
The Bladder Microbiome Is Associated with Epithelial–Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma

 and
and 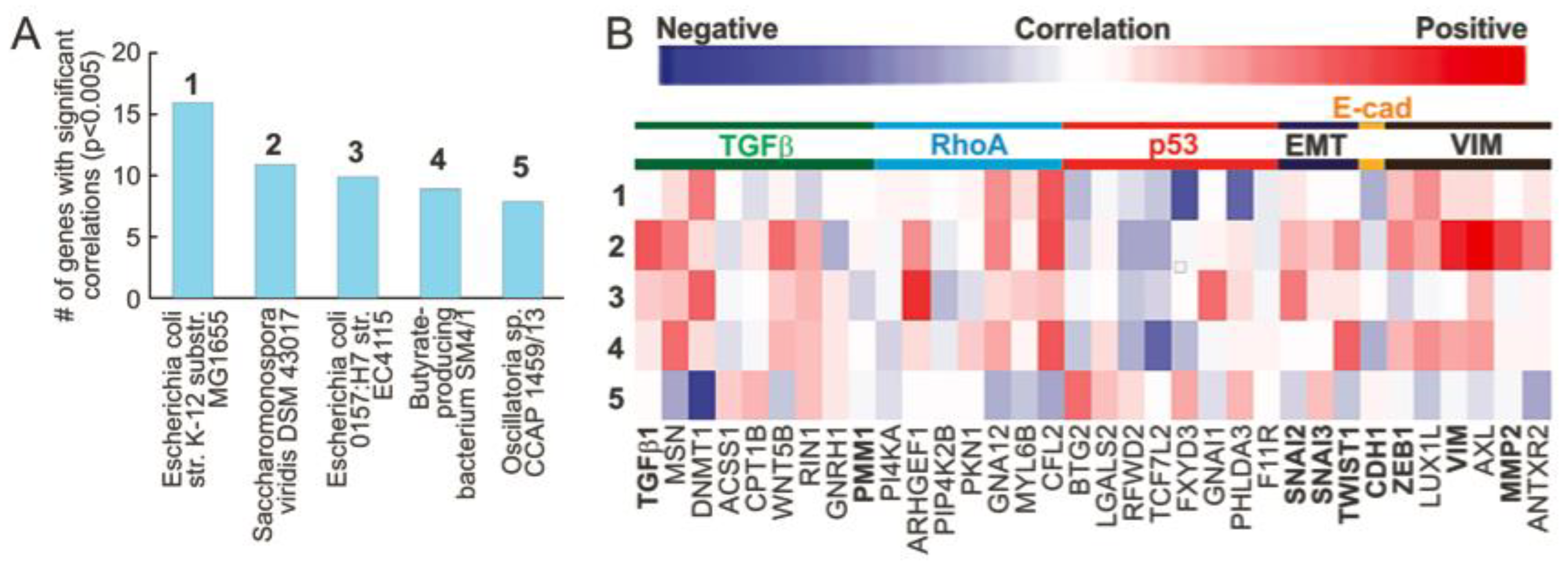{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Download
2.2. Extraction of Microbial Reads and Calculation of Microbial Abundance
2.3. Correlation of Microbial Reads to EMT-Related Genes and Pathways
2.4. Correlations of Microbial Reads to Genes Coding for ECM Proteins and Pathways
2.5. Correlation of Microbial Reads to Elastin and Elastin-Related Genes
2.6. Assessing the Clinical Relevance of Significant Microbes
2.7. Contamination Screening
3. Results
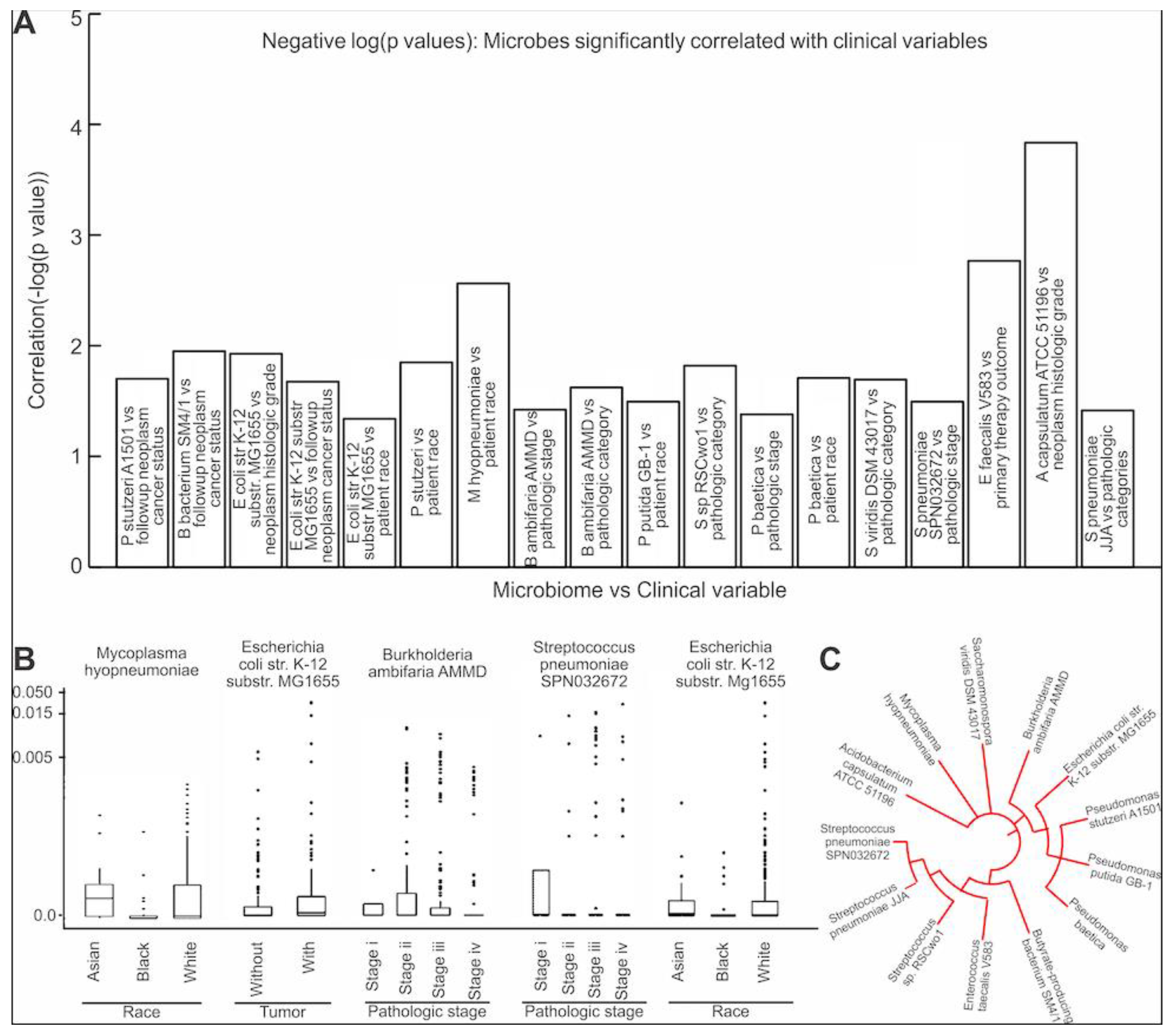
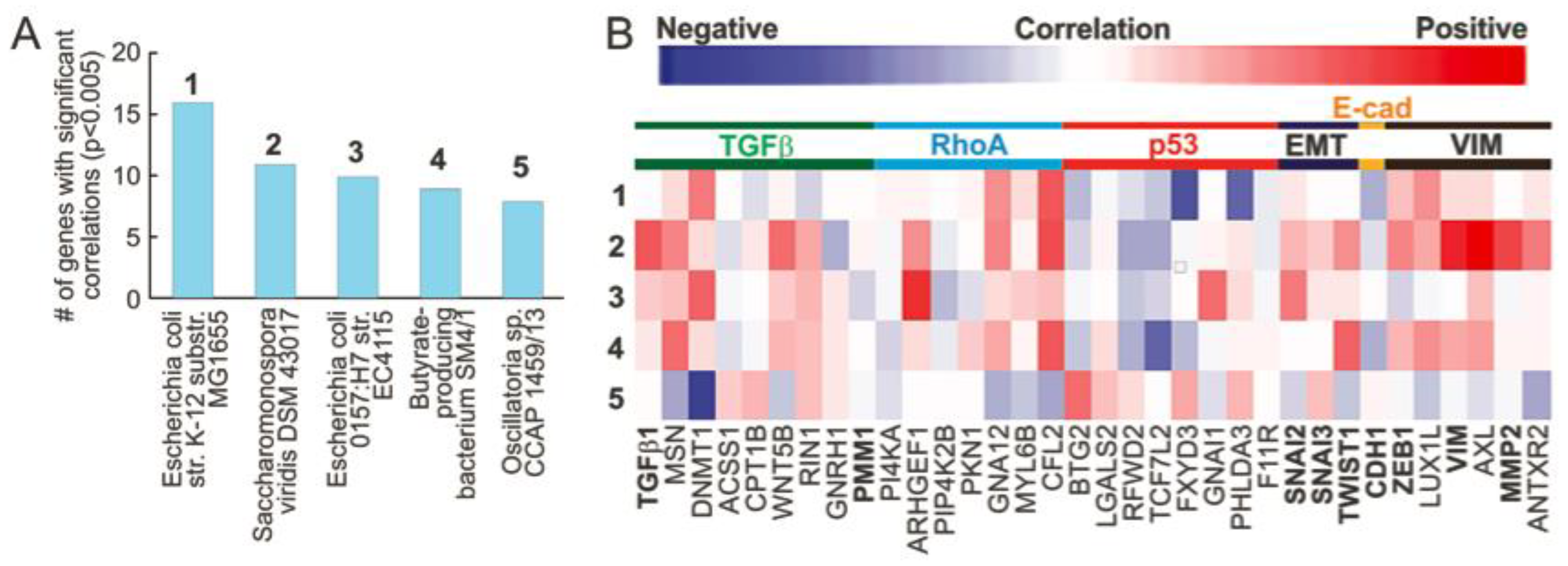
3.1. Microbial Abundance in MIBC Is Significantly Correlated with EMT Related Genes
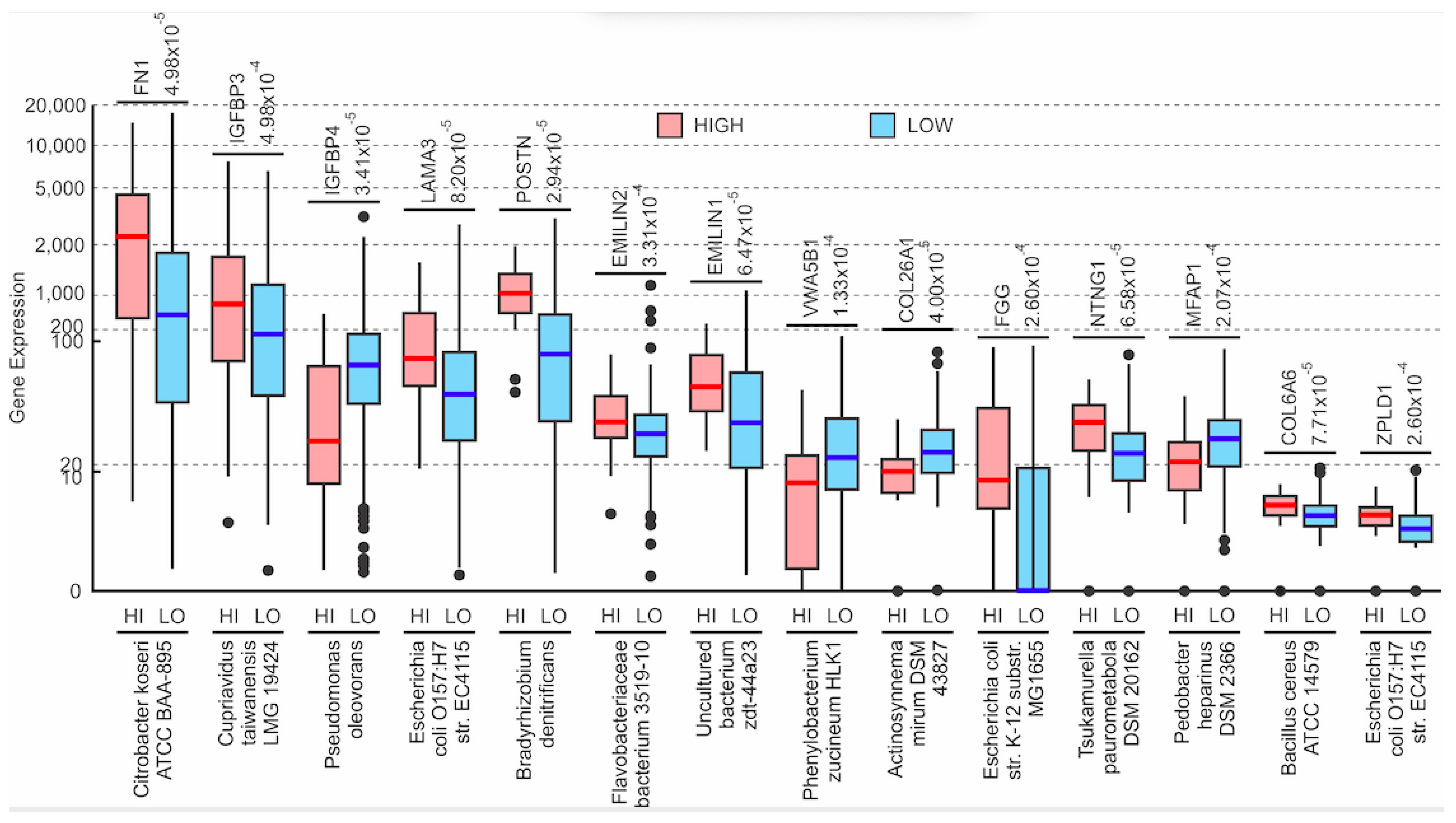
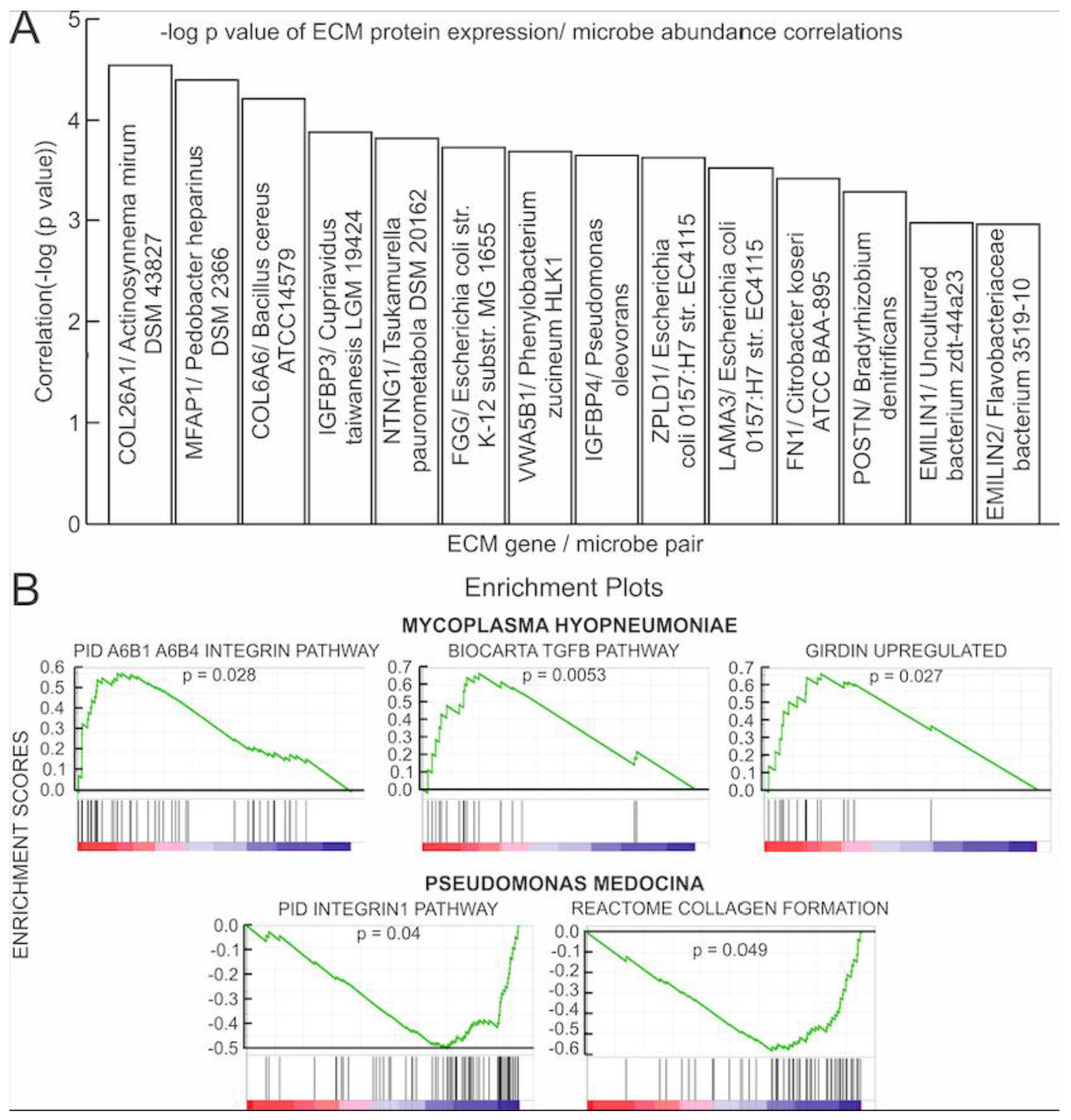
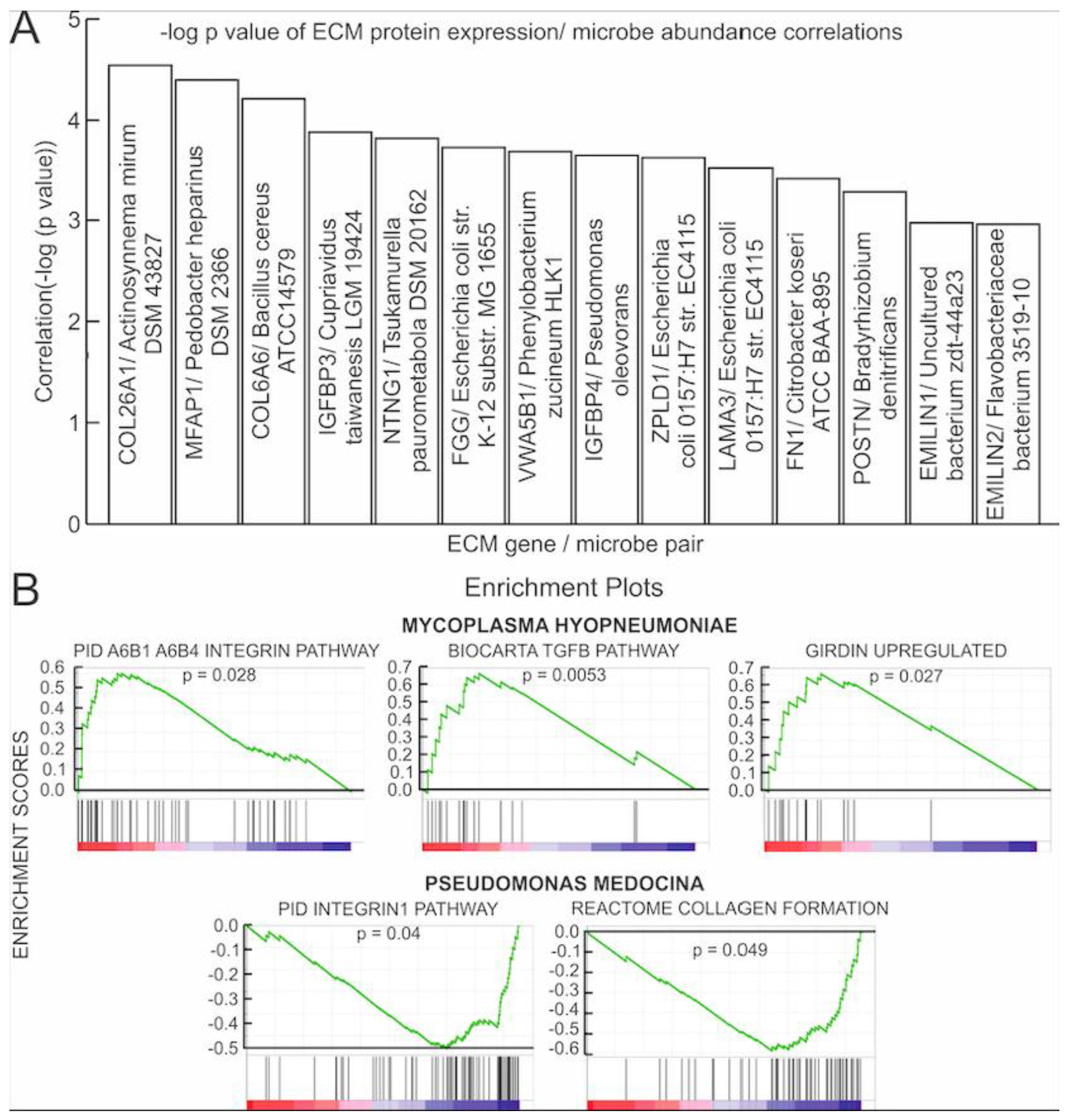
3.2. Correlations of Microbial Reads to Genes Coding for ECM Proteins and Pathways
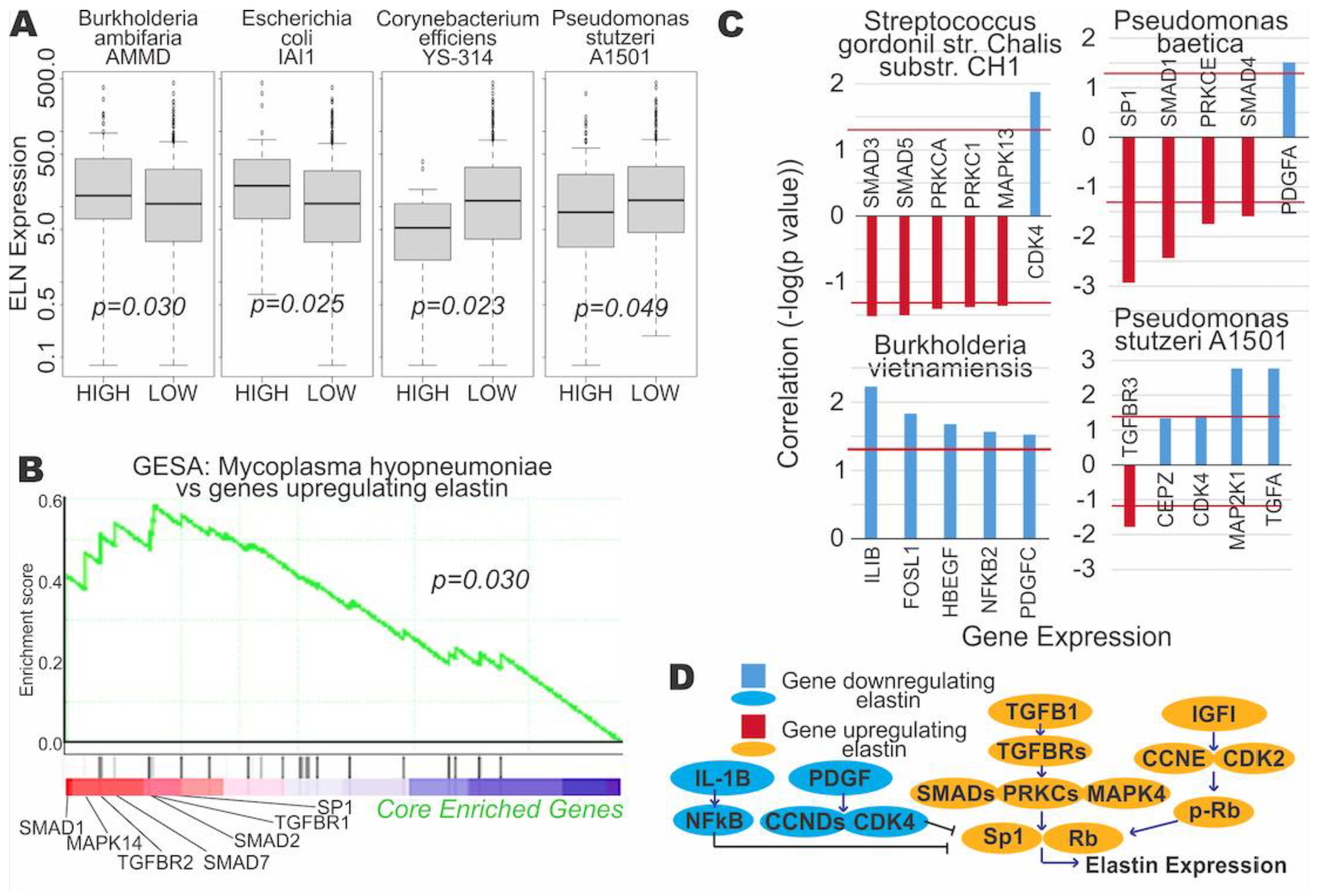
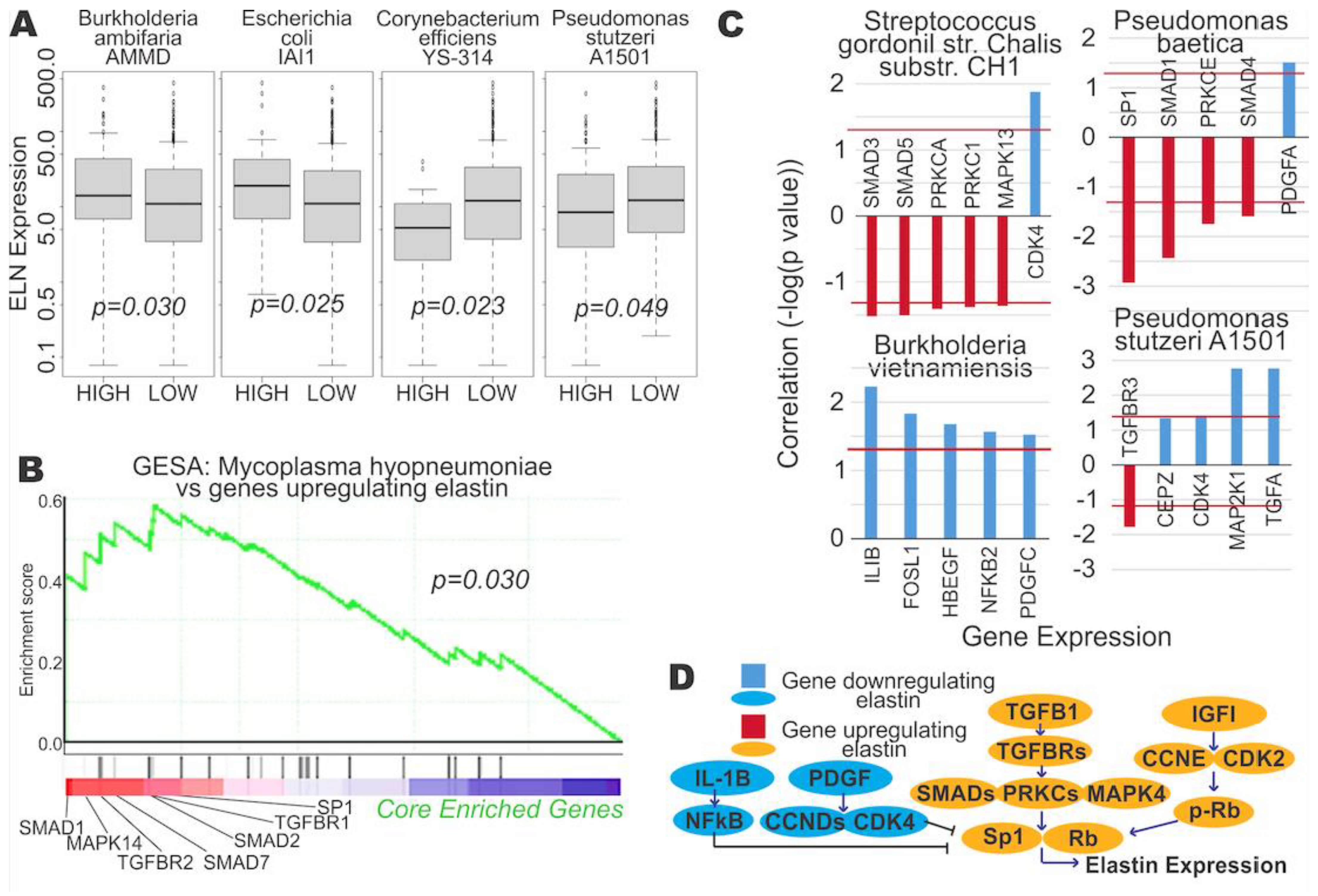
3.3. Microbial Expression Is Significantly Correlated to Elastin Expression
