newly synthesized histone marks during DNA replication. Oncotarget 2016, 7, 70323–70335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obi, J.O.; Lubula, M.Y.; Cornilescu, G.; Henrickson, A.; McGuire, K.; Evans, C.M.; Phillips, M.; Boyson, S.P.; Demeler, B.; Markley, J.L.; et al. The BRPF1 bromodomain is a molecular reader of di-acetyllysine. Curr. Res. Struct. Biol. 2020, 2, 104–115. [Google Scholar] [CrossRef]
- Flynn, E.M.; Huang, O.W.; Poy, F.; Oppikofer, M.; Bellon, S.F.; Tang, Y.; Cochran, A.G. A subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure 2015, 23, 1801–1814. [Google Scholar] [CrossRef] [Green Version]
- Moriniere, J.; Rousseaux, S.; Steuerwald, U.; Soler-Lopez, M.; Curtet, S.; Vitte, A.L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J.; et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668. [Google Scholar] [CrossRef]
- Chen, Y.; Sprung, R.; Tang, Y.; Ball, H.; Sangras, B.; Kim, S.C.; Falck, J.R.; Peng, J.; Gu, W.; Zhao, Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell Proteom. 2007, 6, 812–819. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unoki, M.; Masuda, A.; Dohmae, N.; Arita, K.; Yoshimatsu, M.; Iwai, Y.; Fukui, Y.; Ueda, K.; Hamamoto, R.; Shirakawa, M.; et al. Lysyl 5-hydroxylation, a novel histone modification, by Jumonji domain containing 6 (JMJD6). J. Biol. Chem. 2013, 288, 6053–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhou, X.; Taghizadeh, K.; Dong, M.; Dedon, P.C. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Vollmuth, F.; Geyer, M. Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew. Chem. 2010, 49, 6768–6772. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.P.; Picaud, S.; Fujisawa, T.; Hou, H.; Savitsky, P.; Uuskula-Reimand, L.; Gupta, G.D.; Abdouni, H.; Lin, Z.Y.; Tucholska, M.; et al. Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol. Cell 2019, 73, 621–638.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell Biol. 2011, 31, 2632–2640. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Li, Y.; Lv, J.; Zheng, X.; Wen, H.; Shen, H.; Zhu, G.; Chen, T.Y.; Dhar, S.S.; Kan, P.Y.; et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol. Cell 2016, 63, 470–484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Cai, Z.; Kong, M.; Wu, A.; Hu, Z.; Wang, F.; Wang, H. Prognostic significance of TRIM28 expression in patients with breast carcinoma. Open Med. 2021, 16, 472–480. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytola, V.; Itkonen, H.M.; Coleman, I.M.; Vodak, D.; Sjoblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Maruschke, M.; Hakenberg, O.; Zimmermann, W.; Stief, C.G.; Buchner, A. TOP2A, HELLS, ATAD2, and TET3 Are Novel Prognostic Markers in Renal Cell Carcinoma. Urology 2017, 102, 265.e261–265.e267. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, C.; Du, W.; Yang, X.; Chen, Z. ATAD2 is overexpressed in gastric cancer and serves as an independent poor prognostic biomarker. Clin. Transl. Oncol. 2016, 18, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, B.; Bao, L.; Jin, L.; Yang, M.; Peng, Y.; Kumar, A.; Wang, J.E.; Wang, C.; Zou, X.; et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Investig. 2018, 128, 1937–1955. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pena, J.; Paez, R.; Nieto-Jimenez, C.; Sanchez, V.C.; Galan-Moya, E.M.; Pandiella, A.; Gyorffy, B.; Ocana, A. Mapping Bromodomains in breast cancer and association with clinical outcome. Sci. Rep. 2019, 9, 5734. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute. The Cancer Genome Atlas. Available online: https://www.cancer.gov/tcga (accessed on 6 August 2020).
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Raeder, M.B.; Birkeland, E.; Trovik, J.; Krakstad, C.; Shehata, S.; Schumacher, S.; Zack, T.I.; Krohn, A.; Werner, H.M.; Moody, S.E.; et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS ONE 2013, 8, e54873. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Li, T.; Zhang, Y.; Guo, Y.; Yao, J.; Dou, L.; Guo, K. Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol. Rep. 2015, 33, 2337–2344. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.N.; Zhang, Y.X.; Wang, X.M.; Liu, Y.J.; Zhang, Y.Q.; Que, Y.H.; Zhao, W.J. ATAD2 is highly expressed in ovarian carcinomas and indicates poor prognosis. Asian Pac. J. Cancer Prev. 2014, 15, 2777–2783. [Google Scholar] [CrossRef]
- Luo, Y.; Ye, G.Y.; Qin, S.L.; Yu, M.H.; Mu, Y.F.; Zhong, M. ATAD2 Overexpression Identifies Colorectal Cancer Patients with Poor Prognosis and Drives Proliferation of Cancer Cells. Gastroenterol. Res. Pract. 2015, 2015, 936564. [Google Scholar] [CrossRef] [Green Version]
- Ciro, M.; Prosperini, E.; Quarto, M.; Grazini, U.; Walfridsson, J.; McBlane, F.; Nucifero, P.; Pacchiana, G.; Capra, M.; Christensen, J.; et al. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009, 69, 8491–8498. [Google Scholar] [CrossRef] [Green Version]
- Caron, C.; Lestrat, C.; Marsal, S.; Escoffier, E.; Curtet, S.; Virolle, V.; Barbry, P.; Debernardi, A.; Brambilla, C.; Brambilla, E.; et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene 2010, 29, 5171–5181. [Google Scholar] [CrossRef] [Green Version]
- Kalashnikova, E.V.; Revenko, A.S.; Gemo, A.T.; Andrews, N.P.; Tepper, C.G.; Zou, J.X.; Cardiff, R.D.; Borowsky, A.D.; Chen, H.W. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010, 70, 9402–9412. [Google Scholar] [CrossRef] [Green Version]
- Leachman, N.T.; Brellier, F.; Ferralli, J.; Chiquet-Ehrismann, R.; Tucker, R.P. ATAD2B is a phylogenetically conserved nuclear protein expressed during neuronal differentiation and tumorigenesis. Dev. Growth Differ. 2010, 52, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wei, T.; Si, X.; Wang, Q.; Li, Y.; Leng, Y.; Deng, A.; Chen, J.; Wang, G.; Zhu, S.; et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J. Biol. Chem. 2013, 288, 14510–14521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majaz, S.; Tong, Z.; Peng, K.; Wang, W.; Ren, W.; Li, M.; Liu, K.; Mo, P.; Li, W.; Yu, C. Histone acetyl transferase GCN5 promotes human hepatocellular carcinoma progression by enhancing AIB1 expression. Cell Biosci. 2016, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Reisman, D.N.; Sciarrotta, J.; Wang, W.; Funkhouser, W.K.; Weissman, B.E. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: Correlation with poor prognosis. Cancer Res. 2003, 63, 560–566. [Google Scholar]
- Wu, J.; He, K.; Zhang, Y.; Song, J.; Shi, Z.; Chen, W.; Shao, Y. Inactivation of SMARCA2 by promoter hypermethylation drives lung cancer development. Gene 2019, 687, 193–199. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Sandoval, G.J.; Pulice, J.L.; Pakula, H.; Schenone, M.; Takeda, D.Y.; Pop, M.; Boulay, G.; Williamson, K.E.; McBride, M.J.; Pan, J.; et al. Binding of TMPRSS2-ERG to BAF Chromatin Remodeling Complexes Mediates Prostate Oncogenesis. Mol. Cell 2018, 71, 554–566.e7. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Alexe, G.; Dharia, N.V.; Ross, L.; Iniguez, A.B.; Conway, A.S.; Wang, E.J.; Veschi, V.; Lam, N.; Qi, J.; et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Investig. 2018, 128, 446–462. [Google Scholar] [CrossRef]
- Cancer Dependency Map (DepMap) Portal. Available online: https://depmap.org/portal/depmap (accessed on 7 July 2021).
