
Ras Family of Small GTPases in CRC: New Perspectives for Overcoming Drug Resistance



Abstract
:Simple Summary
Abstract
1. Introduction
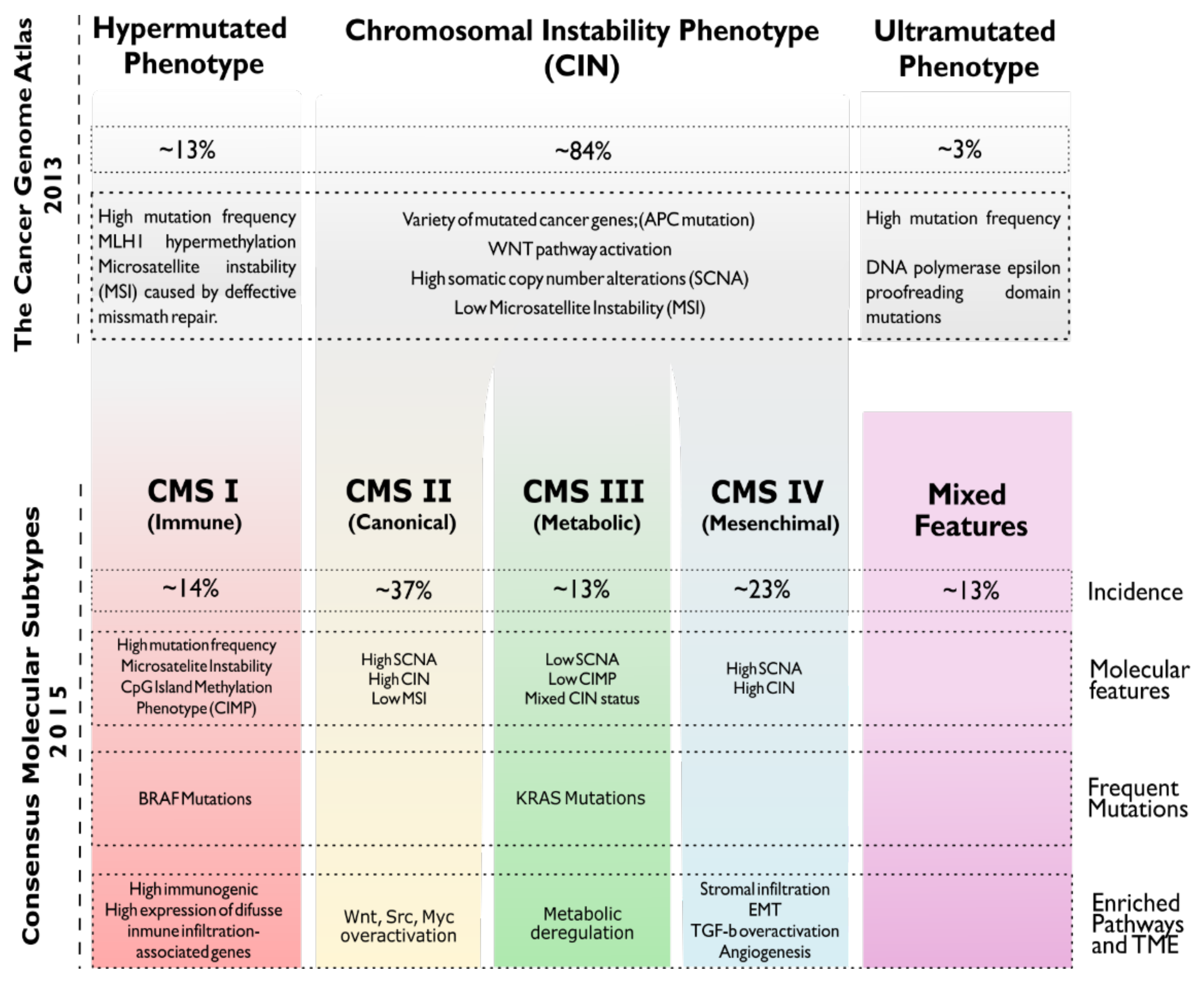
1.1. Colorectal Carcinogenesis
1.2. Standard of Care for Managing CRC Patients. Status of Targeted Therapies
2. Ras-GTPase Family
2.1. Regulation of Ras-GTPase Activity
2.1.1. Regulating GTP/GDP Exchange
2.1.2. Regulating Subcellular Location
3. The Role of Ras-GTPases on CRC
3.1. Status of Ras-Targeted Therapy
3.1.1. Targeting Ras-GTPase Location
3.1.2. Direct Targeting of Ras-GTPases
3.1.3. Targeting Ras Upstream Effectors
3.1.4. Targeting Ras Downstream Effectors
3.1.5. Targeting Ras-Mediated Metabolic Reprogramming
3.1.6. Novel Alternatives for Ras Targeted Therapies
3.2. Anti-Ras Strategies: A New Ally for Improving Current EFGR-Targeted Therapies?
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and Familial Colon Cancer. Gastrojournal 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [Green Version]
- Medina Pabón, M.A.; Babiker, H.M. A Review of Hereditary Colorectal Cancers; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Keum, N.N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Duong, H.Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy (review). Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Bolhaqueiro, A.C.F.; Ponsioen, B.; Bakker, B.; Klaasen, S.J.; Kucukkose, E.; van Jaarsveld, R.H.; Vivié, J.; Verlaan-Klink, I.; Hami, N.; Spierings, D.C.J.; et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 2019, 51, 824–834. [Google Scholar] [CrossRef]
- De Palma, F.D.E.; D’argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.C.; Salvatore, F. The molecular hallmarks of the serrated pathway in colorectal cancer. Cancers 2019, 11, 1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, Y.; Diaz-Meco, M.T.; Moscat, J. Serrated Colorectal Cancer: The Road Less Travelled? Trends Cancer 2019, 5, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef]
- Rustgi, A.K. BRAF: A Driver of the Serrated Pathway in Colon Cancer. Cancer Cell 2013, 24, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.L.; Zhao, W.; Leung, S.Y.; Yuen, S.T. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003, 63, 4878–4881. [Google Scholar] [PubMed]
- Werner, J.; Heinemann, V. Standards and Challenges of Care for Colorectal Cancer Today. Visc. Med. 2016, 32, 156–157. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakedis, J.; Schmidt, C.R. Surgical Treatment of Metastatic Colorectal Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Venook, A. Critical Evaluation of Current Treatments in Metastatic Colorectal Cancer. Oncologist 2005, 10, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D.; The ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii1–iii9. [Google Scholar] [CrossRef]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Linnekamp, J.F.; Wang, X.; Medema, J.P.; Vermeulen, L. Colorectal cancer heterogeneity and targeted therapy: A case for molecular disease subtypes. Cancer Res. 2015, 75, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, M.A.; Bosch, L.J.W.; Bounova, G.; Bolijn, A.S.; Delis-van Diemen, P.M.; Rausch, C.; Hoogstrate, Y.; Stubbs, A.P.; de Jong, M.; Jenster, G.; et al. Consensus molecular subtype classification of colorectal adenomas. J. Pathol. 2018, 246, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [PubMed]
- Luu, C.; Arrington, A.K.; Schoellhammer, H.F.; Singh, G.; Kim, J. Targeted therapies in colorectal cancer: Surgical considerations. J. Gastrointest. Oncol. 2013, 4, 328–336. [Google Scholar]
- Mody, K.; Baldeo, C.; Bekaii-Saab, T. Antiangiogenic therapy in colorectal cancer. Cancer J. 2018, 24, 165–170. [Google Scholar] [CrossRef]
- McCormack, P.L.; Keam, S.J. Bevacizumab: A review of its use in metastatic colorectal cancer. Drugs 2008, 68, 487–506. [Google Scholar] [CrossRef] [PubMed]
- García-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, S.M.; Lee, J.; Kim, S.T.; Hur, J.Y.; Ebert, P.J.; Calley, J.N.; Wulur, I.H.; Gopalappa, T.; Wong, S.S.; Qian, H.R.; et al. Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci. Rep. 2019, 9, 15365. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Dominguez, N.; Parnell, C.; Teng, Y. Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells 2019, 8, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.J.; Li, J.A.; Bai, D.M.; Song, Y. miR-223-RhoB signaling pathway regulates the proliferation and apoptosis of colon adenocarcinoma. Chem. Biol. Interact. 2018, 289, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kinbara, K.; Goldfinger, L.E.; Hansen, M.; Chou, F.L.; Ginsberg, M.H. Ras GTPases: Integrins’ friends or foes? Nat. Rev. Mol. Cell Biol. 2003, 4, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Bilan, P.J.; Moyers, J.S.; Kahn, C.R. The Ras-related protein Rad associates with the cytoskeleton in a non- lipid-dependent manner. Exp. Cell Res. 1998, 242, 391–400. [Google Scholar] [CrossRef]
- Correll, R.N.; Pang, C.; Niedowicz, D.M.; Finlin, B.S.; Andres, D.A. The RGK family of GTP-binding proteins: Regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell. Signal. 2008, 20, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, P.; Adam, S.; Kendall, S.; Lim, K.-H.; Counter, C.; Tyler, D. Defining a role for RalA and RalB in melanoma tumorigenesis and metastasis. Cancer Res. 2008, 68, 5244. [Google Scholar]