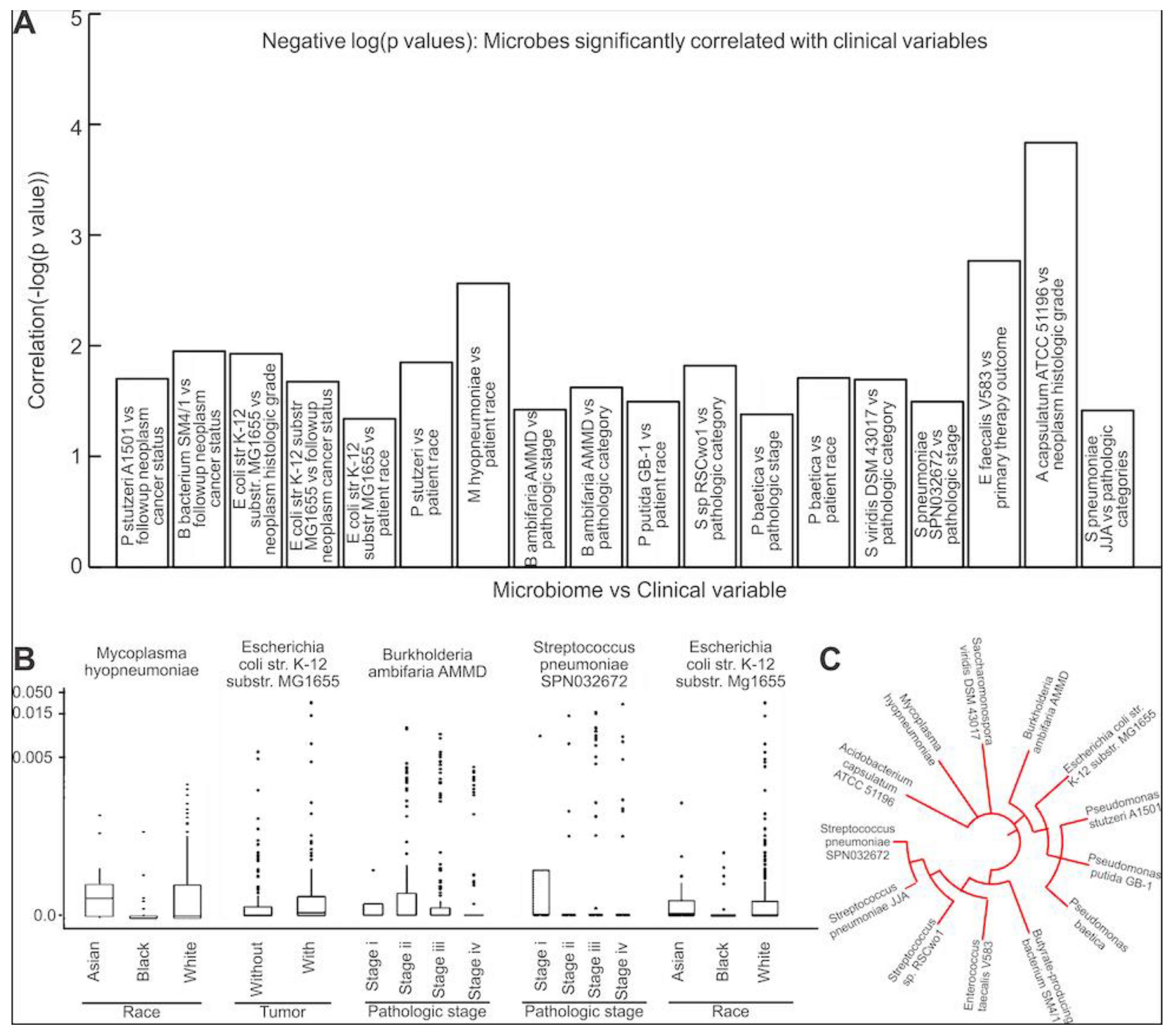
3.4. Assessing the Clinical Relevance of the Microbes That Are Significantly Correlated to EMT and ECM-Related Genes
3.5. Contamination Screening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dick, B.; Olubowale, O.; Kim, J.; Krane, S. Urothelial Carcinoma: Highlights and Reviews on Various Pathologies. EMJ Urol. 2020, 8, 46–53. [Google Scholar] [CrossRef]
- Smith, A.B.; Deal, A.M.; Woods, M.E.; Wallen, E.M.; Pruthi, R.S.; Chen, R.C.; Milowsky, M.I.; Nielsen, M.E. Muscle-invasive bladder cancer: Evaluating treatment and survival in the National Cancer Data Base. BJU Int. 2014, 114, 719–726. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3. [Google Scholar] [CrossRef]
- Ogawa, K.; Shimizu, Y.; Uketa, S.; Utsunomiya, N.; Kanamaru, S. Prognosis of patients with muscle invasive bladder cancer who are intolerable to receive any anti-cancer treatment. Cancer Treat. Res. Commun. 2020, 24, 100195. [Google Scholar] [CrossRef] [PubMed]
- Franzen, C.A.; Blackwell, R.H.; Todorovic, V.; Greco, K.A.; Foreman, K.E.; Flanigan, R.C.; Kuo, P.C.; Gupta, G.N. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis 2015, 4, e163. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.J.; Kim, W.-J. Role of the Epithelial-Mesenchymal Transition in Bladder Cancer: From Prognosis to Therapeutic Target. Korean J. Urol. 2013, 54, 645–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-Mesenchymal Transition: General Principles and Pathological Relevance with Special Emphasis on the Role of Matrix Metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012, 4, a011908. [Google Scholar] [CrossRef] [PubMed]
- Halper, J.; Kjaer, M. Basic Components of Connective Tissues and Extracellular Matrix: Elastin, Fibrillin, Fibulins, Fibrinogen, Fibronectin, Laminin, Tenascins and Thrombospondins. In Progress in Heritable Soft Connective Tissue Diseases; Springer: Dordrecht, The Netherlands, 2014; pp. 31–47. [Google Scholar] [CrossRef]
- Brooks, M.; Mo, Q.; Krasnow, R.; Ho, P.L.; Lee, Y.-C.; Xiao, J.; Kurtova, A.; Lerner, S.; Godoy, G.; Jian, W.; et al. Positive association of collagen type I with non-muscle invasive bladder cancer progression. Oncotarget 2016, 7, 82609–82619. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.-G.; Ha, Y.-R.; Ko, Y.-H.; Kang, S.-H.; Joo, K.-J.; Cho, H.-Y.; Park, H.-S.; Kim, C.-H.; Kwon, S.-Y.; Kim, J.-J.; et al. Effect of laminin 332 on motility and invasion in bladder cancer. Kaohsiung J. Med. Sci. 2013, 29, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Xu, X.; Jiang, Y.; Hansbro, N.G.; Hansbro, P.M.; Xu, J.; Liu, G. Elastin is a key factor of tumor development in colorectal cancer. BMC Cancer 2020, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Simeone, P.; Damato, M.; Maffia, M.; Lanuti, P.; Trerotola, M. The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J. Oncol. 2019, 2019, 1253727. [Google Scholar] [CrossRef] [PubMed]
- The Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Andolfi, C.; Bloodworth, J.C.; Papachristos, A.; Sweis, R.F. The Urinary Microbiome and Bladder Cancer: Susceptibility and Immune Responsiveness. Bladder Cancer 2020, 6, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Gnanasekar, A.; Castaneda, G.; Iyangar, A.; Magesh, S.; Perez, D.; Chakladar, J.; Li, W.T.; Bouvet, M.; Chang, E.Y.; Ongkeko, W.M. The intratumor microbiome predicts prognosis across gender and subtypes in papillary thyroid carcinoma. Comput. Struct. Biotechnol. J. 2021, 19, 1986–1997. [Google Scholar] [CrossRef] [PubMed]
