
Circadian Clock and Liver Cancer


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
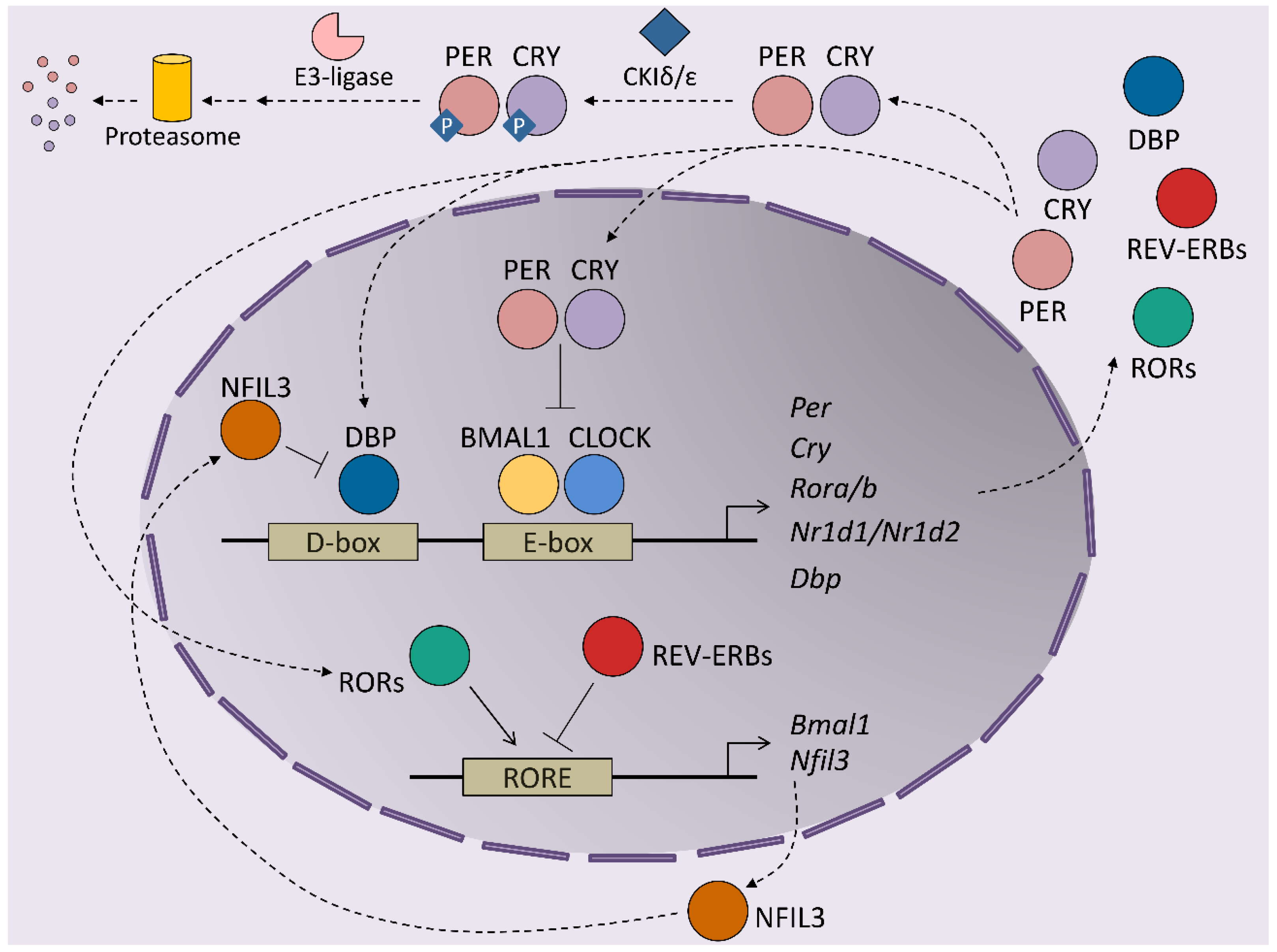
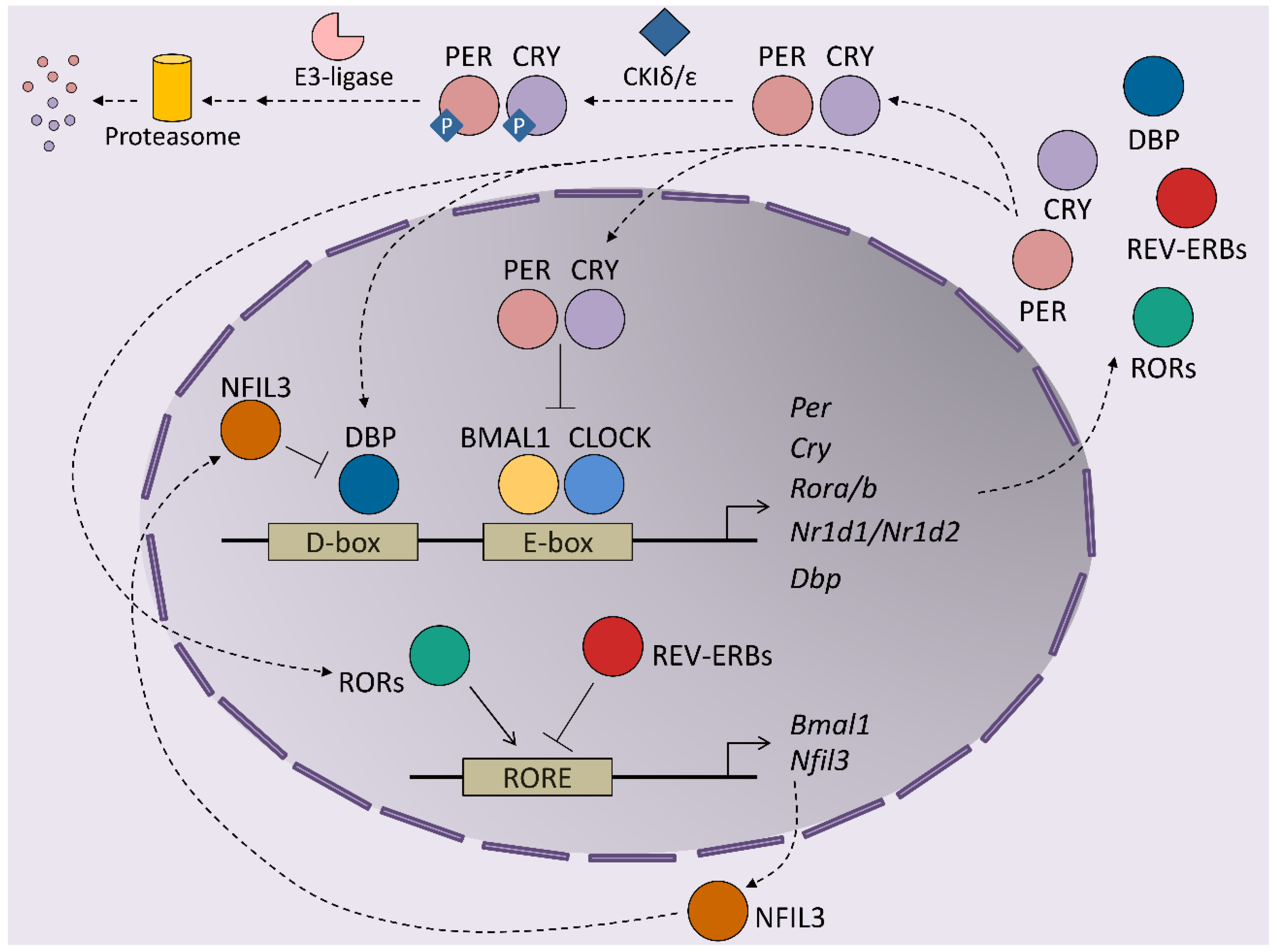
1. Introduction to Circadian Rhythms
2. Modulators of Hepatic Circadian Rhythms
3. Circadian Rhythms and Hepatic Metabolism
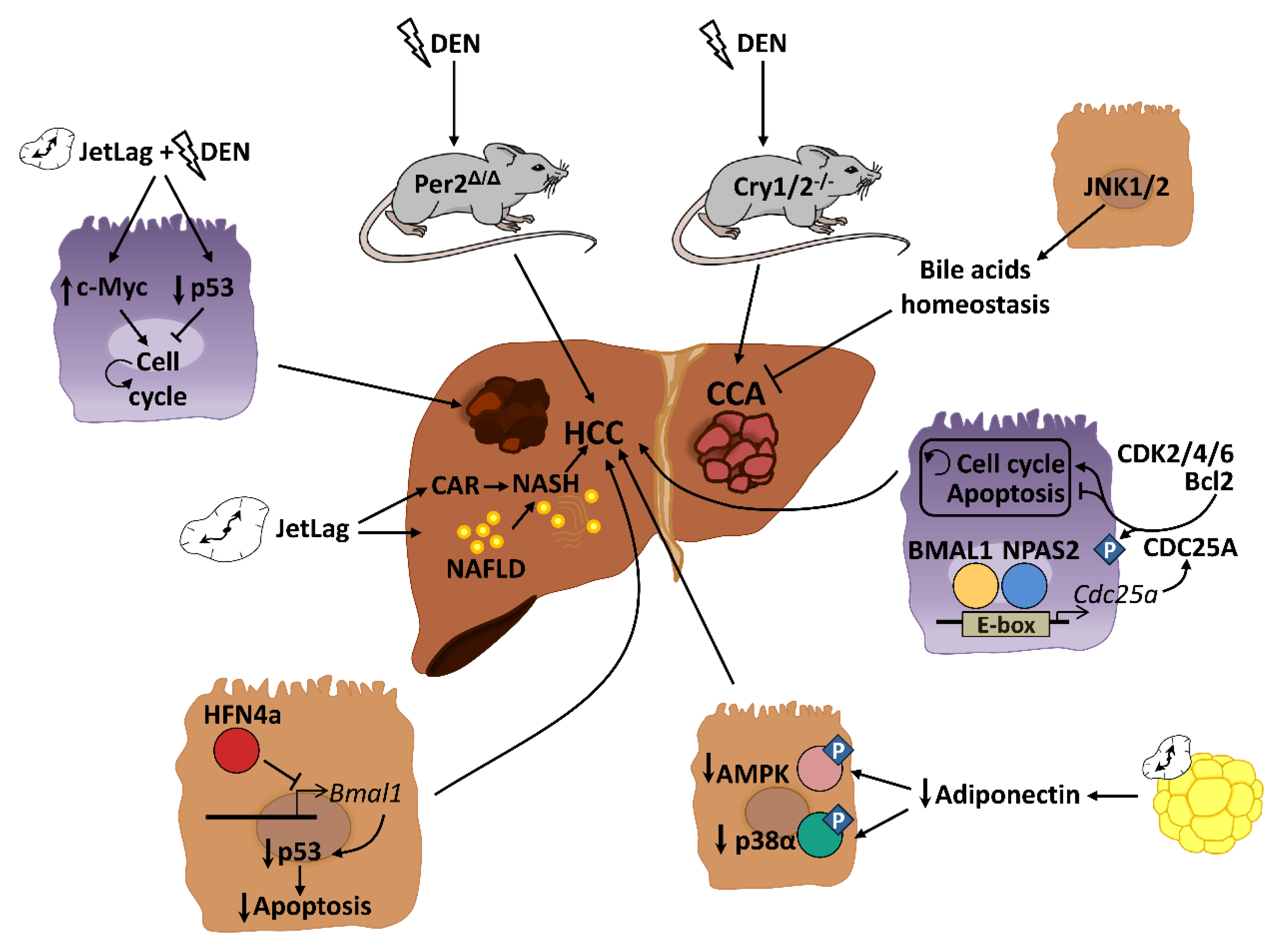
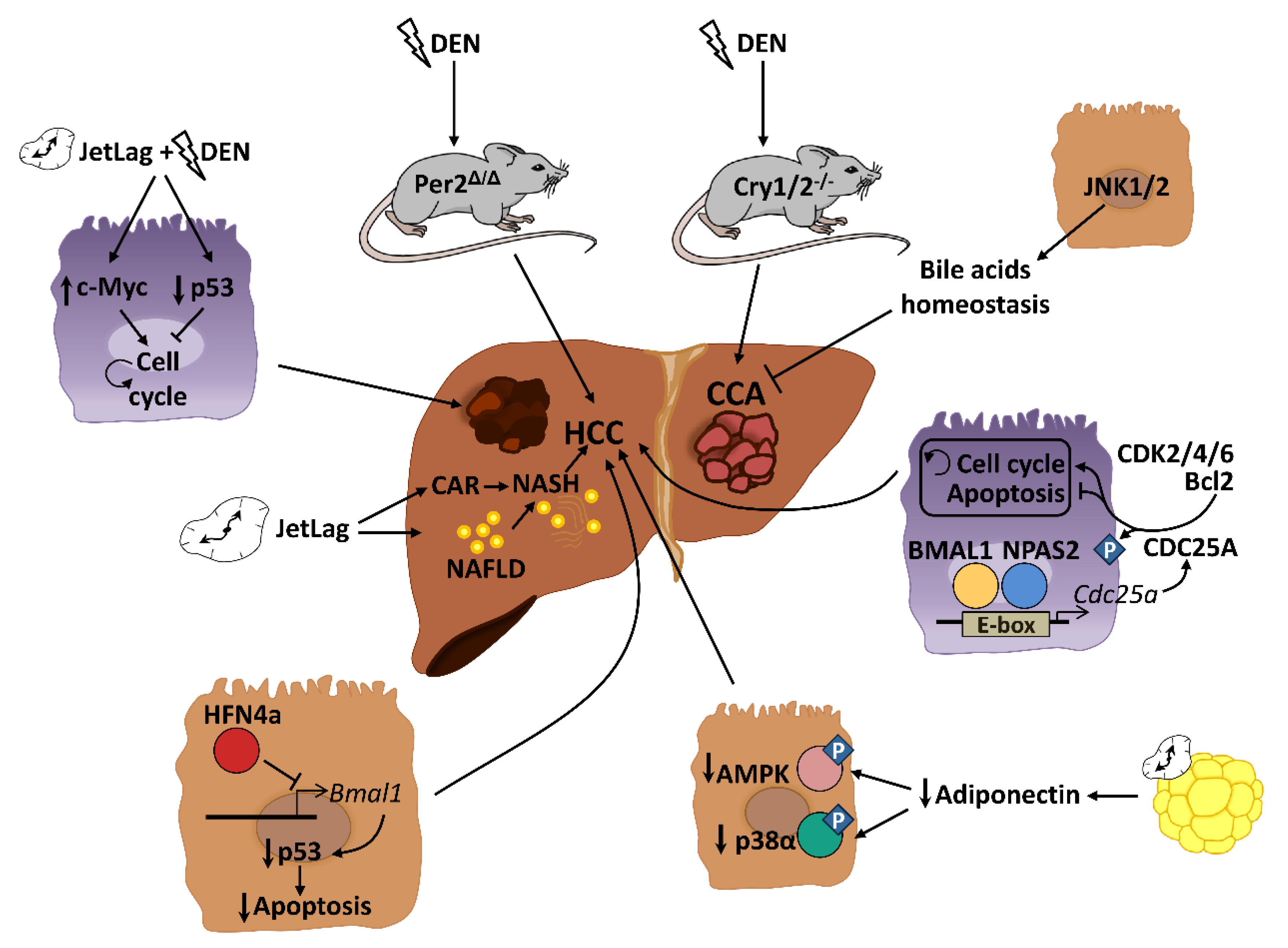
4. Role of Circadian Rhythms in Liver Cancer
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dibner, C.; Schibler, U.; Albrecht, U. The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Brewer, J.M.; Lehman, M.N.; Bittman, E.L. Suprachiasmatic Regulation of Circadian Rhythms of Gene Expression in Hamster Peripheral Organs: Effects of Transplanting the Pacemaker. J. Neurosci. 2006, 26, 6406–6412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujović, N.; Davidson, A.J.; Menaker, M. Sympathetic input modulates, but does not determine, phase of peripheral circadian oscillators. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R355–R360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsalobre, A.; Brown, S.A.; Marcacci, L.; Tronche, F.; Kellendonk, C.; Reichardt, H.M.; Schütz, G.; Schibler, U. Resetting of Circadian Time in Peripheral Tissues by Glucocorticoid Signaling. Science 2000, 289, 2344–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiola, F.; Le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000, 14, 2950–2961. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.A.; Zumbrunn, G.; Fleury-Olela, F.; Preitner, N.; Schibler, U. Rhythms of Mammalian Body Temperature Can Sustain Peripheral Circadian Clocks. Curr. Biol. 2002, 12, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Pezuk, P.; Mohawk, J.A.; Yoshikawa, T.; Sellix, M.T.; Menaker, M. Circadian Organization Is Governed by Extra-SCN Pacemakers. J. Biol. Rhythm. 2010, 25, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Sangoram, A.M.; Saez, L.; Antoch, M.P.; Gekakis, N.; Staknis, D.; Whiteley, A.; Fruechte, E.M.; Vitaterna, M.H.; Shimomura, K.; King, D.P.; et al. Mammalian Circadian Autoregulatory Loop: A Timeless Ortholog and mPer1 Interact and Negatively Regulate CLOCK-BMAL1-Induced Transcription. Neuron 1998, 21, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Kume, K.; Zylka, M.J.; Sriram, S.; Shearman, L.P.; Weaver, D.; Jin, X.; Maywood, E.S.; Hastings, M.H.; Reppert, S.M. mCRY1 and mCRY2 Are Essential Components of the Negative Limb of the Circadian Clock Feedback Loop. Cell 1999, 98, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Shearman, L.P.; Sriram, S.; Weaver, D.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; Van Der Horst, G.T.; Hastings, M.H.; et al. Interacting Molecular Loops in the Mammalian Circadian Clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef]