- Neel, N.F.; Stratford, J.K.; Shinde, V.; Ecsedy, J.A.; Martin, T.D.; Der, C.J.; Yeh, J.J. Response to MLN8237 in pancreatic cancer is not dependent on RalA phosphorylation. Mol. Cancer Ther. 2013, 13, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipfel, P.A.; Brady, D.C.; Kashatus, D.F.; Ancrile, B.D.; Tyler, D.S.; Counter, C.M. Ral activation promotes melanomagenesis. Oncogene 2010, 29, 4859–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Owens, C.; Chandra, N.; Conaway, M.R.; Brautigan, D.L.; Theodorescu, D. Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 2010, 70, 8760–8769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoroghi, S.; Mary, B.; Larnicol, A.; Asokan, N.; Klein, A.; Osmani, N.; Busnelli, I.; Delalande, F.; Paul, N.; Halary, S.; et al. Ral GTPases promote breast cancer metastasis by controlling biogenesis and organ targeting of exosomes. eLife 2021, 10, e61539. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Chuang, H.C.; Huang, C.C.; Fang, F.M.; Huang, H.Y.; Tsai, H.T.; Su, L.J.; Shiu, L.Y.; Leu, S.; Chien, C.Y. Overexpression of rap-1A indicates a poor prognosis for oral cavity squamous cell carcinoma and promotes tumor cell invasion via aurora-A modulation. Am. J. Pathol. 2013, 182, 516–528. [Google Scholar] [CrossRef]
- Liu, M.; Banerjee, R.; Rossa, C.; D’Silva, N.J. RAP1-RAC1 Signaling Has an Important Role in Adhesion and Migration in HNSCC. J. Dent. Res. 2020, 99, 959–968. [Google Scholar] [CrossRef]
- McSherry, E.A.; Brennan, K.; Hudson, L.; Hill, A.D.K.; Hopkins, A.M. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res. 2011, 13, R31. [Google Scholar] [CrossRef] [Green Version]
- Di, J.; Huang, H.; Qu, D.; Tang, J.; Cao, W.; Lu, Z.; Cheng, Q.; Yang, J.; Bai, J.; Zhang, Y.; et al. Rap2B promotes proliferation, migration, and invasion of human breast cancer through calcium-related ERK1/2 signaling pathway. Sci. Rep. 2015, 5, 12363. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yan, X.; Wu, D.; Yang, Y.; Li, M.; Su, Y.; Yang, W.; Shan, Z.; Gao, Y.; Jin, Z. High expression of Ras-related protein 1A promotes an aggressive phenotype in colorectal cancer via PTEN/FOXO3/CCND1 pathway. J. Exp. Clin. Cancer Res. 2018, 37, 178. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, H.; Duan, Y.; Liu, B. MicroRNA.101 suppresses colorectal cancer progression by negative regulation of Rap1b. Oncol. Lett. 2020, 20, 2225–2231. [Google Scholar] [CrossRef]
- Sayyah, J.; Bartakova, A.; Nogal, N.; Quilliam, L.A.; Stupack, D.G.; Brown, J.H. The Ras-related protein, Rap1A, mediates thrombin-stimulated, integrin-dependent glioblastoma cell proliferation and tumor growth. J. Biol. Chem. 2014, 289, 17689–17698. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Lan, X.; Shi, X.; Zhao, K.; Wang, D.; Wang, X.; Li, F.; Huang, H.; Liu, J. Cytoplasmic RAP1 mediates cisplatin resistance of non-small cell lung cancer. Cell Death Dis. 2017, 8, e2803. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Fang, X.; He, C.; Hu, X. The mechanisms of DIRAS family members in role of tumor suppressor. J. Cell. Physiol. 2019, 234, 5564–5577. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jin, L.; Liu, G.; Wang, P.; Sun, Z.; Cao, Y.; Shi, H.; Liu, X.; Shi, Q.; Zhou, X.; et al. Overexpression of RASD1 inhibits glioma cell migration/invasion and inactivates the AKT/mTOR signaling pathway. Sci. Rep. 2017, 7, 3202. [Google Scholar] [CrossRef] [Green Version]
- Louro, R.; Nakaya, H.I.; Paquola, A.C.M.; Martins, E.A.L.; Da Silva, A.M.; Verjovski-Almeida, S.; Reis, E.M. RASL11A, member of a novel small monomeric GTPase gene family, is down-regulated in prostate tumors. Biochem. Biophys. Res. Commun. 2004, 316, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ma, N.; Wang, S.; Mo, Y.; Zhang, Z.; Huang, G.; Midorikawa, K.; Hiraku, Y.; Oikawa, S.; Murata, M.; et al. RERG suppresses cell proliferation, migration and angiogenesis through ERK/NF-κB signaling pathway in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 1–15. [Google Scholar]
- Patel, P.H.; Thapar, N.; Guo, L.; Martinez, M.; Maris, J.; Gau, C.L.; Lengyel, J.A.; Tamanoi, F. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J. Cell Sci. 2003, 116, 3601–3610. [Google Scholar] [CrossRef] [Green Version]