- Chakladar, J.; Kuo, S.Z.; Castaneda, G.; Li, W.T.; Gnanasekar, A.; Yu, M.A.; Chang, E.Y.; Wang, X.Q.; Ongkeko, W.M. The Pancreatic Microbiome is Associated with Carcinogenesis and Worse Prognosis in Males and Smokers. Cancers 2020, 12, 2672. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd-El-Raouf, R.; Ouf, S.A.; Gabr, M.M.; Zakaria, M.M.; El-Yasergy, K.F.; Ali-El-Dein, B. Escherichia coli foster bladder cancer cell line progression via epithelial mesenchymal transition, stemness and metabolic reprogramming. Sci. Rep. 2020, 10, 18024. [Google Scholar] [CrossRef]
- Wang, W.; Fang, D.; Zhang, H.; Xue, J.; Wangchuk, D.; Du, J.; Jiang, L. Sodium Butyrate Selectively Kills Cancer Cells and Inhibits Migration in Colorectal Cancer by Targeting Thioredoxin-1. OncoTargets Ther. 2020, 13, 4691–4704. [Google Scholar] [CrossRef]
- Ahmad, A.; Mrkvicova, A.; Chmelarova, M.; Peterova, E.; Havelek, R.; Baranova, I.; Kazimirova, P.; Rudolf, E.; Rezacova, M. The effect of sodium butyrate and cisplatin on expression of EMT markers. PLoS ONE 2019, 14, e0210889. [Google Scholar] [CrossRef]