- Eide, E.J.; Woolf, M.F.; Kang, H.; Woolf, P.; Hurst, W.; Camacho, F.; Vielhaber, E.L.; Giovanni, A.; Virshup, D.M. Control of Mammalian Circadian Rhythm by CKIε-Regulated Proteasome-Mediated PER2 Degradation. Mol. Cell. Biol. 2005, 25, 2795–2807. [Google Scholar] [CrossRef] [Green Version]
- Shirogane, T.; Jin, J.; Ang, X.L.; Harper, J.W. SCF -TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 2005, 280, 26863–26872. [Google Scholar] [CrossRef] [Green Version]
- Lamia, K.A.; Sachdeva, U.M.; Di Tacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440. [Google Scholar] [CrossRef] [Green Version]
- Preitner, N.; Damiola, F.; Molina, L.-L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The Orphan Nuclear Receptor REV-ERBα Controls Circadian Transcription within the Positive Limb of the Mammalian Circadian Oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, D.; McNamara, P.; Naik, K.A.; FitzGerald, G.A.; Kay, S.A.; Hogenesch, J.B. A Functional Genomics Strategy Reveals Rora as a Component of the Mammalian Circadian Clock. Neuron 2004, 43, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, S.; Yamaguchi, S.; Matsuo, T.; Ishida, Y.; Okamura, H. Antagonistic role of E4BP4 and PAR proteins in the circadian oscillatory mechanism. Genes Dev. 2001, 15, 995–1006. [Google Scholar] [CrossRef] [Green Version]
- Eide, E.J.; Vielhaber, E.L.; Hinz, W.A.; Virshup, D.M. The Circadian Regulatory Proteins BMAL1 and Cryptochromes Are Substrates of Casein Kinase Iε. J. Biol. Chem. 2002, 277, 17248–17254. [Google Scholar] [CrossRef] [Green Version]
- Sanada, K.; Okano, T.; Fukada, Y. Mitogen-activated Protein Kinase Phosphorylates and Negatively Regulates Basic Helix-Loop-Helix-PAS Transcription Factor BMAL1. J. Biol. Chem. 2002, 277, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Robles, M.; Humphrey, S.; Mann, M. Phosphorylation Is a Central Mechanism for Circadian Control of Metabolism and Physiology. Cell Metab. 2017, 25, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; HogenEsch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S. Circadian physiology of metabolism. Science 2016, 354, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrell, J.; Chiang, J.Y. Circadian rhythms in liver metabolism and disease. Acta Pharm. Sin. B 2015, 5, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokkan, K.-A.; Yamazaki, S.; Tei, H.; Sakaki, Y.; Menaker, M. Entrainment of the Circadian Clock in the Liver by Feeding. Science 2001, 291, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel-Mahan, K.; Patel, V.R.; De Mateo, S.; Orozco-Solis, R.; Ceglia, N.J.; Sahar, S.; Dilag-Penilla, S.A.; Dyar, K.; Baldi, P.; Sassone-Corsi, P. Reprogramming of the Circadian Clock by Nutritional Challenge. Cell 2013, 155, 1464–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458. [Google Scholar] [CrossRef] [Green Version]