- Campos, T.; Ziehe, J.; Palma, M.; Escobar, D.; Tapia, J.C.; Pincheira, R.; Castro, A.F. Rheb promotes cancer cell survival through p27Kip1-dependent activation of autophagy. Mol. Carcinog. 2016, 55, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Shen, L.; Li, F.; Yang, J.; Wan, X.; Ouyang, M. Silencing of RHEB inhibits cell proliferation and promotes apoptosis in colorectal cancer cells via inhibition of the mTOR signaling pathway. J. Cell. Physiol. 2020, 235, 442–453. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.X.; Cai, W.; Andres, D.A. Rit subfamily small GTPases: Regulators in neuronal differentiation and survival. Cell. Signal. 2013, 25, 2060–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuichet, K.; Søgaard-Andersen, L. Evolution and diversity of the ras superfamily of small GTPases in Prokaryotes. Genome Biol. Evol. 2014, 7, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, K.F.; Baron, R.; Ali, B.R.; Magee, A.I.; Seabra, M.C. Rab GTPases Containing a CAAX Motif Are Processed Post-geranylgeranylation by Proteolysis and Methylation. J. Biol. Chem. 2006, 282, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2012, 13, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hancock, J.F. A novel prenyl-polybasic domain code determines lipid-binding specificity of the K-Ras membrane anchor. Small GTPases 2020, 11, 220–224. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Cherfils, J.; Zeghouf, M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casalou, C.; Ferreira, A.; Barral, D.C. The Role of ARF Family Proteins and Their Regulators and Effectors in Cancer Progression: A Therapeutic Perspective. Front. Cell Dev. Biol. 2020, 8, 217. [Google Scholar] [CrossRef] [Green Version]
- Boyartchuk, V.L.; Ashby, M.N.; Rine, J. Modulation of ras and a-factor function by carboxyl-terminal proteolysis. Science 1997, 275, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Freije, J.M.P.; Blay, P.; Pendás, A.M.; Cadiñanos, J.; Crespo, P.; López-Otín, C. Identification and chromosomal location of two human genes encoding enzymes potentially involved in proteolytic maturation of farnesylated proteins. Genomics 1999, 58, 270–280. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I.H. An acylation cycle regulates localization and activity of palmitoylated ras isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Campi, G.; Du, G.; Zheng, Y.; Foster, D.A.; Dustin, M.L.; Philips, M.R. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat. Cell Biol. 2007, 9, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; Rocks, O.; Vartak, N.; Menninger, S.; Hedberg, C.; Balamurugan, R.; Wetzel, S.; Renner, S.; Gerauer, M.; Schölermann, B.; et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 2010, 6, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Uechi, Y.; Bayarjargal, M.; Umikawa, M.; Oshiro, M.; Takei, K.; Yamashiro, Y.; Asato, T.; Endo, S.; Misaki, R.; Taguchi, T.; et al. Rap2 function requires palmitoylation and recycling endosome localization. Biochem. Biophys. Res. Commun. 2009, 378, 732–737. [Google Scholar] [CrossRef]
- Gentry, L.R.; Nishimura, A.; Cox, A.D.; Martin, T.D.; Tsygankov, D.; Nishida, M.; Elston, T.C.; Der, C.J. Divergent roles of CAAX motif-signaled posttranslational modifications in the regulation and subcellular localization of Ral GTPases. J. Biol. Chem. 2015, 290, 22851–22861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Nakagawa, M.; Young, S.G.; Yamanaka, S. Differential Membrane Localization of ERas and Rheb, Two Ras-related Proteins Involved in the Phosphatidylinositol 3-Kinase/mTOR Pathway. J. Biol. Chem. 2005, 280, 32768–32774. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; Mccormick, F. RAS Proteins and Their Regulators in Human Disease HHS Public Access. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Quatela, S.E.; Sung, P.J.; Ahearn, I.M.; Bivona, T.G.; Philips, M.R. Analysis of K-Ras Phosphorylation, Translocation, and Induction of Apoptosis. Methods Enzymol. 2008, 439, 87–102. [Google Scholar]
- Barceló, C.; Paco, N.; Morell, M.; Alvarez-Moya, B.; Bota-Rabassedas, N.; Jaumot, M.; Vilardell, F.; Capella, G.; Agell, N. Phosphorylation at ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014, 74, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.-H.; Brady, D.C.; Kashatus, D.F.; Ancrile, B.B.; Der, C.J.; Cox, A.D.; Counter, C.M. Aurora-A Phosphorylates, Activates, and Relocalizes the Small GTPase RalA. Mol. Cell. Biol. 2010, 30, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Angarola, B.; Ferguson, S.M. Weak membrane interactions allow Rheb to activate mTORC1 signaling without major lysosome enrichment. Mol. Biol. Cell 2019, 30, 2750–2760. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. Ras interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.-J.; Ro, E.J.; Choi, K.-Y. Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. NPJ Precis. Oncol. 2018, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Gao, D.; He, T.; Zhang, M.; Zhang, X.; Linghu, E.; Wei, L.; Guo, M. Methylation of DIRAS1 promotes colorectal cancer progression and may serve as a marker for poor prognosis. Clin. Epigenet. 2017, 9, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Neel, N.F.; Martin, T.D.; Stratford, J.K.; Zand, T.P.; Reiner, D.J.; Der, C.J. The RalGEF-ral effector signaling network: The road less traveled for anti-ras drug discovery. Genes Cancer 2011, 2, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Guin, S.; Theodorescu, D. The RAS-RAL axis in cancer: Evidence for mutationspecific selectivity in non-small cell lung cancer. Acta Pharmacol. Sin. 2015, 36, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Male, H.; Patel, V.; Jacob, M.A.; Borrego-Diaz, E.; Wang, K.; Young, D.A.; Wise, A.L.; Huang, C.; Van Veldhuizen, P.; O’Brien-Ladner, A.; et al. Inhibition of RalA signaling pathway in treatment of non-small cell lung cancer. Lung Cancer 2012, 77, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Rybko, V.A.; Knizhnik, A.V.; Komelkov, A.V.; Aushev, V.N.; Trukhanova, L.S.; Tchevkina, E.M. Different metastasis promotive potency of small G-proteins RalA and RalB in in vivo hamster tumor model. Cancer Cell Int. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Stelniec-Klotz, I.; Sigler, C.; Kasack, K.; Redmer, T.; Qian, Y.; Schäfer, R. Effects of RAL signal transduction in KRAS-and BRAF-mutated cells and prognostic potential of the RAL signature in colorectal cancer. Oncotarget 2015, 6, 13334–13346. [Google Scholar] [CrossRef] [Green Version]
