
Natural History of Untreated Retinoblastoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Eligibility
2.2. Data Collection and Ethics
2.3. Statistical Analysis
3. Results
3.1. Study Findings
3.1.1. Patient Characteristics
3.1.2. Orbital Extension
3.1.3. Metastasis
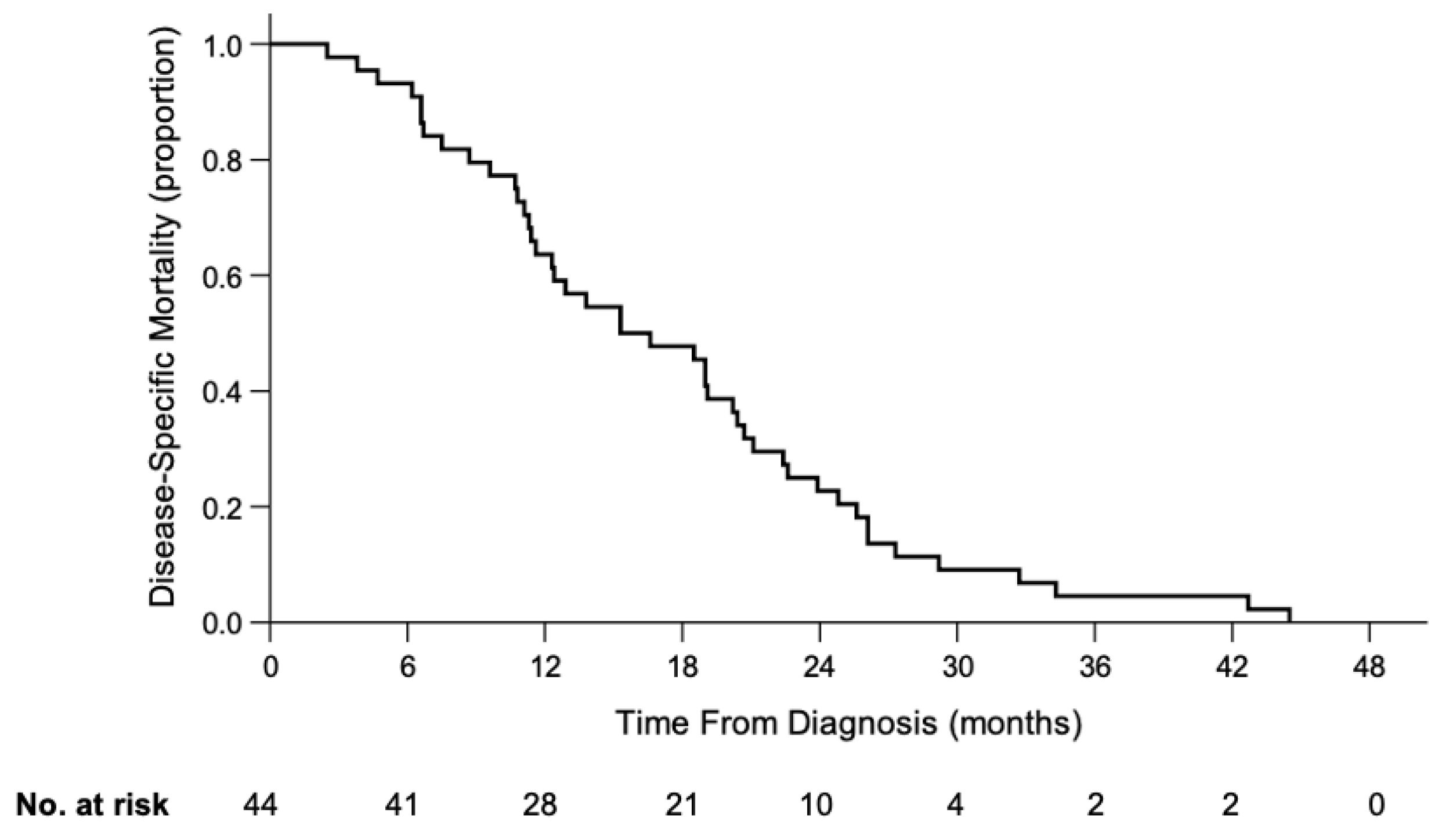
3.1.4. Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kivela, T. The epidemiological challenge of the most frequent eye cancer: Retinoblastoma, an issue of birth and death. Br. J. Ophthalmol. 2009, 93, 1129–1131. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.E.; Francis, J.H.; Dunkel, I.J.; Shields, C.L.; Yu, M.D.; Berry, J.L.; Kogachi, K.; Skalet, A.H.; Miller, A.K.; Santapuram, P.R.; et al. Metastases and death rates after primary enucleation of unilateral retinoblastoma in the USA 2007-2017. Br. J. Ophthalmol. 2019, 103, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Feng, Z.X.; Wei, M.; Liu, G.; Solarte, C.E.; Li, B.; Wang, Y.; Zhang, C.; Gallie, B.L. Impact of Systemic Chemotherapy and Delayed Enucleation on Survival of Children with Advanced Intraocular Retinoblastoma. Ophthalmol. Retin. 2020, 4, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 15021. [Google Scholar] [CrossRef] [Green Version]
- Gallie, B.L.; Budning, A.; DeBoer, G.; Thiessen, J.J.; Koren, G.; Verjee, Z.; Ling, V.; Chan, H.S. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiotherapy. Arch. Ophthalmol. 1996, 114, 1321–1328. [Google Scholar] [CrossRef]
- Francis, J.H.; Levin, A.M.; Zabor, E.C.; Gobin, Y.P.; Abramson, D.H. Ten-year experience with ophthalmic artery chemosurgery: Ocular and recurrence-free survival. PLoS ONE 2018, 13, e0197081. [Google Scholar] [CrossRef]
- Munier, F.L.; Mosimann, P.; Puccinelli, F.; Gaillard, M.C.; Stathopoulos, C.; Houghton, S.; Bergin, C.; Beck-Popovic, M. First-line intra-arterial versus intravenous chemotherapy in unilateral sporadic group D retinoblastoma: Evidence of better visual outcomes, ocular survival and shorter time to success with intra-arterial delivery from retrospective review of 20 years of treatment. Br. J. Ophthalmol. 2017, 101, 1086–1093. [Google Scholar] [CrossRef]
- Munier, F.L.; Soliman, S.; Moulin, A.P.; Gaillard, M.C.; Balmer, A.; Beck-Popovic, M. Profiling safety of intravitreal injections for retinoblastoma using an anti-reflux procedure and sterilisation of the needle track. Br. J. Ophthalmol. 2012, 96, 1084–1087. [Google Scholar] [CrossRef]
- Berry, J.L.; Shah, S.; Bechtold, M.; Zolfaghari, E.; Jubran, R.; Kim, J.W. Long-term outcomes of Group D retinoblastoma eyes during the intravitreal melphalan era. Pediatr. Blood Cancer 2017. [Google Scholar] [CrossRef]