- Crespo, M.; Gonzalez-Teran, B.; Nikolic, I.; Mora, A.; Folgueira, C.; Rodríguez, E.; Leiva-Vega, L.; Pintor-Chocano, A.; Fernández-Chacón, M.; Ruiz-Garrido, I.; et al. Neutrophil infiltration regulates clock-gene expression to organize daily hepatic metabolism. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Tuccinardi, D.; Ernesti, I.; Basciani, S.; Mariani, S.; Genco, A.; Manfrini, S.; Lubrano, C.; Gnessi, L. Scientific evidence underlying contraindications to the ketogenic diet: An update. Obes. Rev. 2020, 21. [Google Scholar] [CrossRef]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538.e5. [Google Scholar] [CrossRef]
- Lamia, K.A.; Storch, K.-F.; Weitz, C.J. Physiological significance of a peripheral tissue circadian clock. Proc. Natl. Acad. Sci. USA 2008, 105, 15172–15177. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010, 16, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.; Kinouchi, K.; Welz, P.-S.; Smith, J.; Zinna, V.M.; Shi, J.; Samad, M.; Chen, S.; Magnan, C.N.; Kinchen, J.; et al. Defining the Independence of the Liver Circadian Clock. Cell 2019, 177, 1448–1462.e14. [Google Scholar] [CrossRef] [PubMed]
- Sinturel, F.; Gos, P.; Petrenko, V.; Hagedorn, C.; Kreppel, F.; Storch, K.-F.; Knutti, D.; Liani, A.; Weitz, C.; Emmenegger, Y.; et al. Circadian hepatocyte clocks keep synchrony in the absence of a master pacemaker in the suprachiasmatic nucleus or other extrahepatic clocks. Genes Dev. 2021, 35, 329–334. [Google Scholar] [CrossRef]
- Ray, S.; Valekunja, U.K.; Stangherlin, A.; Howell, S.A.; Snijders, A.P.; Damodaran, G.; Reddy, A.B. Circadian rhythms in the absence of the clock gene Bmal1. Science 2020, 367, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Everett, L.J.; Lazar, M.A. Nuclear receptor Rev-erbα: Up, down, and all around. Trends Endocrinol. Metab. 2014, 25, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.; Xiong, Y.; Trinh, T.M.; Xiao, Y.; Hu, W.; Jiang, C.; Dierickx, P.; Jang, C.; Rabinowitz, J.D.; Lazar, M.A. The hepatocyte clock and feeding control chronophysiology of multiple liver cell types. Science 2020, 369, eaba8984. [Google Scholar] [CrossRef]
- Reinke, H.; Asher, G. Circadian Clock Control of Liver Metabolic Functions. Gastroenterol. 2016, 150, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Robles, M.; Cox, J.; Mann, M. In-Vivo Quantitative Proteomics Reveals a Key Contribution of Post-Transcriptional Mechanisms to the Circadian Regulation of Liver Metabolism. PLoS Genet. 2014, 10, e1004047. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nat. Cell Biol. 2007, 450, 1086–1090. [Google Scholar] [CrossRef]