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [Green Version]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar] [PubMed]
- Hohmann, A.F.; Martin, L.J.; Minder, J.L.; Roe, J.S.; Shi, J.; Steurer, S.; Bader, G.; McConnell, D.; Pearson, M.; Gerstberger, T.; et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016, 12, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Helming, K.C.; Wang, X.; Kim, Y.; Vazquez, F.; Jagani, Z.; Hahn, W.C.; Roberts, C.W. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol. Cell Biol. 2014, 34, 1136–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017, 45, D408–D414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Mehta, V.; Trinkle-Mulcahy, L. Recent advances in large-scale protein interactome mapping. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.W.; Veschambre, P.; Erdjument-Bromage, H.; Tempst, P.; Conaway, J.W.; Conaway, R.C.; Kornberg, R.D. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 8538–8543. [Google Scholar] [CrossRef] [Green Version]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, Y.; Suzuki, H.; Ohtsuka, M.; Kikuchi, N.; Kimura, M.; Inoko, H. Isolation and characterization of three genes paralogous to mouse Ring3. Nucleic Acids Res. Suppl. 2001, 247–248. [Google Scholar] [CrossRef] [Green Version]
- Shang, E.; Salazar, G.; Crowley, T.E.; Wang, X.; Lopez, R.A.; Wang, X.; Wolgemuth, D.J. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr. Patterns 2004, 4, 513–519. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Wang, L.; Guarnieri, P.; Wolgemuth, D.J. The testis-specific double bromodomain-containing protein BRDT forms a complex with multiple spliceosome components and is required for mRNA splicing and 3′-UTR truncation in round spermatids. Nucleic Acids Res. 2012, 40, 7162–7175. [Google Scholar] [CrossRef] [Green Version]
- Cochran, A.G.; Conery, A.R.; Sims, R.J., III. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Letson, C.; Padron, E. Non-canonical transcriptional consequences of BET inhibition in cancer. Pharm. Res. 2019, 150, 104508. [Google Scholar] [CrossRef]
- Lochrin, S.E.; Price, D.K.; Figg, W.D. BET bromodomain inhibitors—A novel epigenetic approach in castration-resistant prostate cancer. Cancer Biol. Ther. 2014, 15, 1583–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.M.; Goulet, D.R.; Johnson, G.L. Targeting the Breast Cancer Kinome. J. Cell. Physiol. 2017, 232, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G. Targeting BET bromodomain proteins in solid tumors. Oncotarget 2016, 7, 53997–54009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedin, S.M.; Boddy, C.S.; Munshi, H.G. BET inhibitors in the treatment of hematologic malignancies: Current insights and future prospects. Onco Targets Ther. 2016, 9, 5943–5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Ku, A.F.; Anglin, J.L.; Sharma, R.; Ucisik, M.N.; Faver, J.C.; Li, F.; Nyshadham, P.; Simmons, N.; Sharma, K.L.; et al. Discovery and characterization of bromodomain 2-specific inhibitors of BRDT. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Kumar, R.; Li, D.Q.; Muller, S.; Knapp, S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016, 35, 4423–4436. [Google Scholar] [CrossRef]
- Pan, J.; Meyers, R.M.; Michel, B.C.; Mashtalir, N.; Sizemore, A.E.; Wells, J.N.; Cassel, S.H.; Vazquez, F.; Weir, B.A.; Hahn, W.C.; et al. Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell Syst. 2018, 6, 555–568.e7. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, M.D.; Aslanian, A.; Dong, M.Q.; Yates, J.R., III; Emerson, B.M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 2008, 283, 32254–32263. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.T.; Chen, S.M.; Pan, L.B.; Yao, J.; Ma, H.T. Loss of function of SWI/SNF chromatin remodeling genes leads to genome instability of human lung cancer. Oncol. Rep. 2015, 33, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Xu, C.; Huang, W.; Zhang, J.; Carlson, J.E.; Tu, X.; Wu, J.; Shi, Y. Solution structure of human Brg1 bromodomain and its specific binding to acetylated histone tails. Biochemistry 2007, 46, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Popowicz, G.M.; Krajewski, M.; Holak, T.A. Structural ramification for acetyl-lysine recognition by the bromodomain of human BRG1 protein, a central ATPase of the SWI/SNF remodeling complex. Chembiochem. A Eur. J. Chem. Biol. 2007, 8, 1308–1316. [Google Scholar] [CrossRef]
- Morrison, E.A.; Sanchez, J.C.; Ronan, J.L.; Farrell, D.P.; Varzavand, K.; Johnson, J.K.; Gu, B.X.; Crabtree, G.R.; Musselman, C.A. DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nat. Commun. 2017, 8, 16080. [Google Scholar] [CrossRef]
- Aravind, L.; Landsman, D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998, 26, 4413–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, J.C.; Zhang, L.; Evoli, S.; Schnicker, N.J.; Nunez-Hernandez, M.; Yu, L.; Wereszczynski, J.; Pufall, M.A.; Musselman, C.A. The molecular basis of selective DNA binding by the BRG1 AT-hook and bromodomain. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194566. [Google Scholar] [CrossRef] [PubMed]
- Weaver, T.M.; Morrison, E.A.; Musselman, C.A. Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains. Molecules 2018, 23, 2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorov, O.; Castex, J.; Tallant, C.; Owen, D.R.; Martin, S.; Aldeghi, M.; Monteiro, O.; Filippakopoulos, P.; Picaud, S.; Trzupek, J.D.; et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 2015, 1, e1500723. [Google Scholar] [CrossRef] [Green Version]
- Philpott, M.; Rogers, C.M.; Yapp, C.; Wells, C.; Lambert, J.P.; Strain-Damerell, C.; Burgess-Brown, N.A.; Gingras, A.C.; Knapp, S.; Muller, S. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenet. Chromatin 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherell, C.L.; Tallant, C.; Monteiro, O.P.; Yapp, C.; Fuchs, J.E.; Fedorov, O.; Siejka, P.; Muller, S.; Knapp, S.; Brenton, J.D.; et al. Identification and Development of 2,3-Dihydropyrrolo[1,2-a]quinazolin-5(1H)-one Inhibitors Targeting Bromodomains within the Switch/Sucrose Nonfermenting Complex. J. Med. Chem. 2016, 59, 5095–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourachot, B.; Yaniv, M.; Muchardt, C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J. 2003, 22, 6505–6515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchardt, C.; Reyes, J.C.; Bourachot, B.; Leguoy, E.; Yaniv, M. The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J. 1996, 15, 3394–3402. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. SMARCA2/4 PROTAC for Targeted Protein Degradation and Cancer Therapy. ACS Med. Chem. Lett. 2020, 11, 1797–1798. [Google Scholar] [CrossRef]
- Marquez-Vilendrer, S.B.; Rai, S.K.; Gramling, S.J.; Lu, L.; Reisman, D.N. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Oncoscience 2016, 3, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Martinez, J.A.; Reyes, J.C. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci. Rep. 2018, 8, 2043. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Hu, J.C.; Lu, W.C.; Han, J.; Ding, H.; Jiang, H.; Zhang, Y.Y.; Yue, L.Y.; Chen, S.J.; Jiang, H.L.; et al. Identification of small molecule inhibitors targeting the SMARCA2 bromodomain from a high-throughput screening assay. Acta Pharm. Sin. 2018, 39, 1544–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, J.; Liu, W.; Li, G.; Yang, Z.; Qin, P.; Xu, L. The ISWI remodeler in plants: Protein complexes, biochemical functions, and developmental roles. Chromosoma 2017, 126, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama, T.; Wu, C. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 1995, 83, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Tsukiyama, T.; Daniel, C.; Tamkun, J.; Wu, C. ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell 1995, 83, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Barak, O.; Lazzaro, M.A.; Lane, W.S.; Speicher, D.W.; Picketts, D.J.; Shiekhattar, R. Isolation of human NURF: A regulator of Engrailed gene expression. EMBO J. 2003, 22, 6089–6100. [Google Scholar] [CrossRef] [Green Version]