- Khawaja, H.; Campbell, A.; Roberts, J.Z.; Javadi, A.; O’Reilly, P.; McArt, D.; Allen, W.L.; Majkut, J.; Rehm, M.; Bardelli, A.; et al. RALB GTPase: A critical regulator of DR5 expression and TRAIL sensitivity in KRAS mutant colorectal cancer. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. mTOR pathway in colorectal cancer: An update. Oncotarget 2014, 5, 49–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [Green Version]
- To, J.Y.; Smrcka, A.V. Activated heterotrimeric G protein αi subunits inhibit Rap-dependent cell adhesion and promote cell migration. J. Biol. Chem. 2018, 293, 1570–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, C.; Serpente, N.; Cogram, P.; Hosking, C.R.; Bialucha, C.U.; Feller, S.M.; Braga, V.M.M.; Birchmeier, W.; Fujita, Y. Rap1 Regulates the Formation of E-Cadherin-Based Cell-Cell Contacts. Mol. Cell. Biol. 2004, 24, 6690–6700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90–99. [Google Scholar]
- Freeman, S.A.; McLeod, S.J.; Dukowski, J.; Austin, P.; Lee, C.C.Y.; Millen-Martin, B.; Kubes, P.; McCafferty, D.M.; Gold, M.R.; Roskelley, C.D. Preventing the activation or cycling of the Rap1 GTPase alters adhesion and cytoskeletal dynamics and blocks metastatic melanoma cell extravasation into the lungs. Cancer Res. 2010, 70, 4590–4601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Feng, Y.; Bowers, R.; Becker-Hapak, M.; Gardner, J.; Council, L.; Linette, G.; Zhao, H.; Cornelius, L.A. Ras-associated protein-1 regulates extracellular signal-regulated kinase activation and migration in melanoma cells: Two processes important to melanoma tumorigenesis and metastasis. Cancer Res. 2006, 66, 7880–7888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M.; Mitra, R.S.; Liu, M.; Lee, J.; Henson, B.S.; Carey, T.; Bradford, C.; Prince, M.; Wang, C.Y.; Fearon, E.R.; et al. Rap1 stabilizes β-catenin and enhances β-catenin-dependent transcription and invasion in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2010, 16, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Anand, S.; Murphy, E.A.; Desgrosellier, J.S.; Stupack, D.G.; Shattil, S.J.; Schlaepfer, D.D.; Cheresh, D.A. EGFR-dependent pancreatic carcinoma cell metastasis through Rap1 activation. Oncogene 2012, 31, 2783–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, K.; Ye, L.; Toms, A.M.; Hargest, R.; Martin, T.A.; Ruge, F.; Ji, J.; Jiang, W.G. Expression of signal-induced proliferation-associated gene 1 (SIPA1), a RapGTPase-activating protein, is increased in colorectal cancer and has diverse effects on functions of colorectal cancer cells. Cancer Genom. Proteom. 2012, 9, 321–328. [Google Scholar]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Wang, J.; Yao, X.; Huang, J. New tricks for human farnesyltransferase inhibitor: Cancer and beyond. Medchemcomm 2017, 8, 841–854. [Google Scholar] [CrossRef]
- Rao, S.; Cunningham, D.; de Gramont, A.; Scheithauer, W.; Smakal, M.; Humblet, Y.; Kourteva, G.; Iveson, T.; Andre, T.; Dostalova, J.; et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J. Clin. Oncol. 2004, 22, 3950–3957. [Google Scholar] [CrossRef]
- Hanrahan, E.O.; Kies, M.S.; Glisson, B.S.; Khuri, F.R.; Feng, L.; Tran, H.T.; Ginsberg, L.E.; Truong, M.T.; Hong, W.K.; Kim, E.S. A phase II study of lonafarnib (SCH66336) in patients with chemorefractory, advanced squamous cell carcinoma of the head and neck. Am. J. Clin. Oncol. Cancer Clin. Trials 2009, 32, 274–279. [Google Scholar] [CrossRef]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Zonder, J.A.; Shields, A.F.; Zalupski, M.; Chaplen, R.; Heilbrun, L.K.; Arlauskas, P.; Philip, P.A. A Phase II trial of bryostatin 1 in the treatment of metastatic colorectal cancer. Clin. Cancer Res. 2001, 7, 38–42. [Google Scholar] [PubMed]
- Morgan, M.A.; Onono, F.O.; Spielmann, H.P.; Subramanian, T.; Scherr, M.; Venturini, L.; Dallmann, I.; Ganser, A.; Reuter, C.W.M. Modulation of anthracycline-induced cytotoxicity by targeting the prenylated proteome in myeloid leukemia cells. J. Mol. Med. 2012, 90, 149–161. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Baron, R.A.; Wong, W.; Dela Cruz, J.; York, J.D.; Gooden, D.M.; Bergo, M.O.; Young, S.G.; Toone, E.J.; Casey, P.J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4336–4341. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.Y.; Ramanujulu, P.M.; Guo, D.; Yang, T.; Wirawan, M.; Casey, P.J.; Go, M.L.; Wang, M. An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol. Ther. 2014, 15, 1280–1291. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.L.; Luo, L.X.; Li, Y.; Liu, Z.; Li, L.L.; Shi, D.F.; Xie, Y.; Huang, M.; Lu, L.L.; Duan, F.G.; et al. Identification of a new inhibitor of KRAS-PDEδ interaction targeting KRAS mutant nonsmall cell lung cancer. Int. J. Cancer 2019, 145, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.H.; Truxius, D.C.; Vogel, H.A.; Harizanova, J.; Murarka, S.; Martín-Gago, P.; Bastiaens, P.I.H. PDEδ inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic KRas. Int. J. Cancer 2019, 144, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martín-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Mologni, L.; Brussolo, S.; Ceccon, M.; Gambacorti-Passerini, C. Synergistic Effects of Combined Wnt/KRAS Inhibition in Colorectal Cancer Cells. PLoS ONE 2012, 7, 51449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makovski, V.; Jacob-Hirsch, J.; Gefen-Dor, C.; Shai, B.; Ehrlich, M.; Rechavi, G.; Kloog, Y. Analysis of gene expression array in TSC2-deficient AML cells reveals IRF7 as a pivotal factor in the Rheb/mTOR pathway. Cell Death Dis. 2014, 5, e1557. [Google Scholar] [CrossRef] [Green Version]
- Furuse, J.; Kurata, T.; Okano, N.; Fujisaka, Y.; Naruge, D.; Shimizu, T.; Kitamura, H.; Iwasa, T.; Nagashima, F.; Nakagawa, K. An early clinical trial of Salirasib, an oral RAS inhibitor, in Japanese patients with relapsed/refractory solid tumors. Cancer Chemother. Pharmacol. 2018, 82, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, C.R.; Blackhall, F.H. Direct Ras G12C inhibitors: Crossing the rubicon. Br. J. Cancer 2019, 121, 197–198. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef]
- Ryan, M.B.; de la Cruz, F.F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to KrasG12C inhibition. Clin. Cancer Res. 2020, 26, 1617–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Liu, D.; Li, L.; Wempe, M.F.; Guin, S.; Khanna, M.; Meier, J.; Hoffman, B.; Owens, C.; Wysoczynski, C.L.; et al. Discovery and characterization of small molecules that target the GTPase Ral. Nature 2014, 515, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Jang, H.; Zhang, J.; Nussinov, R. Inhibitors of Ras-SOS Interactions. ChemMedChem 2016, 11, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Gmachl, M.; Ramharter, J.; Teh, J.; Fu, S.-C.; Trapani, F.; Kessler, D.; Rumpel, K.; Botesteanu, D.-A.; Ettmayer, P.; et al. Abstract 1091: BI-3406 and BI 1701963: Potent and selective SOS1::KRAS inhibitors induce regressions in combination with MEK inhibitors or irinotecan. In Proceedings of the AACR Annual Meeting 2020, Philadelphia, PA, USA, 22–24 June 2020; Volume 80, p. 1091. [Google Scholar]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. Bi-3406, a potent and selective sos1–kras interaction inhibitor, is effective in kras-driven cancers through combined mek inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Inchanalkar, S.; Deshpande, N.U.; Kasherwal, V.; Jayakannan, M.; Balasubramanian, N. Polymer Nanovesicle-Mediated Delivery of MLN8237 Preferentially Inhibits Aurora Kinase A to Target RalA and Anchorage-Independent Growth in Breast Cancer Cells. Mol. Pharm. 2018, 15, 3046–3059. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.; Bosch, L.J.W.; Martens-De Kemp, S.R.; Carvalho, B.; Sillars-Hardebol, A.H.; Dobson, R.J.; De Rinaldis, E.; Meijer, G.A.; Abeln, S.; Heringa, J.; et al. Aurora kinase A (AURKA) interaction with Wnt and Ras-MAPK signalling pathways in colorectal cancer. Sci. Rep. 2018, 8, 7522. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, T.; Izawa, N.; Nakajima, T.E.; Sunakawa, Y. Targeting EGFR and RAS/RAF Signaling in the Treatment of Metastatic Colorectal Cancer: From Current Treatment Strategies to Future Perspectives. Drugs 2019, 79, 633–645. [Google Scholar] [CrossRef]
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Cappellini, G.C.A.; D’Atri, S. Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating effect of dabrafenib on the invasive behavior of melanoma cells with acquired resistance to the BRAF inhibitor. Int. J. Oncol. 2016, 49, 1164–1174. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Bradley, W.D.; Lee, R.J.; Schostack, K.; Simcox, M.E.; Kopetz, S.; Heimbrook, D.; et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012, 72, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, L.; Barlesi, F.; Martinez-Garcia, M.; Dieras, V.; Schellens, J.H.M.; Spano, J.P.; Middleton, M.R.; Calvo, E.; Paz-Ares, L.; Larkin, J.; et al. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin. Cancer Res. 2014, 20, 4251–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaikuad, A.; Tacconi, E.M.C.; Zimmer, J.; Liang, Y.; Gray, N.S.; Tarsounas, M.; Knapp, S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S.; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, e92352. [Google Scholar] [CrossRef]
- Boga, S.B.; Deng, Y.; Zhu, L.; Nan, Y.; Cooper, A.B.; Shipps, G.W.; Doll, R.; Shih, N.Y.; Zhu, H.; Sun, R.; et al. MK-8353: Discovery of an Orally Bioavailable Dual Mechanism ERK Inhibitor for Oncology. ACS Med. Chem. Lett. 2018, 9, 761–767. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Soria, J.-C.; Sharma, S.; Banerji, U.; Azaro, A.; Desai, J.; Ringeisen, F.P.; Kaag, A.; Radhakrishnan, R.; Hourcade-Potelleret, F.; et al. A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. J. Clin. Oncol. 2014, 32, 9051. [Google Scholar] [CrossRef]