- Feng, D.; Liu, T.; Sun, Z.; Bugge, A.; Mullican, S.E.; Alenghat, T.; Liu, X.S.; Lazar, M.A. A Circadian Rhythm Orchestrated by Histone Deacetylase 3 Controls Hepatic Lipid Metabolism. Science 2011, 331, 1315–1319. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Solanas, G.; Peixoto, F.O.; Bee, L.; Symeonidi, A.; Schmidt, M.; Brenner, C.; Masri, S.; Benitah, S.A.; Sassone-Corsi, P. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 2017, 170, 664–677.e11. [Google Scholar] [CrossRef]
- Edwards, P.A.; Muroya, H.; Gould, R.G. In vivo demonstration of the circadian thythm of cholesterol biosynthesis in the liver and intestine of the rat. J. Lipid Res. 1972, 13, 396–401. [Google Scholar] [CrossRef]
- Yamada, M.; Nagatomo, J.; Setoguchi, Y.; Kuroki, N.; Higashi, S.; Setoguchi, T. Circadian Rhythms of Sterol 12α-Hydroxylase, Cholesterol 7α-Hydroxylase and DBP Involved in Rat Cholesterol Catabolism. Biol. Chem. 2000, 381, 1149–1153. [Google Scholar] [CrossRef]
- Gälman, C.; Angelin, B.; Rudling, M. Bile Acid Synthesis in Humans Has a Rapid Diurnal Variation That is Asynchronous with Cholesterol Synthesis. Gastroenterology 2005, 129, 1445–1453. [Google Scholar] [CrossRef]
- Duez, H.; Van Der Veen, J.N.; Duhem, C.; Pourcet, B.; Touvier, T.; Fontaine, C.; Derudas, B.; Baugé, E.; Havinga, R.; Bloks, V.W.; et al. Regulation of Bile Acid Synthesis by the Nuclear Receptor Rev-erbα. Gastroenterology 2008, 135, 689–698.e5. [Google Scholar] [CrossRef] [PubMed]
- Manieri, E.; Folgueira, C.; Rodríguez, M.E.; Leiva-Vega, L.; Esteban-Lafuente, L.; Chen, C.; Cubero, F.J.; Barrett, T.; Cavanagh-Kyros, J.; Seruggia, D.; et al. JNK-mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 16492–16499. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Xiao, R.; Tseng, H.-T.; Shan, L.; Fu, L.; Moore, D.D. Circadian Dysregulation Disrupts Bile Acid Homeostasis. PLoS ONE 2009, 4, e6843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrell, J.; Chiang, J.Y. Short-Term Circadian Disruption Impairs Bile Acid and Lipid Homeostasis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Adamovich, Y.; Aviram, R.; Asher, G. The emerging roles of lipids in circadian control. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2015, 1851, 1017–1025. [Google Scholar] [CrossRef]
- Chen, H.; Gao, L.; Yang, D.; Xiao, Y.; Zhang, M.; Li, C.; Wang, A.; Jin, Y. Coordination between the circadian clock and androgen signaling is required to sustain rhythmic expression of Elovl3 in mouse liver. J. Biol. Chem. 2019, 294, 7046–7056. [Google Scholar] [CrossRef] [PubMed]