- Alkhatib, S.G.; Landry, J.W. The nucleosome remodeling factor. FEBS Lett. 2011, 585, 3197–3207. [Google Scholar] [CrossRef] [Green Version]
- Olson, N.M.; Kroc, S.; Johnson, J.A.; Zahid, H.; Ycas, P.D.; Chan, A.; Kimbrough, J.R.; Kalra, P.; Schonbrunn, E.; Pomerantz, W.C.K. NMR Analyses of Acetylated H2A.Z Isoforms Identify Differential Binding Interactions with the Bromodomain of the NURF Nucleosome Remodeling Complex. Biochemistry 2020, 59, 1871–1880. [Google Scholar] [CrossRef]
- Ycas, P.D.; Zahid, H.; Chan, A.; Olson, N.M.; Johnson, J.A.; Talluri, S.K.; Schonbrunn, E.; Pomerantz, W.C.K. New inhibitors for the BPTF bromodomain enabled by structural biology and biophysical assay development. Org. Biomol. Chem. 2020, 18, 5174–5182. [Google Scholar] [CrossRef]
- Li, X.; Ding, D.; Yao, J.; Zhou, B.; Shen, T.; Qi, Y.; Ni, T.; Wei, G. Chromatin remodeling factor BAZ1A regulates cellular senescence in both cancer and normal cells. Life Sci. 2019, 229, 225–232. [Google Scholar] [CrossRef]
- Gu, L.; Frommel, S.C.; Oakes, C.C.; Simon, R.; Grupp, K.; Gerig, C.Y.; Bar, D.; Robinson, M.D.; Baer, C.; Weiss, M.; et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat. Genet. 2015, 47, 22–30. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, Y.; Zhong, L. Discovery of BAZ1A bromodomain inhibitors with the aid of virtual screening and activity evaluation. Bioorg. Med. Chem. Lett 2021, 33, 127745. [Google Scholar] [CrossRef]
- Bevill, S.M.; Olivares-Quintero, J.F.; Sciaky, N.; Golitz, B.T.; Singh, D.; Beltran, A.S.; Rashid, N.U.; Stuhlmiller, T.J.; Hale, A.; Moorman, N.J.; et al. GSK2801, a BAZ2/BRD9 Bromodomain Inhibitor, Synergizes with BET Inhibitors to Induce Apoptosis in Triple-Negative Breast Cancer. Mol. Cancer Res. 2019, 17, 1503–1518. [Google Scholar] [CrossRef] [Green Version]
- Drouin, L.; McGrath, S.; Vidler, L.R.; Chaikuad, A.; Monteiro, O.; Tallant, C.; Philpott, M.; Rogers, C.; Fedorov, O.; Liu, M.; et al. Structure enabled design of BAZ2-ICR, a chemical probe targeting the bromodomains of BAZ2A and BAZ2B. J. Med. Chem. 2015, 58, 2553–2559. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Shen, J. BRD7: A novel tumor suppressor gene in different cancers. Am. J. Transl. Res. 2016, 8, 742–748. [Google Scholar]
- Drost, J.; Mantovani, F.; Tocco, F.; Elkon, R.; Comel, A.; Holstege, H.; Kerkhoven, R.; Jonkers, J.; Voorhoeve, P.M.; Agami, R.; et al. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat. Cell Biol. 2010, 12, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Harte, M.T.; O’Brien, G.J.; Ryan, N.M.; Gorski, J.J.; Savage, K.I.; Crawford, N.T.; Mullan, P.B.; Harkin, D.P. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010, 70, 2538–2547. [Google Scholar] [CrossRef] [Green Version]
- Carlson, S.; Glass, K.C. The MOZ histone acetyltransferase in epigenetic signaling and disease. J. Cell. Physiol. 2014, 229, 1571–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syntichaki, P.; Topalidou, I.; Thireos, G. The Gcn5 bromodomain co-ordinates nucleosome remodelling. Nature 2000, 404, 414–417. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, Q.; Gerona-Navarro, G.; Moshkina, N.; Zhou, M.M. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure 2008, 16, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ghazawi, F.M.; Li, Q. Interplay of bromodomain and histone acetylation in the regulation of p300-dependent genes. Epigenet. Off. J. DNA Methylation Soc. 2010, 5, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucconi, B.E.; Makofske, J.L.; Meyers, D.J.; Hwang, Y.; Wu, M.; Kuroda, M.I.; Cole, P.A. Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry 2019, 58, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Rebel, V.I.; Bronson, R.T.; Ch’ng, L.E.; Sieff, C.A.; Livingston, D.M.; Yao, T.P. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000, 14, 272–277. [Google Scholar] [PubMed]
- He, Z.X.; Wei, B.F.; Zhang, X.; Gong, Y.P.; Ma, L.Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 2021, 209, 112861. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbert, M.A.; Sikorski, T.; Carten, J.; Snowflack, D.; Hodawadekar, S.; Marmorstein, R. The human monocytic leukemia zinc finger histone acetyltransferase domain contains DNA-binding activity implicated in chromatin targeting. J. Biol. Chem. 2007, 282, 36603–36613. [Google Scholar] [CrossRef] [Green Version]
- Champagne, N.; Pelletier, N.; Yang, X.J. The monocytic leukemia zinc finger protein MOZ is a histone acetyltransferase. Oncogene 2001, 20, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Carapeti, M.; Aguiar, R.C.; Goldman, J.M.; Cross, N.C. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 1998, 91, 3127–3133. [Google Scholar] [CrossRef]
- Kitabayashi, I.; Aikawa, Y.; Nguyen, L.A.; Yokoyama, A.; Ohki, M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 2001, 20, 7184–7196. [Google Scholar] [CrossRef] [Green Version]
- Kitabayashi, I.; Aikawa, Y.; Yokoyama, A.; Hosoda, F.; Nagai, M.; Kakazu, N.; Abe, T.; Ohki, M. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 2001, 15, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, I.; Fioretos, T.; Isaksson, M.; Samuelsson, U.; Billstrom, R.; Strombeck, B.; Mitelman, F.; Johansson, B. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum. Mol. Genet. 2001, 10, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.; Pelletier, N.; Xiao, L.; Zhao, S.P.; Wang, K.; Degerny, C.; Tahmasebi, S.; Cayrou, C.; Doyon, Y.; Goh, S.L.; et al. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 2008, 28, 6828–6843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, W.S.; Poncet-Montange, G.; Liu, G.; Petrocchi, A.; Reyna, N.; Subramanian, G.; Theroff, J.; Yau, A.; Kost-Alimova, M.; Bardenhagen, J.P.; et al. Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J. Med. Chem. 2016, 59, 1440–1454. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhou, C.; Caflisch, A. Structure-based discovery of selective BRPF1 bromodomain inhibitors. Eur. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Meier, J.C.; Tallant, C.; Fedorov, O.; Witwicka, H.; Hwang, S.Y.; van Stiphout, R.G.; Lambert, J.P.; Rogers, C.; Yapp, C.; Gerstenberger, B.S.; et al. Selective targeting of bromodomains of the bromodomain-PHD fingers family impairs osteoclast differentiation. ACS Chem. Biol. 2017, 12, 2619–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, T.W.; Cook, J.G.; Asano, M.; Nevins, J.R. Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J. Biol. Chem. 2001, 276, 15397–15408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Andrews, N.P.; Chen, C.Z.; Fan, M.; Wang, J.; Shen, J.; Li, J.J.; Chen, H.W. Targeting bromodomain protein ANCCA/ATAD2 enhances the efficacy of DNAdamaging chemotherapy agents and radiation. Oncol. Rep. 2020, 43, 318–327. [Google Scholar] [CrossRef]
- Mishima, Y.; Miyagi, S.; Saraya, A.; Negishi, M.; Endoh, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Katsumoto, T.; Chiba, T.; et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 2011, 118, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Bouche, L.; Christ, C.D.; Siegel, S.; Fernandez-Montalvan, A.E.; Holton, S.J.; Fedorov, O.; Ter Laak, A.; Sugawara, T.; Stockigt, D.; Tallant, C.; et al. Benzoisoquinolinediones as Potent and Selective Inhibitors of BRPF2 and TAF1/TAF1L Bromodomains. J. Med. Chem. 2017, 60, 4002–4022. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Vlassis, A.; Roques, C.; Lalonde, M.E.; Gonzalez-Aguilera, C.; Lambert, J.P.; Lee, S.B.; Zhao, X.; Alabert, C.; Johansen, J.V.; et al. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016, 35, 176–192. [Google Scholar] [CrossRef]
- Cho, H.I.; Kim, M.S.; Lee, J.; Yoo, B.C.; Kim, K.H.; Choe, K.M.; Jang, Y.K. BRPF3-HUWE1-mediated regulation of MYST2 is required for differentiation and cell-cycle progression in embryonic stem cells. Cell Death Differ. 2020, 27, 3273–3288. [Google Scholar] [CrossRef]