- Shapiro, G.I.; LoRusso, P.; Kwak, E.; Pandya, S.; Rudin, C.M.; Kurkjian, C.; Cleary, J.M.; Pilat, M.J.; Jones, S.; de Crespigny, A.; et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Dai, J.; Kang, Z.; Yang, T.; Zhao, Q.; Zheng, J.; Zhang, X.; Zhang, J.; Xu, J.; Sun, G.; et al. A combinatorial strategy for overcoming primary and acquired resistance of MEK inhibition in colorectal cancer. Exp. Cell Res. 2020, 393, 112060. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Moore, C.; Rana, S.; Van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11, eaaw7999. [Google Scholar] [CrossRef]
- Kawada, K.; Toda, K.; Sakai, Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int. J. Clin. Oncol. 2017, 22, 651–659. [Google Scholar] [CrossRef]
- Nowak, E.M.; Poczeta, M.; Bieg, D.; Bednarek, I. DNA methyltransferase inhibitors influence on the DIRAS3 and STAT3 expression and in vitro migration of ovarian and breast cancer cells. Ginekol. Pol. 2017, 88, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226. [Google Scholar] [CrossRef]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef]
- Cenigaonandia-Campillo, A.; Serna-Blasco, R.; Gómez-Ocabo, L.; Solanes-Casado, S.; Baños-Herraiz, N.; Puerto-Nevado, L.D.; Cañas, J.A.; Aceñero, M.J.; García-Foncillas, J.; Aguilera, Ó. Vitamin C activates pyruvate dehydrogenase (PDH) targeting the mitochondrial tricarboxylic acid (TCA) cycle in hypoxic KRAS mutant colon cancer. Theranostics 2021, 11, 3595–3606. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhove, K.; Derveaux, E.; Graulus, G.-J.; Mesotten, L.; Thomeer, M.; Noben, J.-P.; Guedens, W.; Adriaensens, P. Molecular Sciences Glutamine Addiction and Therapeutic Strategies in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 252. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; Van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef]
- Miyo, M.; Konno, M.; Nishida, N.; Sueda, T.; Noguchi, K.; Matsui, H.; Colvin, H.; Kawamoto, K.; Koseki, J.; Haraguchi, N.; et al. Metabolic Adaptation to Nutritional Stress in Human Colorectal Cancer. Sci. Rep. 2016, 6, 38415. [Google Scholar] [CrossRef] [Green Version]
- Toda, K.; Nishikawa, G.; Iwamoto, M.; Itatani, Y.; Takahashi, R.; Sakai, Y.; Kawada, K. Clinical role of ASCT2 (SLC1A5) in KRAS-mutated colorectal cancer. Int. J. Mol. Sci. 2017, 18, 1632. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Li, X.; Lu, Y.; Qiu, S.; Fan, Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Lett. 2016, 381, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Toda, K.; Kawada, K.; Iwamoto, M.; Inamoto, S.; Sasazuki, T.; Shirasawa, S.; Hasegawa, S.; Sakai, Y. Metabolic Alterations Caused by KRAS Mutations in Colorectal Cancer Contribute to Cell Adaptation to Glutamine Depletion by Upregulation of Asparagine Synthetase. Neoplasia 2016, 18, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Hahn, W.C. Synthetic lethal vulnerabilities in kras-mutant cancers. Cold Spring Harb. Perspect. Med. 2018, 8, a031518. [Google Scholar] [CrossRef]
- Ku, A.A.; Hu, H.M.; Zhao, X.; Shah, K.N.; Kongara, S.; Wu, D.; McCormick, F.; Balmain, A.; Bandyopadhyay, S. Integration of multiple biological contexts reveals principles of synthetic lethality that affect reproducibility. Nat. Commun. 2020, 11, 2375. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A Genome-wide RNAi Screen Identifies Multiple Synthetic Lethal Interactions with the Ras Oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hu, K.; Guo, J.; Cheng, F.; Lv, J.; Jiang, W.; Lu, W.; Liu, J.; Pang, X.; Liu, M. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat. Commun. 2016, 7, 11363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarthy, A.V.; Morgan-Lappe, S.E.; Zakula, D.; Vernetti, L.; Schurdak, M.; Packer, J.C.L.; Anderson, M.G.; Shirasawa, S.; Sasazuki, T.; Fesik, S.W. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol. Cancer Ther. 2007, 6, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ngo, V.N.; Marani, M.; Yang, Y.; Wright, G.; Staudt, L.M.; Downward, J. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene 2010, 29, 4658–4670. [Google Scholar] [CrossRef]
- Steckel, M.; Molina-Arcas, M.; Weigelt, B.; Marani, M.; Warne, P.H.; Kuznetsov, H.; Kelly, G.; Saunders, B.; Howell, M.; Downward, J.; et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012, 22, 1227–1245. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012, 148, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Gu, L.; Liao, S.; Zheng, Y.; Zhang, S.; Cao, Y.; Zhang, J.; Wang, Y. NG25, a novel inhibitor of TAK1, suppresses KRAS-mutant colorectal cancer growth in vitro and in vivo. Apoptosis 2019, 24, 83–94. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Cheng, K.A.; Hata, A.N.; Faber, A.C.; Ebi, H.; Coffee, E.M.; Greninger, P.; Brown, R.D.; Godfrey, J.T.; Cohoon, T.J.; et al. Synthetic Lethal Interaction of Combined BCL-XL and MEK Inhibition Promotes Tumor Regressions in KRAS Mutant Cancer Models. Cancer Cell 2013, 23, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Costa-Cabral, S.; Brough, R.; Konde, A.; Aarts, M.; Campbel, J.; Marinari, E.; Riffell, J.; Bardelli, A.; Torrance, C.; Lord, C.J.; et al. CDK1 Is a synthetic lethal target for KRAS mutant tumours. PLoS ONE 2016, 11, e0149099. [Google Scholar]