- Shin, J.; Song, I.-S.; Pak, J.H.; Jang, S.-W. Upregulation of annexin A1 expression by butyrate in human melanoma cells induces invasion by inhibiting E-cadherin expression. Tumor Biol. 2016, 37, 14577–14584. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A new genomic blueprint of the human gut microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, B.; Wu, J.-T. Production of Natural Butylated Hydroxytoluene as an Antioxidant by Freshwater Phytoplankton1. J. Phycol. 2008, 44, 1447–1454. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [Green Version]
- Brunner, A.; Tzankov, A. The role of structural extracellular matrix proteins in urothelial bladder cancer (review). Biomark Insights 2007, 2, 418–427. [Google Scholar] [CrossRef]
- Alfano, M.; Canducci, F.; Nebuloni, M.; Clementi, M.; Montorsi, F.; Salonia, A. The interplay of extracellular matrix and microbiome in urothelial bladder cancer. Nat. Rev. Urol. 2015, 13, 77–90. [Google Scholar] [CrossRef]
- Soltermann, A.; Tischler, V.; Arbogast, S.; Braun, J.; Probst-Hensch, N.; Weder, W.; Moch, H.; Kristiansen, G. Prognostic significance of epithelial-mesenchymal and mesenchymal-epithelial transition protein