- Fulop, V.; Bocskei, Z.; Polgar, L. Prolyl oligopeptidase: An unusual beta-propeller domain regulates proteolysis. Cell 1998, 94, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.K.; Chan, N.L.; Wang, A.H. The many blades of the beta-propeller proteins: Conserved but versatile. Trends Biochem. Sci. 2011, 36, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Stirnimann, C.U.; Petsalaki, E.; Russell, R.B.; Muller, C.W. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010, 35, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Hamel, K.M.; Maienschein-Cline, M.; Tanaka, A.; Teng, G.; Tuteja, J.H.; Bunker, J.J.; Bahroos, N.; Eppig, J.J.; Schatz, D.G.; et al. Histone reader BRWD1 targets and restricts recombination to the Igk locus. Nat. Immunol. 2015, 16, 1094–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.; Maienschein-Cline, M.; Maffucci, P.; Veselits, M.; Kennedy, D.E.; McLean, K.C.; Okoreeh, M.K.; Karki, S.; Cunningham-Rundles, C.; Clark, M.R. BRWD1 orchestrates epigenetic landscape of late B lymphopoiesis. Nat. Commun. 2018, 9, 3888. [Google Scholar] [CrossRef] [Green Version]
- Suh, E.J.; Kabir, M.H.; Kang, U.B.; Lee, J.W.; Yu, J.; Noh, D.Y.; Lee, C. Comparative profiling of plasma proteome from breast cancer patients reveals thrombospondin-1 and BRWD3 as serological biomarkers. Exp. Mol. Med. 2012, 44, 36–44. [Google Scholar] [CrossRef] [Green Version]
- de Semir, D.; Bezrookove, V.; Nosrati, M.; Dar, A.A.; Wu, C.; Shen, J.; Rieken, C.; Venkatasubramanian, M.; Miller, J.R., III; Desprez, P.Y.; et al. PHIP as a therapeutic target for driver-negative subtypes of melanoma, breast, and lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5766–E5775. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.; Jang, J.; Kang, Y.; Watanabe, H.; Uchihashi, T.; Kim, S.J.; Kato, K.; Lee, J.Y.; Song, J.J. Structural basis of nucleosome assembly by the Abo1 AAA+ ATPase histone chaperone. Nat. Commun. 2019, 10, 5764. [Google Scholar] [CrossRef]
- Poncet-Montange, G.; Zhan, Y.; Bardenhagen, J.P.; Petrocchi, A.; Leo, E.; Shi, X.; Lee, G.R.t.; Leonard, P.G.; Geck Do, M.K.; Cardozo, M.G.; et al. Observed bromodomain flexibility reveals histone peptide- and small molecule ligand-compatible forms of ATAD2. Biochem. J. 2015, 466, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morozumi, Y.; Boussouar, F.; Tan, M.; Chaikuad, A.; Jamshidikia, M.; Colak, G.; He, H.; Nie, L.; Petosa, C.; de Dieuleveult, M.; et al. Atad2 is a generalist facilitator of chromatin dynamics in embryonic stem cells. J. Mol. Cell Biol. 2016, 8, 349–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.X.; Guo, L.; Revenko, A.S.; Tepper, C.G.; Gemo, A.T.; Kung, H.J.; Chen, H.W. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009, 69, 3339–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.X.; Revenko, A.S.; Li, L.B.; Gemo, A.T.; Chen, H.W. ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification. Proc. Natl. Acad. Sci. USA 2007, 104, 18067–18072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussouar, F.; Jamshidikia, M.; Morozumi, Y.; Rousseaux, S.; Khochbin, S. Malignant genome reprogramming by ATAD2. Biochim. Biophys. Acta 2013, 1829, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Tangen, I.L.; Hoivik, E.A.; Halle, M.K.; Berg, A.; Werner, H.M.; Raeder, M.B.; Kusonmano, K.; Zou, J.X.; Oyan, A.M.; et al. ATAD2 overexpression links to enrichment of B-MYB-translational signatures and development of aggressive endometrial carcinoma. Oncotarget 2015, 6, 28440–28452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Zou, J.X.; Yang, P.; Wang, Y.; Borowsky, A.D.; Gao, A.C.; Chen, H.W. Developmental and androgenic regulation of chromatin regulators EZH2 and ANCCA/ATAD2 in the prostate Via MLL histone methylase complex. Prostate 2013, 73, 455–466. [Google Scholar] [CrossRef]
- Hong, S.; Bi, M.; Yan, Z.; Sun, D.; Ling, L.; Zhao, C. Silencing of ATPase family AAA domain-containing protein 2 inhibits migration and invasion of colorectal cancer cells. Neoplasma 2016, 63, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Gay, J.C.; Eckenroth, B.E.; Evans, C.M.; Langini, C.; Carlson, S.; Lloyd, J.T.; Caflisch, A.; Glass, K.C. Disulfide bridge formation influences ligand recognition by the ATAD2 bromodomain. Proteins 2019, 87, 157–167. [Google Scholar] [CrossRef]
- Demont, E.H.; Chung, C.W.; Furze, R.C.; Grandi, P.; Michon, A.M.; Wellaway, C.; Barrett, N.; Bridges, A.M.; Craggs, P.D.; Diallo, H.; et al. Fragment-Based Discovery of Low-Micromolar ATAD2 Bromodomain Inhibitors. J. Med. Chem. 2015, 58, 5649–5673. [Google Scholar] [CrossRef]
- Bamborough, P.; Chung, C.W.; Furze, R.C.; Grandi, P.; Michon, A.M.; Sheppard, R.J.; Barnett, H.; Diallo, H.; Dixon, D.P.; Douault, C.; et al. Structure-based optimization of naphthyridones into potent ATAD2 bromodomain inhibitors. J. Med. Chem. 2015, 58, 6151–6178. [Google Scholar] [CrossRef]
- Bamborough, P.; Chung, C.W.; Demont, E.H.; Furze, R.C.; Bannister, A.J.; Che, K.H.; Diallo, H.; Douault, C.; Grandi, P.; Kouzarides, T.; et al. A Chemical Probe for the ATAD2 Bromodomain. Angew. Chem. 2016, 55, 11382–11386. [Google Scholar] [CrossRef]
- Yao, D.; Zhang, J.; Wang, J.; Pan, D.; He, Z. Discovery of novel ATAD2 bromodomain inhibitors that trigger apoptosis and autophagy in breast cells by structure-based virtual screening. J. Enzym. Inhib. Med. Chem. 2020, 35, 713–725. [Google Scholar] [CrossRef]
- Fernandez-Montalvan, A.E.; Berger, M.; Kuropka, B.; Koo, S.J.; Badock, V.; Weiske, J.; Puetter, V.; Holton, S.J.; Stockigt, D.; Ter Laak, A.; et al. Isoform-Selective ATAD2 Chemical Probe with Novel Chemical Structure and Unusual Mode of Action. ACS Chem. Biol. 2017, 12, 2730–2736. [Google Scholar] [CrossRef]
- Ziemin-van der Poel, S.; McCabe, N.R.; Gill, H.J.; Espinosa, R., III; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; LeBeau, M.M.; Rowley, J.D.; et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [Green Version]
- Canaani, E.; Nakamura, T.; Rozovskaia, T.; Smith, S.T.; Mori, T.; Croce, C.M.; Mazo, A. ALL-1/MLL1, a homologue of Drosophila TRITHORAX, modifies chromatin and is directly involved in infant acute leukaemia. Br. J. Cancer 2004, 90, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Ernst, P.; Wang, J.; Huang, M.; Goodman, R.H.; Korsmeyer, S.J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell Biol. 2001, 21, 2249–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Hom, R.A.; Blakeslee, W.; Ikenouye, L.; Kutateladze, T.G. Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochim. Biophys. Acta 2014, 1843, 366–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobulo, O.M.; Borrow, J.; Tomek, R.; Reshmi, S.; Harden, A.; Schlegelberger, B.; Housman, D.; Doggett, N.A.; Rowley, J.D.; Zeleznik-Le, N.J. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc. Natl. Acad. Sci. USA 1997, 94, 8732–8737. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, Q.; Wong, S.H.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Herquel, B.; Ouararhni, K.; Davidson, I. The TIF1alpha-related TRIM cofactors couple chromatin modifications to transcriptional regulation, signaling and tumor suppression. Transcription 2011, 2, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, Z.; Guo, X.; Li, F.; Wei, Q.; Chen, X.; Gong, D.; Xu, Y.; Chen, W.; Liu, Y.; et al. TRIM66 reads unmodified H3R2K4 and H3K56ac to respond to DNA damage in embryonic stem cells. Nat. Commun. 2019, 10, 4273. [Google Scholar] [CrossRef] [Green Version]
- Stevens, R.V.; Esposito, D.; Rittinger, K. Characterisation of class VI TRIM RING domains: Linking RING activity to C-terminal domain identity. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Fiorentini, F.; Esposito, D.; Rittinger, K. Does it take two to tango? RING domain self-association and activity in TRIM E3 ubiquitin ligases. Biochem. Soc. Trans. 2020, 48, 2615–2624. [Google Scholar] [CrossRef]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sun, L.; Tang, Z.; Fu, L.; Xu, Y.; Li, Z.; Luo, W.; Qiu, X.; Wang, E. Overexpression of TRIM24 correlates with tumor progression in non-small cell lung cancer. PLoS ONE 2012, 7, e37657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambon, M.; Orsetti, B.; Berthe, M.L.; Bascoul-Mollevi, C.; Rodriguez, C.; Duong, V.; Gleizes, M.; Thenot, S.; Bibeau, F.; Theillet, C.; et al. Prognostic significance of TRIM24/TIF-1alpha gene expression in breast cancer. Am. J. Pathol. 2011, 178, 1461–1469. [Google Scholar] [CrossRef]