- Cretenet, G.; Le Clech, M.; Gachon, F. Circadian Clock-Coordinated 12 Hr Period Rhythmic Activation of the IRE1α Pathway Controls Lipid Metabolism in Mouse Liver. Cell Metab. 2010, 11, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Ruiter, M.; La Fleur, S.E.; Van Heijningen, C.; Van Der Vliet, J.; Kalsbeek, A.; Buijs, R.M. The daily rhythm in plasma glucagon concentrations in the rat is modulated by the biological clock and by feeding behavior. Diabetes 2003, 52, 1709–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, G.; Ruiz, J.; Urbain, J.L.; Chen, X. Evidence for a circadian rhythm of insulin secretion. Am. J. Physiol. Metab. 1996, 271, E246–E252. [Google Scholar] [CrossRef]
- Doi, R.; Oishi, K.; Ishida, N. CLOCK Regulates Circadian Rhythms of Hepatic Glycogen Synthesis through Transcriptional Activation of Gys. J. Biol. Chem. 2010, 285, 22114–22121. [Google Scholar] [CrossRef] [Green Version]
- Krishnaiah, S.Y.; Wu, G.; Altman, B.; Growe, J.; Rhoades, S.D.; Coldren, F.; Venkataraman, A.; Olarerin-George, A.O.; Francey, L.J.; Mukherjee, S.; et al. Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab. 2017, 25, 961–974.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimba, S.; Ogawa, T.; Hitosugi, S.; Ichihashi, Y.; Nakadaira, Y.; Kobayashi, M.; Tezuka, M.; Kosuge, Y.; Ishige, K.; Ito, Y.; et al. Deficient of a Clock Gene, Brain and Muscle Arnt-Like Protein-1 (BMAL1), Induces Dyslipidemia and Ectopic Fat Formation. PLoS ONE 2011, 6, e25231. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Queiroz, J.; Hussain, M.M. Nonalcoholic fatty liver disease in CLOCK mutant mice. J. Clin. Investig. 2020, 130, 4282–4300. [Google Scholar] [CrossRef]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and Metabolic Syndrome in Circadian Clock Mutant Mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, B.; Bellet, M.M.; Katada, S.; Astarita, G.; Hirayama, J.; Amin, R.H.; Granneman, J.G.; Piomelli, D.; Leff, T.; Sassone-Corsi, P. PER2 Controls Lipid Metabolism by Direct Regulation of PPARγ. Cell Metab. 2010, 12, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Bugge, A.; Feng, D.; Everett, L.J.; Briggs, E.R.; Mullican, S.E.; Wang, F.; Jager, J.; Lazar, M.A. Rev-erb and Rev-erb coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012, 26, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Ünlütürk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m6A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Tong, X.; Arthurs, B.; Guha, A.; Rui, L.; Kamath, A.; Inoki, K.; Yin, L. Liver Clock Protein BMAL1 Promotes de Novo Lipogenesis through Insulin-mTORC2-AKT Signaling. J. Biol. Chem. 2014, 289, 25925–25935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Tong, X.; Nelson, B.B.; Jin, E.; Sit, J.; Charney, N.; Yang, M.; Omary, B.; Yin, L. The hepatic BMAL1/AKT/lipogenesis axis protects against alcoholic liver disease in mice via promoting PPARα pathway. Hepatology 2018, 68, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, C.; Kathale, N.D.; Liu, D.; Lee, C.; Freeman, D.A.; HogenEsch, J.B.; Cao, R.; Liu, A.C. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018, 14, e1007369. [Google Scholar] [CrossRef]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nat. Cell Biol. 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Makishima, M.; Repa, J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Ji, S.; Liu, Q.; Zhang, S.; Chen, Q.; Wang, C.; Zhang, W.; Xiao, C.; Li, Y.; Nian, C.; Li, J.; et al. FGF15 Activates Hippo Signaling to Suppress Bile Acid Metabolism and Liver Tumorigenesis. Dev. Cell 2019, 48, 460–474.e9. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Zhang, R.; Jain, R.; Shi, H.; Zhang, L.; Zhou, G.; Sangwung, P.; Tugal, D.; Atkins, G.B.; Prosdocimo, D.A.; et al. Circadian control of bile acid synthesis by a KLF15-Fgf15 axis. Nat. Commun. 2015, 6, 7231. [Google Scholar] [CrossRef] [Green Version]
- Le Martelot, G.; Claudel, T.; Gatfield, D.; Schaad, O.; Kornmann, B.; Sasso, G.L.; Moschetta, A.; Schibler, U. REV-ERBα Participates in Circadian SREBP Signaling and Bile Acid Homeostasis. PLoS Biol. 2009, 7, e1000181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Fleur, S.E.; Kalsbeek, A.; Wortel, J.; Fekkes, M.L.; Buijs, R.M. A Daily Rhythm in Glucose Tolerance. Diabetes 2001, 50, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Zani, F.; Breasson, L.; Becattini, B.; Vukolic, A.; Montani, J.-P.; Albrecht, U.; Provenzani, A.; Ripperger, J.A.; Solinas, G. PER2 promotes glucose storage to liver glycogen during feeding and acute fasting by inducing Gys2 PTG and GL expression. Mol. Metab. 2013, 2, 292–305. [Google Scholar] [CrossRef]
- Barclay, J.L.; Shostak, A.; Leliavski, A.; Tsang, A.H.; Jöhren, O.; Müller-Fielitz, H.; Landgraf, D.; Naujokat, N.; Van Der Horst, G.T.J.; Oster, H. High-fat diet-induced hyperinsulinemia and tissue-specific insulin resistance in Cry-deficient mice. Am. J. Physiol. Metab. 2013, 304, E1053–E1063. [Google Scholar] [CrossRef] [Green Version]
- Toledo, M.; Batista-Gonzalez, A.; Merheb, E.; Aoun, M.L.; Tarabra, E.; Feng, D.; Sarparanta, J.; Merlo, P.; Botrè, F.; Schwartz, G.J.; et al. Autophagy Regulates the Liver Clock and Glucose Metabolism by Degrading CRY1. Cell Metab. 2018, 28, 268–281.e4. [Google Scholar] [CrossRef] [Green Version]
- Lamia, K.A.; Papp, S.J.; Yu, R.T.; Barish, G.D.; Uhlenhaut, H.; Jonker, J.; Downes, M.; Evans, R.M. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nat. Cell Biol. 2011, 480, 552–556. [Google Scholar] [CrossRef]