- Simpson, E.R.; Gallie, B.; Laperrierre, N.; Beiki-Ardakani, A.; Kivela, T.; Raivio, V.; Heikkonen, J.; Desjardins, L.; Dendale, R.; Mazal, A.; et al. The American Brachytherapy Society consensus guidelines for plaque brachytherapy of uveal melanoma and retinoblastoma. Brachytherapy 2014, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Reese, A.B.; Merriam, G.R., Jr.; Martin, H.E. Treatment of bilateral retinoblastoma by irradiation and surgery; report on 15-year results. Am. J. Ophthalmol. 1949, 32, 175–190. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, Y. Treatment of Retinoblastoma: The Role of External Beam Radiotherapy. Yonsei Med. J. 2015, 56, 1478–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, Q.; Wu, S.; Jin, L.; Ma, X.; Jin, M.; Wang, Y.; Gallie, B. Pars Plana Vitrectomy and Endoresection of Refractory Intraocular Retinoblastoma. Ophthalmology 2018, 125, 320–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseripour, M. “Retinoblastoma survival disparity”: The expanding horizon in developing countries. Saudi J. Ophthalmol. Off. J. Saudi Ophthalmol. Soc. 2012, 26, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Moulik, N.R.; Mishra, R.K.; Kumar, D. Causes, outcome and prevention of abandonment in retinoblastoma in India. Pediatr. Blood Cancer 2013, 60, 771–775. [Google Scholar] [CrossRef]
- Bakhshi, S.; Gupta, S.; Gogia, V.; Ravindranath, Y. Compliance in retinoblastoma. Indian J. Pediatr. 2010, 77, 535–540. [Google Scholar] [CrossRef]
- Vasquez, L.; Diaz, R.; Chavez, S.; Tarrillo, F.; Maza, I.; Hernandez, E.; Oscanoa, M.; Garcia, J.; Geronimo, J.; Rossell, N. Factors associated with abandonment of therapy by children diagnosed with solid tumors in Peru. Pediatr. Blood Cancer 2018, 65, e27007. [Google Scholar] [CrossRef] [PubMed]
- Mostert, S.; Arora, R.S.; Arreola, M.; Bagai, P.; Friedrich, P.; Gupta, S.; Kaur, G.; Koodiyedath, B.; Kulkarni, K.; Lam, C.G.; et al. Abandonment of treatment for childhood cancer: Position statement of a SIOP PODC Working Group. Lancet Oncol. 2011, 12, 719–720. [Google Scholar] [CrossRef]
- Murphree, A.L. Intraocular retinoblastoma: The case for a new group classification. Ophthalmol. Clin. N. Am. 2005, 18, 41–53. [Google Scholar]
- Friedrich, P.; Lam, C.G.; Itriago, E.; Perez, R.; Ribeiro, R.C.; Arora, R.S. Magnitude of Treatment Abandonment in Childhood Cancer. PLoS ONE 2015, 10, e0135230. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Zeng, J.; Guo, B.; He, W.; Chen, J.; Lu, F.; Chen, D. Clinical presentation and treatment outcome of retinoblastoma in children of South Western China. Medicine 2016, 95, e5204. [Google Scholar] [CrossRef]