- Lin, L.; Zhao, W.; Sun, B.; Wang, X.; Liu, Q. Overexpression of TRIM24 is correlated with the progression of human cervical cancer. Am. J. Transl. Res. 2017, 9, 620–628. [Google Scholar]
- Liu, X.; Huang, Y.; Yang, D.; Li, X.; Liang, J.; Lin, L.; Zhang, M.; Zhong, K.; Liang, B.; Li, J. Overexpression of TRIM24 is associated with the onset and progress of human hepatocellular carcinoma. PLoS ONE 2014, 9, e85462. [Google Scholar] [CrossRef] [Green Version]
- Hoflmayer, D.; Fraune, C.; Hube-Magg, C.; Simon, R.; Schroeder, C.; Buscheck, F.; Moller, K.; Dum, D.; Weidemann, S.; Wittmer, C.; et al. Overexpression of the TRIM24 E3 Ubiquitin Ligase is Linked to Genetic Instability and Predicts Unfavorable Prognosis in Prostate Cancer. Appl. Immunohistochem. Mol. Morphol. 2021, 29, e29–e38. [Google Scholar] [CrossRef]
- Fang, Z.; Deng, J.; Zhang, L.; Xiang, X.; Yu, F.; Chen, J.; Feng, M.; Xiong, J. TRIM24 promotes the aggression of gastric cancer via the Wnt/beta-catenin signaling pathway. Oncol. Lett. 2017, 13, 1797–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Kost-Alimova, M.; Shi, X.; Leo, E.; Bardenhagen, J.P.; Shepard, H.E.; Appikonda, S.; Vangamudi, B.; Zhao, S.; Tieu, T.N.; et al. Development of novel cellular histone-binding and chromatin-displacement assays for bromodomain drug discovery. Epigenet. Chromatin. 2015, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J., III. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosmann, P.; Georgiev, O.; Le Douarin, B.; Bourquin, J.P.; Schaffner, W. Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 1996, 24, 4859–4867. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, A.L.; Ortiz, J.A.; You, J.; Oulad-Abdelghani, M.; Khechumian, R.; Gansmuller, A.; Chambon, P.; Losson, R. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 1999, 18, 6385–6395. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Saeki, Y.; Takahashi, H.; Ohtake, F.; Yoshida, Y.; Kasuga, Y.; Kondo, T.; Yaguchi, H.; Suzuki, M.; Ishida, H.; et al. A substrate-trapping strategy to find E3 ubiquitin ligase substrates identifies Parkin and TRIM28 targets. Commun. Biol. 2020, 3, 592. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.X.; Cai, J.J.; Chen, L.C.; Yue, Q.; Gong, Y.; Yao, Y.; Mao, Y. TRIM28 as an independent prognostic marker plays critical roles in glioma progression. J. Neurooncol. 2016, 126, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Z.; Lu, G. TRIM28 promotes cervical cancer growth through the mTOR signaling pathway. Oncol. Rep. 2018, 39, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Lee, G.D.; Nam, S.H.; Lee, T.H.; Kang, D.H.; Yun, J.K.; Lee, P.C. Sequential ubiquitination of p53 by TRIM28, RLIM, and MDM2 in lung tumorigenesis. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, J.; Li, Q.; Ma, H.; Xu, Z.; Gao, Y. KAP1 is overexpressed in hepatocellular carcinoma and its clinical significance. Int. J. Clin. Oncol. 2016, 21, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.P.; Dolle, P.; Mark, M.; Lerouge, T.; Wendling, O.; Chambon, P.; Losson, R. Molecular cloning, genomic structure, and expression analysis of the mouse transcriptional intermediary factor 1 gamma gene. Gene 2004, 334, 3–13. [Google Scholar] [CrossRef]
- Ransom, D.G.; Bahary, N.; Niss, K.; Traver, D.; Burns, C.; Trede, N.S.; Paffett-Lugassy, N.; Saganic, W.J.; Lim, C.A.; Hersey, C.; et al. The zebrafish moonshine gene encodes transcriptional intermediary factor 1gamma, an essential regulator of hematopoiesis. PLoS Biol. 2004, 2, E237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.F.; Yan, K.P.; Treilleux, I.; Gay, F.; Arfi, V.; Kaniewski, B.; Marie, J.C.; Lepinasse, F.; Martel, S.; Goddard-Leon, S.; et al. Inactivation of TIF1gamma cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 2009, 5, e1000575. [Google Scholar] [CrossRef]
- Hesling, C.; Fattet, L.; Teyre, G.; Jury, D.; Gonzalo, P.; Lopez, J.; Vanbelle, C.; Morel, A.P.; Gillet, G.; Mikaelian, I.; et al. Antagonistic regulation of EMT by TIF1gamma and Smad4 in mammary epithelial cells. Embo Rep. 2011, 12, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucagne, R.; Droin, N.; Paggetti, J.; Lagrange, B.; Largeot, A.; Hammann, A.; Bataille, A.; Martin, L.; Yan, K.P.; Fenaux, P.; et al. Transcription intermediary factor 1gamma is a tumor suppressor in mouse and human chronic myelomonocytic leukemia. J. Clin. Investig. 2011, 121, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Agricola, E.; Randall, R.A.; Gaarenstroom, T.; Dupont, S.; Hill, C.S. Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 2011, 43, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Xi, Q.; Wang, Z.; Zaromytidou, A.I.; Zhang, X.H.; Chow-Tsang, L.F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A poised chromatin platform for TGF-beta access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Allton, K.; Duncan, A.D.; Barton, M.C. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol. Cell Biol. 2014, 34, 2695–2709. [Google Scholar] [CrossRef] [Green Version]
- Hemmerich, P. KAP1: A new link between the DNA damage response and PML nuclear bodies. Cell Cycle 2011, 10, 576–577. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Oza, J.; Yao, M.; Sohail, H.; Ginjala, V.; Tomas-Loba, A.; Horejsi, Z.; Tan, A.R.; Boulton, S.J.; Ganesan, S. Tripartite Motif-containing 33 (TRIM33) protein functions in the poly(ADP-ribose) polymerase (PARP)-dependent DNA damage response through interaction with Amplified in Liver Cancer 1 (ALC1) protein. J. Biol. Chem. 2013, 288, 32357–32369. [Google Scholar] [CrossRef] [Green Version]
- McAvera, R.M.; Crawford, L.J. TIF1 Proteins in Genome Stability and Cancer. Cancers 2020, 12, 2094. [Google Scholar] [CrossRef]
- Gong, F.; Chiu, L.Y.; Cox, B.; Aymard, F.; Clouaire, T.; Leung, J.W.; Cammarata, M.; Perez, M.; Agarwal, P.; Brodbelt, J.S.; et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015, 29, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Chiu, L.Y.; Miller, K.M. Acetylation Reader Proteins: Linking Acetylation Signaling to Genome Maintenance and Cancer. PLoS Genet. 2016, 12, e1006272. [Google Scholar] [CrossRef] [PubMed]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Battu, A.; Ray, A.; Wani, A.A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res. 2011, 39, 7931–7945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Pu, M.; Zhang, Z.; Lou, Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 2009, 8, 1747–1753. [Google Scholar] [CrossRef] [Green Version]
- Fraschilla, I.; Jeffrey, K.L. The Speckled Protein (SP) Family: Immunity’s Chromatin Readers. Trends Immunol. 2020, 41, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Bouchier-Hayes, L.; Martin, S.J. CARD games in apoptosis and immunity. EMBO Rep. 2002, 3, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Collard, M.W.; Huggenvik, J.I.; Liu, Z.; Gibson, T.J.; Sattler, M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 2001, 8, 626–633. [Google Scholar] [CrossRef]
- Waterfield, M.; Khan, I.S.; Cortez, J.T.; Fan, U.; Metzger, T.; Greer, A.; Fasano, K.; Martinez-Llordella, M.; Pollack, J.L.; Erle, D.J.; et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat. Immunol. 2014, 15, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Sternsdorf, T.; Guldner, H.H.; Szostecki, C.; Grotzinger, T.; Will, H. Two nuclear dot-associated proteins, PML and Sp100, are often co-autoimmunogenic in patients with primary biliary cirrhosis. Scand. J. Immunol. 1995, 42, 257–268. [Google Scholar] [CrossRef]
- Collados Rodriguez, M. The Fate of Speckled Protein 100 (Sp100) During Herpesviruses Infection. Front. Cell Infect. Microbiol. 2020, 10, 607526. [Google Scholar] [CrossRef]
- ProteomicsDB. Sp100. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/18390 (accessed on 20 April 2021).