- Lamba, S.; Russo, M.; Sun, C.; Lazzari, L.; Cancelliere, C.; Grernrum, W.; Lieftink, C.; Bernards, R.; DiNicolantonio, F.; Bardelli, A. RAF Suppression Synergizes with MEK Inhibition in KRAS Mutant Cancer Cells. Cell Rep. 2014, 8, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crunkhorn, S. Cancer: RNA-based approaches target KRAS. Nat. Rev. Drug Discov. 2017, 16, 529. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Yao, J.; Xie, J.; Xie, B.; Li, Y.; Jiang, L.; Sui, X.; Zhou, X.; Pan, H.; Han, W. Therapeutic effect of hydroxychloroquine on colorectal carcinogenesis in experimental murine colitis. Biochem. Pharmacol. 2016, 115, 51–63. [Google Scholar] [CrossRef]
- O’Hara, M.H.; Karasic, T.B.; Vasilevskaya, I.; Redlinger, M.; Loaiza-Bonilla, A.; Teitelbaum, U.R.; Giantonio, B.J.; Damjanov, N.; Reiss, K.A.; Rosen, M.A.; et al. Phase II trial of the autophagy inhibitor hydroxychloroquine with FOLFOX and bevacizumab in front line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2017, 35, 3545. [Google Scholar] [CrossRef]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The nrf2, thioredoxin, and glutathione system in tumorigenesis and anticancer therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cort, A.; Ozben, T.; Saso, L.; De Luca, C.; Korkina, L. Redox Control of Multidrug Resistance and Its Possible Modulation by Antioxidants. Oxid. Med. Cell. Longev. 2016, 2016, 4251912. [Google Scholar] [CrossRef] [Green Version]
- Montazami, N.; Kheirandish, M.; Majidi, J.; Yousefi, M.; Yousefi, B.; Mohamadnejad, L.; Shanebandi, D.; Estiar, M.A.; Khaze, V.; Mansoori, B.; et al. siRNA-mediated silencing of MDR1 reverses the resistance to oxaliplatin in SW480/OxR colon cancer cells. Cell. Mol. Biol. 2015, 61, 98–103. [Google Scholar]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1α Inhibition Reverses Multidrug Resistance in Colon Cancer Cells via Downregulation of MDR1/P-Glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar]
- Jiang, Y.; Li, S.; Yang, H.; Wu, C.H.; Liu, Y.Y. RalA regulates ROS and ATP production in cancer cells by association with cavelolin-1. Yiyong Shengwu Lixue/J. Med. Biomech. 2017, 32, 458–463. [Google Scholar]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Bebber, C.M.; Müller, F.; Clemente, L.P.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Oh, J.; Kim, M.; Jin, E.-J. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim. Cell. Syst. 2018, 22, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitroulakos, J.; Lorimer, I.A.; Goss, G.; Lynch, T.; Heymach, J.; Eisen, T.; Settleman, J.; Bunn, P.; Jänne, P. Strategies to enhance epidermal growth factor inhibition: Targeting the mevalonate pathway. Clin. Cancer Res. 2006, 12, 4426s–4431s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtkowiak, J.W.; Gibbs, R.A.; Mattingly, R.R. Working together: Farnesyl transferase inhibitors and statins block protein prenylation. Mol. Cell. Pharmacol. 2009, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Baas, J.M.; Krens, L.L.; Ten Tije, A.J.; Erdkamp, F.; Van Wezel, T.; Morreau, H.; Gelderblom, H.; Guchelaar, H.J. Safety and efficacy of the addition of simvastatin to cetuximab in previously treated KRAS mutant metastatic colorectal cancer patients. Investig. New Drugs 2015, 33, 1242–1247. [Google Scholar] [CrossRef] [Green Version]
- McFall, T.; Trogdon, M.; Sisk-Hackworth, L.; Stites, E.C. Inhibition of both mutant and wild-type RAS-GTP in KRAS G12C colorectal cancer through cotreatment with G12C and EGFR inhibitors. bioRxiv 2019, 845263. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Reinmuth, N.; Liu, W.; Ahmad, S.A.; Fan, F.; Stoeltzing, O.; Parikh, A.A.; Bucana, C.D.; Gallick, G.E.; Nickols, M.A.; Westlin, W.F.; et al. αvβ3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res. 2003, 63, 2079–2087. [Google Scholar] [PubMed]
- Vonlaufen, A.; Wiedle, G.; Borisch, B.; Birrer, S.; Luder, P.; Imhof, B.A. Integrin αvβ3 expression in colon carcinoma correlates with survival. Mod. Pathol. 2001, 14, 1126–1132. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β 3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, S.; Jiang, Z.; Liu, Z.; Kaur, K.; Wang, X. The role of EGFR monoclonal antibodies (MoABs) cetuximab/panitumab, and BRAF inhibitors in BRAF mutated colorectal cancer. J. Gastrointest. Oncol. 2013, 4, 72–81. [Google Scholar]