- Chang, C.Y.; Chiou, T.J.; Hwang, B.; Bai, L.Y.; Hsu, W.M.; Hsieh, Y.L. Retinoblastoma in Taiwan: Survival rate and prognostic factors. Jpn. J. Ophthalmol. 2006, 50, 242–249. [Google Scholar] [CrossRef]
- Kaliki, S.; Patel, A.; Iram, S.; Ramappa, G.; Mohamed, A.; Palkonda, V.A.R. RETINOBLASTOMA IN INDIA: Clinical Presentation and Outcome in 1,457 Patients (2,074 Eyes). Retina 2019, 39, 379–391. [Google Scholar] [CrossRef]
- Chung, S.E.; Sa, H.S.; Koo, H.H.; Yoo, K.H.; Sung, K.W.; Ham, D.I. Clinical manifestations and treatment of retinoblastoma in Korea. Br. J. Ophthalmol. 2008, 92, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Rahmat, J.; Rahman, N.A.; Ramasamy, S.; Bhoo-Pathy, N.; Pin, G.P.; Alagaratnam, J. Presentation of retinoblastoma patients in Malaysia. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 7863–7867. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhang, W.; Pan, H.; Li, T.; Liu, B.; Zhao, J.; Wang, B. Retrospective investigation of retinoblastoma in Chinese patients. Oncotarget 2017, 8, 108492–108497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honavar, S.G.; Manjandavida, F.P.; Reddy, V.A.P. Orbital retinoblastoma: An update. Indian J. Ophthalmol. 2017, 65, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kathpalia, V.; Dunkel, I.J.; Wong, R.K.; Riedel, E.; Abramson, D.H. Orbital recurrence of retinoblastoma following enucleation. Br. J. Ophthalmol. 2009, 93, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Chevez-Barrios, P.; Hurwitz, M.Y.; Louie, K.; Marcus, K.T.; Holcombe, V.N.; Schafer, P.; Aguilar-Cordova, C.E.; Hurwitz, R.L. Metastatic and nonmetastatic models of retinoblastoma. Am. J. Pathol. 2000, 157, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Gunduz, K.; Muftuoglu, O.; Gunalp, I.; Unal, E.; Tacyildiz, N. Metastatic retinoblastoma clinical features, treatment, and prognosis. Ophthalmology 2006, 113, 1558–1566. [Google Scholar] [CrossRef]
- Namouni, F.; Doz, F.; Tanguy, M.L.; Quintana, E.; Michon, J.; Pacquement, H.; Bouffet, E.; Gentet, J.C.; Plantaz, D.; Lutz, P.; et al. High-dose chemotherapy with carboplatin, etoposide and cyclophosphamide followed by a haematopoietic stem cell rescue in patients with high-risk retinoblastoma: A SFOP and SFGM study. Eur. J. Cancer 1997, 33A, 2368–2375. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, W.; Wang, Y.; Huang, D.; Shi, J.; Li, B.; Zhang, Y.; Zhou, Y. Characterization, treatment and prognosis of retinoblastoma with central nervous system metastasis. BMC Ophthalmol. 2018, 18, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, R.C., Jr. High-risk features and tumor differentiation in retinoblastoma: A retrospective histopathologic study. Arch. Pathol. Lab. Med. 2009, 133, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Luna-Fineman, S.; Chantada, G.; Alejos, A.; Amador, G.; Barnoya, M.; Castellanos, M.E.; Fu, L.; Fuentes-Alabi, S.; Giron, V.; Goenz, M.A.; et al. Delayed Enucleation With Neoadjuvant Chemotherapy in Advanced Intraocular Unilateral Retinoblastoma: AHOPCA II, a Prospective, Multi-Institutional Protocol in Central America. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Luna-Fineman, S.; Barnoya, M.; Bonilla, M.; Fu, L.; Baez, F.; Rodriguez-Galindo, C. Retinoblastoma in Central America: Report from the Central American Association of Pediatric Hematology Oncology (AHOPCA). Pediatr. Blood Cancer 2012, 58, 545–550. [Google Scholar] [CrossRef]
- Waddell, K.M.; Kagame, K.; Ndamira, A.; Twinamasiko, A.; Picton, S.V.; Simmons, I.G.; Revill, P.; Johnston, W.T.; Newton, R. Improving survival of retinoblastoma in Uganda. Br. J. Ophthalmol. 2015. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Number of Patients | % | |
|---|---|---|---|
| Sex | Male | 17 | 39 |
| Female | 27 | 61 | |
| Laterality | Unilateral | 12 | 27 |
| Bilateral | 32 | 73 | |
| Stage of RB at Presentation | Intraocular | 6 | 14 |
| Extraocular | 38 | 86 | |
| Family History of RB | No | 42 | 95 |
| Yes | 2 | 5 | |
| Year of Diagnosis | 2007–2009 | 13 | 29 |
| 2010–2012 | 15 | 34 | |
| 2013–2015 | 10 | 23 | |
| 2016–2017 | 6 | 14 | |
| Age at first sign (months) | Median (range) | 5.0 (0.2–40.6) | |
| Age at diagnosis (months) | Median (range) | 9.0 (1.0–46.9) | |
| Lag period (months) | Median (range) | 1.3 (0–11.6) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Feng, Z.; Gallie, B.L. Natural History of Untreated Retinoblastoma. Cancers 2021, 13, 3646. https://doi.org/10.3390/cancers13153646
Zhao J, Feng Z, Gallie BL. Natural History of Untreated Retinoblastoma. Cancers. 2021; 13(15):3646. https://doi.org/10.3390/cancers13153646
Chicago/Turabian StyleZhao, Junyang, Zhaoxun Feng, and Brenda L. Gallie. 2021. "Natural History of Untreated Retinoblastoma" Cancers 13, no. 15: 3646. https://doi.org/10.3390/cancers13153646
APA StyleZhao, J., Feng, Z., & Gallie, B. L. (2021). Natural History of Untreated Retinoblastoma. Cancers, 13(15), 3646. https://doi.org/10.3390/cancers13153646