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: Catch me if you can! Nucleic Acids Res. 2020, 48, 11890–11912. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, D.; Xiong, X.; He, Z.; Li, H. Multifaceted Histone H3 Methylation and Phosphorylation Readout by the Plant Homeodomain Finger of Human Nuclear Antigen Sp100C. J. Biol. Chem. 2016, 291, 12786–12798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchelli, C.; Tamburri, S.; Filosa, G.; Ghitti, M.; Quilici, G.; Bachi, A.; Musco, G. Sp140 is a multi-SUMO-1 target and its PHD finger promotes SUMOylation of the adjacent Bromodomain. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.C.; Kuroda, S.; Price, J.A.; Pendleton, J.K.; Freedman, B.I.; Bowden, D.W. Identification and characterization of PRKCBP1, a candidate RACK-like protein. Mamm. Genome 2000, 11, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Hateboer, G.; Gennissen, A.; Ramos, Y.F.; Kerkhoven, R.M.; Sonntag-Buck, V.; Stunnenberg, H.G.; Bernards, R. BS69, a novel adenovirus E1A-associated protein that inhibits E1A transactivation. EMBO J. 1995, 14, 3159–3169. [Google Scholar] [CrossRef]
- Adhikary, S.; Sanyal, S.; Basu, M.; Sengupta, I.; Sen, S.; Srivastava, D.K.; Roy, S.; Das, C. Selective Recognition of H3.1K36 Dimethylation/H4K16 Acetylation Facilitates the Regulation of All-trans-retinoic Acid (ATRA)-responsive Genes by Putative Chromatin Reader ZMYND8. J. Biol. Chem. 2016, 291, 2664–2681. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Li, Y.; Xi, Y.; Jiang, S.; Stratton, S.; Peng, D.; Tanaka, K.; Ren, Y.; Xia, Z.; Wu, J.; et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 2014, 508, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Savitsky, P.; Krojer, T.; Fujisawa, T.; Lambert, J.P.; Picaud, S.; Wang, C.Y.; Shanle, E.K.; Krajewski, K.; Friedrichsen, H.; Kanapin, A.; et al. Multivalent Histone and DNA Engagement by a PHD/BRD/PWWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin. Cell Rep. 2016, 17, 2724–2737. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Xu, X.E.; Jiang, Y.Z.; Liu, Y.R.; Sun, W.; Guo, Y.J.; Ren, Y.X.; Zuo, W.J.; Hu, X.; Huang, S.L.; et al. The endogenous retrovirus-derived long noncoding RNA TROJAN promotes triple-negative breast cancer progression via ZMYND8 degradation. Sci. Adv. 2019, 5, eaat9820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, M.; Khan, M.W.; Chakrabarti, P.; Das, C. Chromatin reader ZMYND8 is a key target of all trans retinoic acid-mediated inhibition of cancer cell proliferation. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 450–459. [Google Scholar] [CrossRef]
- Mukherjee, S.; Adhikary, S.; Gadad, S.S.; Mondal, P.; Sen, S.; Choudhari, R.; Singh, V.; Adhikari, S.; Mandal, P.; Chaudhuri, S.; et al. Suppression of poised oncogenes by ZMYND8 promotes chemo-sensitization. Cell Death Dis. 2020, 11, 1073. [Google Scholar] [CrossRef]
- Tang, B.; Sun, R.; Wang, D.; Sheng, H.; Wei, T.; Wang, L.; Zhang, J.; Ho, T.H.; Yang, L.; Wei, Q.; et al. ZMYND8 preferentially binds phosphorylated EZH2 to promote a PRC2-dependent to -independent function switch in hypoxia-inducible factor-activated cancer. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Chen, Y.; Tsai, Y.H.; Tseng, S.H. Regulation of ZMYND8 to Treat Cancer. Molecules 2021, 26, 1083. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, C.; Zhao, X.; Wu, Q.; Fu, X.; Yu, B.; Shao, Y.; Guan, M.; Zhang, W.; Wan, J.; et al. Analysis of copy number variations of BS69 in multiple types of hematological malignancies. Ann. Hematol. 2010, 89, 959–964. [Google Scholar] [CrossRef] [PubMed]
- De Braekeleer, E.; Auffret, R.; Douet-Guilbert, N.; Basinko, A.; Le Bris, M.J.; Morel, F.; De Braekeleer, M. Recurrent translocation (10;17)(p15;q21) in acute poorly differentiated myeloid leukemia likely results in ZMYND11-MBTD1 fusion. Leuk. Lymphoma 2014, 55, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.D.; van den Heuvel-Eibrink, M.M.; Kollen, W.J.; Sonneveld, E.; Kaspers, G.J.; Beverloo, H.B.; Fornerod, M.; Pieters, R.; Zwaan, C.M. Recurrent translocation t(10;17)(p15;q21) in minimally differentiated acute myeloid leukemia results in ZMYND11/MBTD1 fusion. Genes Chromosomes Cancer 2016, 55, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Li, Y.; Li, H.; Shi, X. ZMYND11: An H3.3-specific reader of H3K36me3. Cell Cycle 2014, 13, 2153–2154. [Google Scholar] [CrossRef]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Shi, Y. Histone H3.3 and cancer: A potential reader connection. Proc. Natl. Acad. Sci. USA 2015, 112, 6814–6819. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovits, B.D.; Wolgemuth, D.J. The role of the double bromodomain-containing BET genes during mammalian spermatogenesis. Curr. Top. Dev. Biol. 2013, 102, 293–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Heidenreich, D.; Zhou, S.; Ackloo, S.; Kramer, A.; Nakka, K.; Lima-Fernandes, E.; Deblois, G.; Duan, S.; Vellanki, R.N.; et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019, 10, 1915. [Google Scholar] [CrossRef] [Green Version]
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Klingbeil, O.; Lesche, R.; Gelato, K.A.; Haendler, B.; Lejeune, P. Inhibition of BET bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis. 2016, 7, e2365. [Google Scholar] [CrossRef] [Green Version]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Vazquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613. [Google Scholar] [CrossRef] [Green Version]
- Odore, E.; Lokiec, F.; Cvitkovic, E.; Bekradda, M.; Herait, P.; Bourdel, F.; Kahatt, C.; Raffoux, E.; Stathis, A.; Thieblemont, C.; et al. Phase I Population Pharmacokinetic Assessment of the Oral Bromodomain Inhibitor OTX015 in Patients with Haematologic Malignancies. Clin. Pharm. 2016, 55, 397–405. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Brana, I.; Cassier, P.A.; Moreno, V.; de Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. Jnci Cancer Spectr. 2020, 4, pkz093. [Google Scholar] [CrossRef]