- Martinelli, E.; Troiani, T.; Morgillo, F.; Rodolico, G.; Vitagliano, D.; Morelli, M.P.; Tuccillo, C.; Vecchione, L.; Capasso, A.; Orditura, M.; et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin. Cancer Res. 2010, 16, 4990–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galal, K.M.; Khaled, Z.; Mourad, A.M.M. Role of cetuximab and sorafenib in treatment of metastatic colorectal cancer. Indian J. Cancer 2011, 48, 47–54. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and Acquired Resistance of Colorectal Cancer Cells to Anti-EGFR Antibodies Converge on MEK/ERK Pathway Activation and Can Be Overcome by Combined MEK/EGFR Inhibition. Clin. Cancer Res. 2014, 20, 3775–3786. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Pang, N.; Wan, T.; Huang, Y.; Wei, T.; Jiang, X.; Zhou, Y.; Huang, Y.; Yang, H.; Zhang, Z.; et al. Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis. J. Cancer 2019, 10, 757. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, Y.; Pan, T.; Fan, Z. Roles of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy 2010, 6, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the Beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeli, J.P.F.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ras Subfamilies | Main Functions | Members | Implications in Cancer | Refs |
|---|---|---|---|---|
| RGK | Cytoskeleton remodelling Voltage-dependent Ca-Channels Insulin-induced glucose uptake | REM1, REM2, RAD, GEM | No consistent evidence of its role in cancer | [33,34] |
| RAL | Mediator of exocytosis Cell proliferation Cell migration | RALA RALB | Mediates Ras oncogenic transformation. Implication in colorectal, pancreatic, bladder, lung, prostate cancer, and melanoma. | [35,36,37,38,39] |
| RAP | Integrin-mediated cell adhesion Formation of cell–cell junctions Establishment of cell polarity Exocytosis Apoptosis Cell proliferation | RAP1A, RAP1B, RAP2A, RAP2B, RAP2C | Implication in squamous cell and head and neck carcinoma, breast, colorectal, brain, lung | [40,41,42,43,44,45,46,47] |
| RAS | Cell proliferation Cell adhesion Cell differentiation Organ development Neuronal plasticity | ERAS, NRAS, HRAS, KRAS, MRAS, RRAS RRAS2, DIRAS, DIRAS2, DIRAS3 NKIRAS1 NKIRAS2 RASD1 RASD2 RASL10A RASL10B RASL11A RASL11B RASL12 RERG | Oncogenic branch of Ras-GTPases (KRAS, HRAS, NRAS) are implicated in almost all types of cancer. DIRAS subgroup counteracts oncogenic Ras acting as a tumour suppressor in breast and ovarian cancer. RASD1, RASL11A and RERG seems to act as tumour suppressors in glioblastoma, prostate cancer, and nasopharyngeal carcinoma, respectively. | [32,48,49,50,51,52,53] |
| RHEB | Cell growth Cell cycle control Autophagy Amino acid uptake | RHEB, RHEBL1 | Related with metastasis in prostate cancer and increasing survival and proliferation in CRC cell lines. | [54,55,56,57,58] |
| RIT | Neuronal differentiation and survival | RIT1, RIT2, RIN, RIC | Not described | [59] |
| Biomarker | Inhibitor | Strategy | Phase | Status | Study Identifier |
|---|---|---|---|---|---|
| PKC | Bryostatin | Targeting Location | II | Completed | NCT00003220 |
| RAS Farnesyl | L-778 123 | Targeting Location | I | Completed | NCT00003430 |
| RAS MBS * | Salirasib | Targeting Location | II | Completed | NCT00531401 |
| KRASG12C | MRTX-849 | Direct Targeting | III | Recruiting | NCT04793958 |
| KRASG12C | LY3499446 | Direct Targeting | I | Terminated | NCT04165031 |
| KRASG12C | JNJ-74699157 | Direct Targeting | I | Completed | NCT04006301 |
| SOS1 | BI-1701963 | Targeting Upstream Elements | I | Recruiting | NCT04111458 |
| MEK | Trametinib | Targeting Downstream Elements | I | Recruiting | NCT03714958 |
| ERK | MK-8353 | Targeting Downstream Elements | I | Active | NCT02972034 |
| - | Vitamin C | Targeting Metabolism | II | Recruiting | NCT03146962 |
| - | Vitamin C + Carbohydrate restriction | Targeting Metabolism | I | Not yet recruiting | NCT04035096 |
| Genes | Type of Screening | Cell Lines Used | Drug Inhibition | Refs |
|---|---|---|---|---|
| PLK1 | Genome scale shRNA screening | DLD1 (KRAS G13D) | BI-2536 | [162,163] |
| Survivin | siRNA screening of ~4000 genes | DLD1 (KRAS G13D) | Not tested | [164] |
| SNAIL2 | shRNA screening of ~2500 cancer-related genes | HCT116 (KRAS G13D) | Not tested | [165] |
| GATA2 CDC16 | siRNA screening of ~7000 genes | HCT116 (KRAS G13D) | Bortezomib (proteosome inhibitor) + GATA2 silencing | [166] |
| TAK1 | Screening of 17 kinases, using 5 shRNAs | SW620 (KRAS G12V) SW837 (KRAS G12C) HCT116 (WT) HCT116 (KRAS G13D) | NG25 (in vitro and in vivo) | [167,168] |
| BCLXL MEK | Screening of genes whose inhibition cooperate with MEK inhibitors | SW620 (KRAS G12V) HCT116 (WT) HCT116 (KRAS G13D) | Selumetinib and navitoclax (MEK inhibitors) | [169] |
| CDK1 | siRNA library targeting 784 genes. | Isogenic LIM1215 (KRAS WT and mutant) | RO-3306 (in vitro and in vivo) | [170] |
| RAF1 | shRNA library targeting 535 kinases and related genes | SW480 (KRAS G12V) | RAF265 or AZ628 (RAF inhibitors) with selumetinib | [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rio-Vilariño, A.; del Puerto-Nevado, L.; García-Foncillas, J.; Cebrián, A. Ras Family of Small GTPases in CRC: New Perspectives for Overcoming Drug Resistance. Cancers 2021, 13, 3757. https://doi.org/10.3390/cancers13153757
Rio-Vilariño A, del Puerto-Nevado L, García-Foncillas J, Cebrián A. Ras Family of Small GTPases in CRC: New Perspectives for Overcoming Drug Resistance. Cancers. 2021; 13(15):3757. https://doi.org/10.3390/cancers13153757
Chicago/Turabian StyleRio-Vilariño, Anxo, Laura del Puerto-Nevado, Jesús García-Foncillas, and Arancha Cebrián. 2021. "Ras Family of Small GTPases in CRC: New Perspectives for Overcoming Drug Resistance" Cancers 13, no. 15: 3757. https://doi.org/10.3390/cancers13153757