- Jang, H.; Lee, G.Y.; Selby, C.P.; Lee, G.; Jeon, Y.G.; Lee, J.H.; Cheng, K.K.Y.; Titchenell, P.; Birnbaum, M.J.; Xu, A.; et al. SREBP1c-CRY1 signalling represses hepatic glucose production by promoting FOXO1 degradation during refeeding. Nat. Commun. 2016, 7, 12180. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Muchnik, M.; Chen, Z.; Patel, M.; Wu, N.; Joshi, S.; Rui, L.; Lazar, M.A.; Yin, L. Transcriptional Repressor E4-binding Protein 4 (E4BP4) Regulates Metabolic Hormone Fibroblast Growth Factor 21 (FGF21) during Circadian Cycles and Feeding. J. Biol. Chem. 2010, 285, 36401–36409. [Google Scholar] [CrossRef] [Green Version]
- Gavrila, A.; Peng, C.-K.; Chan, J.L.; Mietus, J.E.; Goldberger, A.L.; Mantzoros, C.S. Diurnal and Ultradian Dynamics of Serum Adiponectin in Healthy Men: Comparison with Leptin, Circulating Soluble Leptin Receptor, and Cortisol Patterns. J. Clin. Endocrinol. Metab. 2003, 88, 2838–2843. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Bartosch, B. Hepatitis B and C Viruses and Hepatocellular Carcinoma. Viruses 2010, 2, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Cicuéndez, B.; Ruiz-Garrido, I.; Mora, A.; Sabio, G. Stress kinases in the development of liver steatosis and hepatocellular carcinoma. Mol. Metab. 2021, 101190, 101190. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J. Circadian Integration of Metabolism and Energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-W.; Yun, K.E.; Jung, H.-S.; Chang, Y.; Choi, E.-S.; Kwon, M.-J.; Lee, E.-H.; Woo, E.J.; Kim, N.H.; Shin, H.; et al. Sleep duration and quality in relation to non-alcoholic fatty liver disease in middle-aged workers and their spouses. J. Hepatol. 2013, 59, 351–357. [Google Scholar] [CrossRef]
- Kettner, N.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Filipski, E.; Lévi, F. Circadian Disruption in Experimental Cancer Processes. Integr. Cancer Ther. 2009, 8, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Filipski, E.; Subramanian, P.; Carrière, J.; Guettier, C.; Barbason, H.; Lévi, F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 680, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Mteyrek, A.; Filipski, E.; Guettier, C.; Okyar, A.; Lévi, F. Clock gene Per2 as a controller of liver carcinogenesis. Oncotarget 2016, 7, 85832–85847. [Google Scholar] [CrossRef] [Green Version]
- Fleet, T.; Stashi, E.; Zhu, B.; Rajapakshe, K.; Marcelo, K.L.; Kettner, N.M.; Gorman, B.K.; Coarfa, C.; Fu, L.; O’Malley, B.W.; et al. Genetic and Environmental Models of Circadian Disruption Link SRC-2 Function to Hepatic Pathology. J. Biol. Rhythm. 2016, 31, 443–460. [Google Scholar] [CrossRef] [Green Version]