- Xie, F.; Huang, M.; Lin, X.; Liu, C.; Liu, Z.; Meng, F.; Wang, C.; Huang, Q. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci. Rep. 2018, 8, 8102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, J.; Lamotte, Y.; Donche, F.; Bouillot, A.; Mirguet, O.; Gellibert, F.; Nicodeme, E.; Krysa, G.; Kirilovsky, J.; Beinke, S.; et al. Identification of a novel series of BET family bromodomain inhibitors: Binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968–2972. [Google Scholar] [CrossRef]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 5338–5347. [Google Scholar] [CrossRef]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chaikuad, A.; Bamborough, P.; Bantscheff, M.; Bountra, C.; Chung, C.W.; Fedorov, O.; Grandi, P.; Jung, D.; Lesniak, R.; et al. Discovery and Characterization of GSK2801, a Selective Chemical Probe for the Bromodomains BAZ2A and BAZ2B. J. Med. Chem. 2016, 59, 1410–1424. [Google Scholar] [CrossRef]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef] [Green Version]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Cheng, X.; Huang, Z.; Long, D.; Jin, W. BET inhibitor bromosporine enhances 5-FU effect in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2020, 521, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, D.Y.; Hwang, Y.S.; Seo, H.R.; Lee, S.A.; Kwon, J. The Bromodomain Inhibitor PFI-3 Sensitizes Cancer Cells to DNA Damage by Targeting SWI/SNF. Mol. Cancer Res. 2021, 19, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Sachchidanand; Resnick-Silverman, L.; Yan, S.; Mutjaba, S.; Liu, W.J.; Zeng, L.; Manfredi, J.J.; Zhou, M.M. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Hay, D.A.; Fedorov, O.; Martin, S.; Singleton, D.C.; Tallant, C.; Wells, C.; Picaud, S.; Philpott, M.; Monteiro, O.P.; Rogers, C.M.; et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. [Google Scholar] [CrossRef]
- Hammitzsch, A.; Tallant, C.; Fedorov, O.; O’Mahony, A.; Brennan, P.E.; Hay, D.A.; Martinez, F.O.; Al-Mossawi, M.H.; de Wit, J.; Vecellio, M.; et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10768–10773. [Google Scholar] [CrossRef] [Green Version]
- Conery, A.R.; Centore, R.C.; Neiss, A.; Keller, P.J.; Joshi, S.; Spillane, K.L.; Sandy, P.; Hatton, C.; Pardo, E.; Zawadzke, L.; et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 2016, 5. [Google Scholar] [CrossRef]
- Hogg, S.J.; Newbold, A.; Vervoort, S.J.; Cluse, L.A.; Martin, B.P.; Gregory, G.P.; Lefebure, M.; Vidacs, E.; Tothill, R.W.; Bradner, J.E.; et al. BET Inhibition Induces Apoptosis in Aggressive B-Cell Lymphoma via Epigenetic Regulation of BCL-2 Family Members. Mol. Cancer Ther. 2016, 15, 2030–2041. [Google Scholar] [CrossRef] [Green Version]
- Cummin, T.E.C.; Cox, K.L.; Murray, T.D.; Turaj, A.H.; Dunning, L.; English, V.L.; Fell, R.; Packham, G.; Ma, Y.; Powell, B.; et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv. 2020, 4, 3316–3328. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, D.; Li, J.; Wang, W.; Zhong, W.; Fan, W.; Yu, M.; Cheng, H. VDAC1 is regulated by BRD4 and contributes to JQ1 resistance in breast cancer. Oncol. Lett. 2019, 18, 2340–2347. [Google Scholar] [CrossRef]
- Shu, S.; Wu, H.J.; Ge, J.Y.; Zeid, R.; Harris, I.S.; Jovanovic, B.; Murphy, K.; Wang, B.; Qiu, X.; Endress, J.E.; et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 2020. [Google Scholar] [CrossRef]
- Esteve-Arenys, A.; Valero, J.G.; Chamorro-Jorganes, A.; Gonzalez, D.; Rodriguez, V.; Dlouhy, I.; Salaverria, I.; Campo, E.; Colomer, D.; Martinez, A.; et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 2018, 37, 1830–1844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.C.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 2019, 35, 752–766.e9. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.; et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef] [Green Version]
- Saygin, C.; Carraway, H.E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Anwer, F.; Gee, K.M.; Iftikhar, A.; Baig, M.; Russ, A.D.; Saeed, S.; Zar, M.A.; Razzaq, F.; Carew, J.; Nawrocki, S.; et al. Future of Personalized Therapy Targeting Aberrant Signaling Pathways in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing effective combination therapy for pancreatic cancer: An overview. Pharm. Res. 2020, 155, 104740. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulou, A.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. Clinical perspectives of BET inhibition in ovarian cancer. Cell Oncol. 2021, 44, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulou, A.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. The emerging role of BET inhibitors in breast cancer. Breast 2020, 53, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.S.; Dastjerdi, S.; Wuhr, M.; Dhe-Paganon, S.; Armstrong, S.A.; et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. 2017, 56, 5738–5743. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, Y.; Hong, Y.; Chu, X.; Zhang, Z.; Tao, Y.; Fan, Z.; Bai, Z.; Li, X.; Chen, Y.; et al. BRD4 PROTAC degrader ARV-825 inhibits T-cell acute lymphoblastic leukemia by targeting ‘Undruggable’ Myc-pathway genes. Cancer Cell Int. 2021, 21, 230. [Google Scholar] [CrossRef]
- Chew, H.K. Adjuvant therapy for breast cancer: Who should get what? West. J. Med. 2001, 174, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, H.L.; Nagari, A.; Malladi, V.S.; Li, W.; Xi, Y.; Richardson, D.; Allton, K.L.; Tanaka, K.; Li, J.; Murakami, S.; et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018, 28, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.; Bainer, R.; Haverty, P.M.; Benedetti, K.L.; Gascoigne, K.E. Super-enhancer acquisition drives oncogene expression in triple negative breast cancer. PLoS ONE 2020, 15, e0235343. [Google Scholar] [CrossRef] [PubMed]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]