expression in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 7430–7437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, A.A.; Elsayed, A.S.; Durrani, M.; Jing, Z.; Iqbal, U.; Gomez, E.C.; Singh, P.K.; Liu, S.; Smith, G.; Tang, L.; et al. Investigating the association between the urinary microbiome and bladder cancer: An exploratory study. Urol. Oncol. 2021, 39, 370.e9–370.e19. [Google Scholar] [CrossRef]
- Mansour, B.; Monyok, A.; Makra, N.; Gajdacs, M.; Vadnay, I.; Ligeti, B.; Juhasz, J.; Szabo, D.; Ostorhazi, E. Bladder cancer-related microbiota: Examining differences in urine and tissue samples. Sci. Rep. 2020, 10, 11042. [Google Scholar] [CrossRef] [PubMed]
- Neugent, M.L.; Hulyalkar, N.V.; Nguyen, V.H.; Zimmern, P.E.; De Nisco, N.J.; Garsin, D.A. Advances in Understanding the Human Urinary Microbiome and Its Potential Role in Urinary Tract Infection. mBio 2020, 11, e00218-20. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Wang, J.; Wang, J.; Li, Y. Estimate of the sequenced proportion of the global prokaryotic genome. Microbiome 2020, 8, 134. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.T.; Iyangar, A.S.; Reddy, R.; Chakladar, J.; Bhargava, V.; Sakamoto, K.; Ongkeko, W.M.; Rajasekaran, M. The Bladder Microbiome Is Associated with Epithelial–Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma. Cancers 2021, 13, 3649. https://doi.org/10.3390/cancers13153649
Li WT, Iyangar AS, Reddy R, Chakladar J, Bhargava V, Sakamoto K, Ongkeko WM, Rajasekaran M. The Bladder Microbiome Is Associated with Epithelial–Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma. Cancers. 2021; 13(15):3649. https://doi.org/10.3390/cancers13153649
Chicago/Turabian StyleLi, Wei Tse, Anjali S. Iyangar, Rohan Reddy, Jaideep Chakladar, Valmik Bhargava, Kyoko Sakamoto, Weg M. Ongkeko, and Mahadevan Rajasekaran. 2021. "The Bladder Microbiome Is Associated with Epithelial–Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma" Cancers 13, no. 15: 3649. https://doi.org/10.3390/cancers13153649
APA StyleLi, W. T., Iyangar, A. S., Reddy, R., Chakladar, J., Bhargava, V., Sakamoto, K., Ongkeko, W. M., & Rajasekaran, M. (2021). The Bladder Microbiome Is Associated with Epithelial–Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma. Cancers, 13(15), 3649. https://doi.org/10.3390/cancers13153649