- Mteyrek, A.; Filipski, E.; Guettier, C.; Oklejewicz, M.; Van Der Horst, G.T.; Okyar, A.; Lévi, F. Critical cholangiocarcinogenesis control by cryptochrome clock genes. Int. J. Cancer 2017, 140, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Anafi, R.C.; Francey, L.J.; Hogenesch, J.B.; Kim, J. CYCLOPS reveals human transcriptional rhythms in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, 5312–5317. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-L.; Yu, C.; Jiang, J.-X.; Liu, L.-P.; Fang, X.; Wu, C. Hepatitis B virus X protein disrupts the balance of the expression of circadian rhythm genes in hepatocellular carcinoma. Oncol. Lett. 2014, 8, 2715–2720. [Google Scholar] [CrossRef]
- Lin, Y.; Chang, J.H.; Yeh, K.; Yang, M.; Liu, T.; Lin, S.; Su, W.; Chang, J. Disturbance of circadian gene expression in hepatocellular carcinoma. Mol. Carcinog. 2008, 47, 925–933. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, K.; Zhao, M.; Chen, H.; Ji, G.; Zhang, Y.; Wang, T.; Cao, H.; Li, Y.; Qu, L. Analysis of miRNA expression profiles in the liver ofClockΔ19mutant mice. PeerJ 2019, 7, e8119. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Li, J.; Zhou, F.; Huang, Q.; Zhang, J.; Guo, X.; Lyu, Z.; Zhang, H.; Xing, J. NPAS2 promotes cell survival of hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis. 2017, 8, e2704. [Google Scholar] [CrossRef]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Deans, J.R.; Kwok, C.; Patel, P.; Fu, L.; Berdeaux, R.; Sun, K.; Kolonin, M.G.; et al. Incompatibility of the circadian protein BMAL1 and HNF4α in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Manieri, E.; Herrera-Melle, L.; Mora, A.; Tomás-Loba, A.; Leiva-Vega, L.; Fernández, D.I.; Rodríguez, E.; Morán, L.; Cosido, L.H.; Torres, J.L.; et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J. Exp. Med. 2019, 216, 1108–1119. [Google Scholar] [CrossRef]
- Singhal, G.; Kumar, G.; Chan, S.; Fisher, F.M.; Ma, Y.; Vardeh, H.G.; Nasser, I.A.; Flier, J.S.; Maratos-Flier, E. Deficiency of fibroblast growth factor 21 (FGF21) promotes hepatocellular carcinoma (HCC) in mice on a long term obesogenic diet. Mol. Metab. 2018, 13, 56–66. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crespo, M.; Leiva, M.; Sabio, G. Circadian Clock and Liver Cancer. Cancers 2021, 13, 3631. https://doi.org/10.3390/cancers13143631
Crespo M, Leiva M, Sabio G. Circadian Clock and Liver Cancer. Cancers. 2021; 13(14):3631. https://doi.org/10.3390/cancers13143631
Chicago/Turabian StyleCrespo, María, Magdalena Leiva, and Guadalupe Sabio. 2021. "Circadian Clock and Liver Cancer" Cancers 13, no. 14: 3631. https://doi.org/10.3390/cancers13143631
APA StyleCrespo, M., Leiva, M., & Sabio, G. (2021). Circadian Clock and Liver Cancer. Cancers, 13(14), 3631. https://doi.org/10.3390/cancers13143631