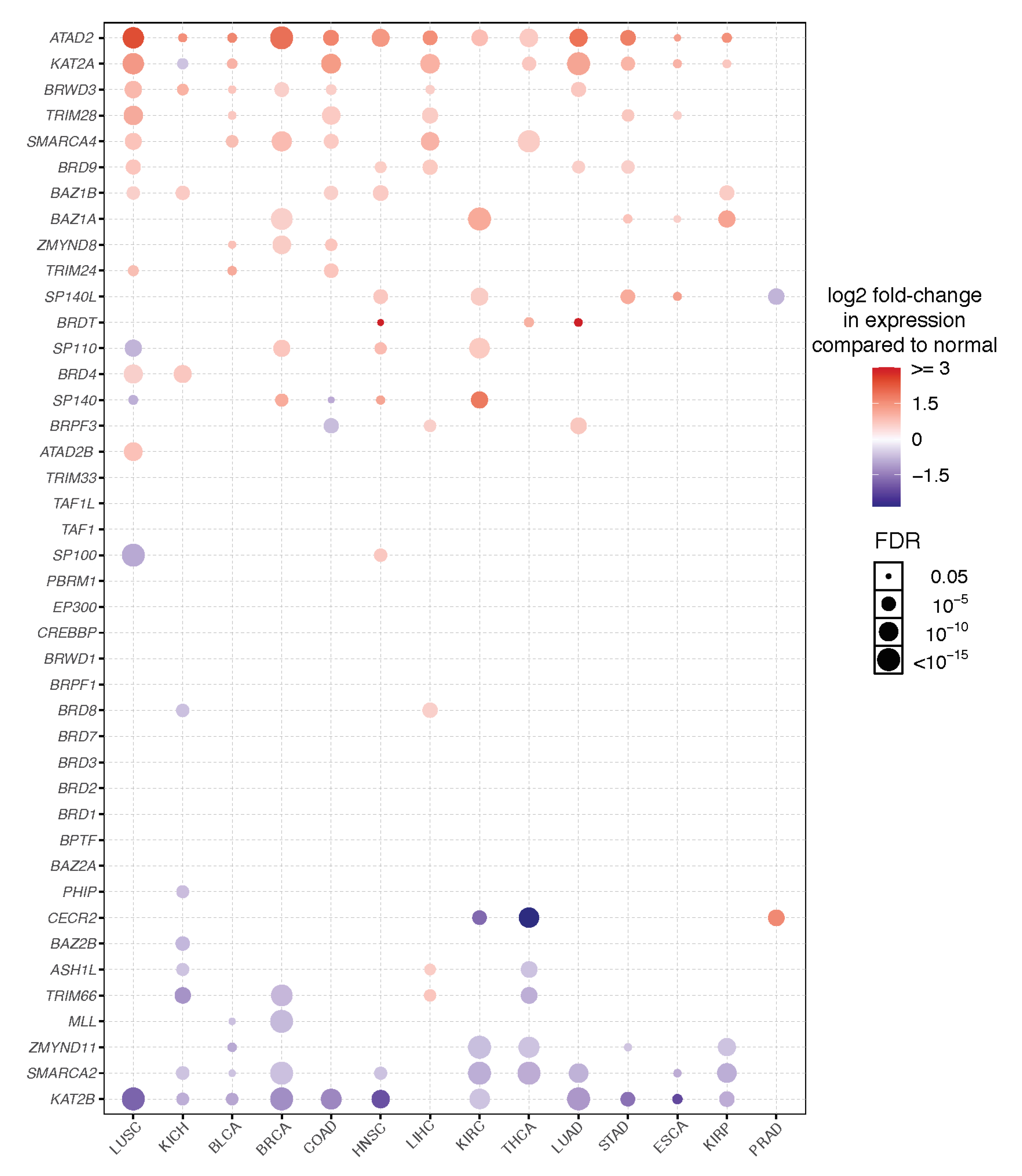



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRD Inhibitor (Molecule Images Created with JSME [279]) | Target | Cancer Type/Results | Clinical Trial ID/Reference | |
|---|---|---|---|---|
 | JQ1 | BET’s | The first generation of BET inhibitors which has proven to be a valuable tool for understanding BET BRDs in numerous cancers, but demonstrated toxicities in the clinic. As a result derivatives of JQ1 have had greater clinical success. | [27,280,281] |
 | OTX015* (Birabresib) | BET’s | Identifiers and resulting publications for ongoing and completed clinical trials for OTX015 as an exclusive therapy or in combination treatment. This JQ1 derivative used in combination with PROTACs has shown promise in cell models of prostate cancer, lymphoma, and leukemia. | NCT02698176, NCT02259114, NCT01713582, NCT02698189, NCT02296476 [282,283,284,285,286,287,288] |
 | I-BET762 GSK525762A, (Molibresib) | BET’s | Phase 1 clinical trial of this orally available compound initially showed that daily dosing with molibresib was well tolerated and showed efficacy for patients with nuclear protein in testis (NUT) carcinoma. | [28,288,289,290] |
 | I-BET151 GSK1210151A | BET’s | This BET inhibitor demonstrates strong anti-proliferative effects, and xenograft models indicate repression of proliferation in myeloma cells. However, this drug has not made progressed to clinical trials. | [291] |
 | ABBV-744 | Pan-BET (Selective for the 2nd bromodo-main) | Selective for the 2nd bromodomain of the BET-bromodomain proteins, and has demonstrated anti-proliferative effects for numerous acute myeloid leukemia and prostate cancer cell lines. | NCT04454658 [110,111] |
| Not available | ZEN-3694 | BET’s | A current phase 2 clinical trial using ZEN-3694 in combination with the enzalutamide is recruiting for castration resistant prostate cancer (CRPC), where the compound has shown efficacy in a phase 1 clinical trial. | NCT04471974 [292] |
 | ACBI1 | SMARCA2/4 | This PROTAC degrader resulted in reduced protein levels and apoptosis of acute myeloid leukemia (AML) cells. | [293] |
 | GSK2801 | BAZ2A/B | The selective acetyl-lysine competitive inhibitor induces apoptosis in triple negative breast cancer (TNBC) cells in combination with BET inhibitors. | [144,294] |
 | CCS1477 | CBP/p300 | Current Phase 1 & 2 clinical trials are recruiting patients for treatment of hematological malignancies and advanced prostate cancer. | NCT03568656, NCT04068597 |
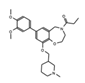 | I-CBP112 | CBP/p300 | Combination therapy with the p300/CBP active site inhibitor (A-485) resulted in reduced p300 chromatin enrichment, and decreased expression of androgen-dependent and pro-oncogenic genes in leukemia and prostate cancer. | [154,295] |
 | IACS-9571 | BRPF1/TRIM24 | This selective inhibitor has provided insights into cellular functions, and may be useful as a potential therapeutic for acute myeloid leukemia (AML) and breast cancer (BCa). | [165] |
 | IACS-9571 | TRIM24 | When developed into a bifunctional degrader linked to the VHL E3 ligase, TRIM24 protein degradation resulted in a greater negative impact on proliferation in leukemia cell lines. | [296] |
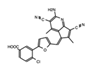 | AM879 | ATAD2 | Treatment with AM879, prevented cell proliferation, and induced apoptosis in triple negative breast cancer (TNBC) cells. | [195] |
 | Bromospo-rine | Multi-BRD (BET) | Shows promise as a therapeutic for colorectal cancer (CRC) when administered in combination with 5-Fluorouracil (5-FU). | [12,297] |
 | I-BRD9 | BRD9 | The selective inhibitor identified cancer associated and immune response genes as possible targets of BRD9 regulation in leukemia cells. | [298] |
 BI-7273 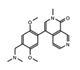 BI-9564 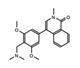 BI-7271  BI-7189 | BI-7273/BI-9564 BI-7271/BI-7273/BI-7189 | BRD9 | These small molecule inhibitors display anti-tumor activity in xenograft models of AML. | [299] |
 | PFI-3 | SMARCA2/4 and PB1(5) | Treatment with PFI-3 has been shown to sensitize cancer cells to chemotherapeutic agents. | http://www.thesgc.org/chemical-probes/PFI-3, accessed on 7 July 2021 [300] |
 MS2126  MS7972 | MS2126/MS7972 | CBP/p300 | Cell based assays in osteosarcoma cells, demonstrated that these molecules can modulate the p53 response to DNA damage. | [301] |
 | SGC-CBP30 | CBP/p300 | Inhibition of CBP is suggested to be a potential method for targeting transcriptional dependencies in multiple myeloma. | [302,303,304] |
 OF-1  PFI-4 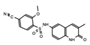 NI-57 | OF-1, PFI-4, NI-57 | pan-BRPF | While these inhibitors have not be linked to anti-proliferative or anti-cancer therapies, they have been suggested as potential therapeutics in bone malignancies. | [167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyson, S.P.; Gao, C.; Quinn, K.; Boyd, J.; Paculova, H.; Frietze, S.; Glass, K.C. Functional Roles of Bromodomain Proteins in Cancer. Cancers 2021, 13, 3606. https://doi.org/10.3390/cancers13143606
Boyson SP, Gao C, Quinn K, Boyd J, Paculova H, Frietze S, Glass KC. Functional Roles of Bromodomain Proteins in Cancer. Cancers. 2021; 13(14):3606. https://doi.org/10.3390/cancers13143606
Chicago/Turabian StyleBoyson, Samuel P., Cong Gao, Kathleen Quinn, Joseph Boyd, Hana Paculova, Seth Frietze, and Karen C. Glass. 2021. "Functional Roles of Bromodomain Proteins in Cancer" Cancers 13, no. 14: 3606. https://doi.org/10.3390/cancers13143606
APA StyleBoyson, S. P., Gao, C., Quinn, K., Boyd, J., Paculova, H., Frietze, S., & Glass, K. C. (2021). Functional Roles of Bromodomain Proteins in Cancer. Cancers, 13(14), 3606. https://doi.org/10.3390/cancers13143606