
Plasma BRAF Mutation Detection for the Diagnostic and Monitoring Trajectory of Patients with LDH-High Stage IV Melanoma

 ,
, 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Study Design
2.2. Cell-Free DNA Isolation and ctDNA Quantification
2.3. Statistical Analysis
3. Results
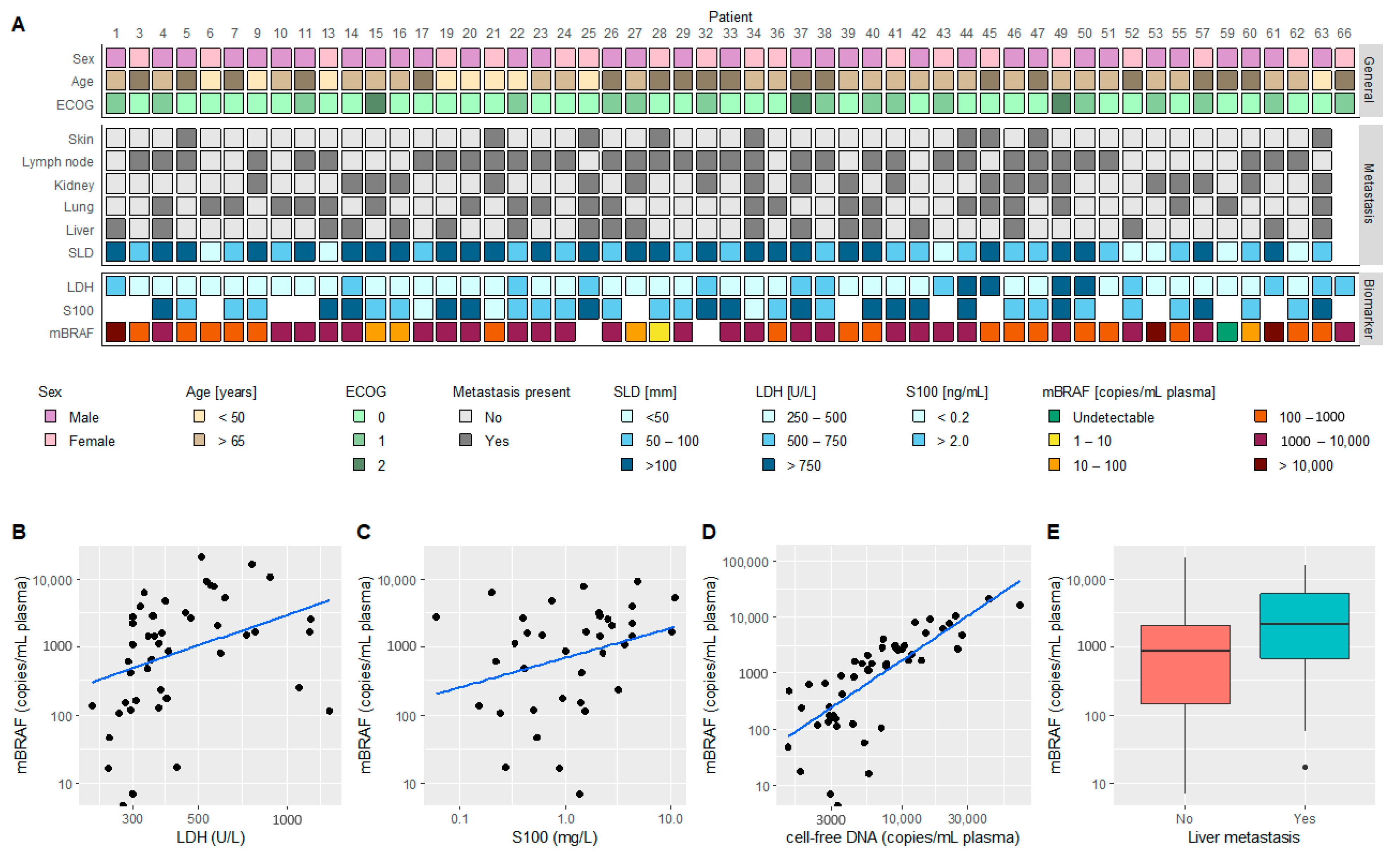
3.1. Patient Characteristics
3.2. ctDNA-Based mBRAF Assessment
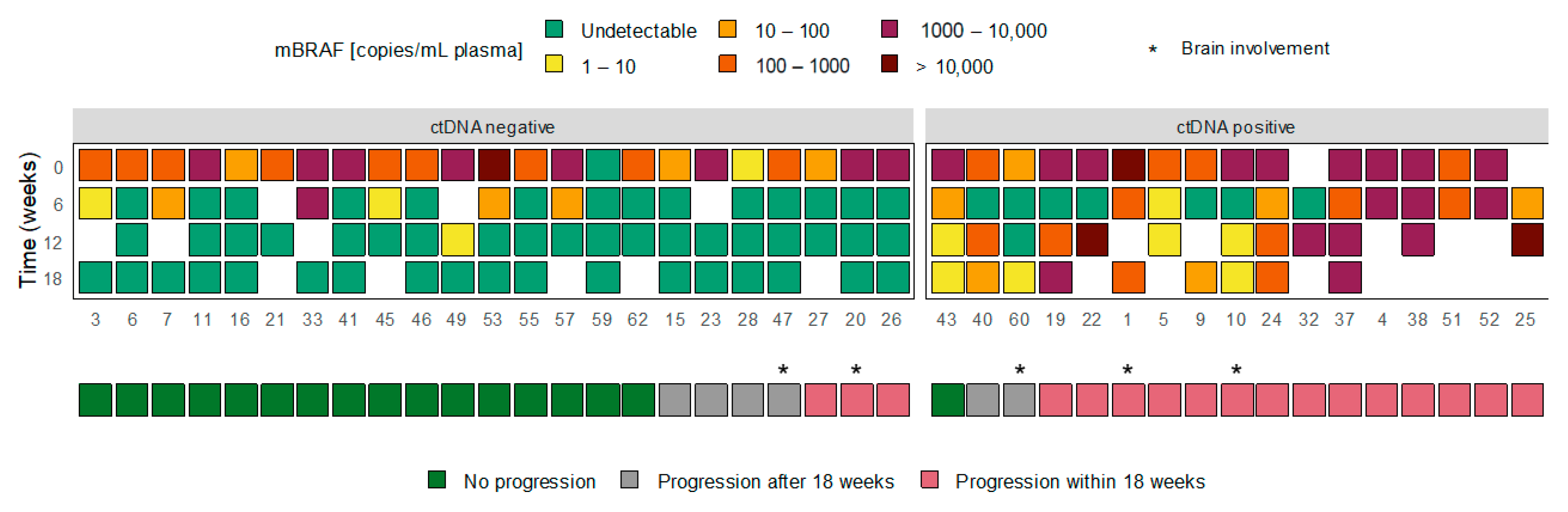
3.3. ctDNA Dynamics and Treatment Response
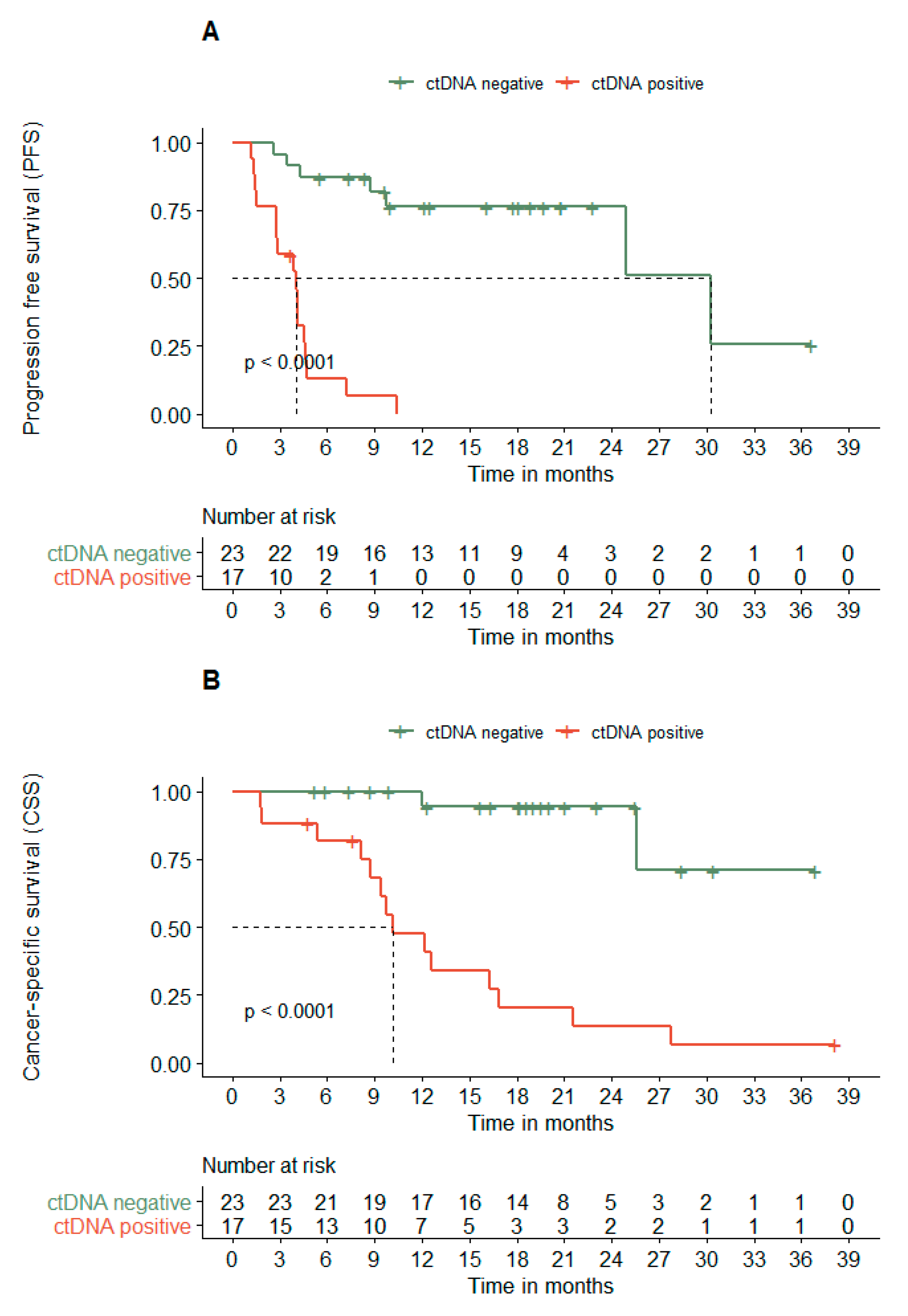
3.4. ctDNA Dynamics Associates with the PFS and CSS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rigel, D.S.; Carucci, J.A. Malignant melanoma: Prevention, early detection, and treatment in the 21st century. CA Cancer J. Clin. 2000, 50, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Guy, G.P., Jr.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982-2030. MMWR Morb. Mortal Wkly. Rep. 2015, 64, 591–596. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; Gibney, G.; Sullivan, R.J.; Sosman, J.A.; Slingluff, C.L., Jr.; Lawrence, D.P.; Logan, T.F.; Schuchter, L.M.; Nair, S.; Fecher, L.; et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol. 2016, 17, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Hamid, O.; Puzanov, I.; Dummer, R.; Schachter, J.; Daud, A.; Schadendorf, D.; Blank, C.; Cranmer, L.D.; Robert, C.; Pavlick, A.C.; et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur. J. Cancer 2017, 86, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Weber, J.S.; Daud, A.; Hamid, O.; Patnaik, A.; Ribas, A.; Robert, C.; Gangadhar, T.C.; et al. Evaluation of Immune-Related Response Criteria and RECIST v1.1 in Patients With Advanced Melanoma Treated With Pembrolizumab. J. Clin. Oncol. 2016, 34, 1510–1517. [Google Scholar] [CrossRef]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet. Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef] [Green Version]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release. Clinica. Chimica. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Garlan, F.; Blanchet, B.; Kramkimel, N.; Puszkiel, A.; Golmard, J.L.; Noe, G.; Dupin, N.; Laurent-Puig, P.; Vidal, M.; Taly, V.; et al. Circulating Tumor DNA Measurement by Picoliter Droplet-Based Digital PCR and Vemurafenib Plasma Concentrations in Patients with Advanced BRAF-Mutated Melanoma. Target Oncol. 2017, 12, 365–371. [Google Scholar] [CrossRef]
- Kozak, K.; Kowalik, A.; Gos, A.; Wasag, B.; Lugowska, I.; Jurkowska, M.; Krawczynska, N.; Kosela-Paterczyk, H.; Switaj, T.; Teterycz, P.; et al. Cell-free DNA BRAF V600E measurements during BRAF inhibitor therapy of metastatic melanoma: Long-term analysis. Tumori. J. 2020, 106, 241–248. [Google Scholar] [CrossRef]
- Braune, J.; Keller, L.; Schiller, F.; Graf, E.; Rafei-Shamsabadi, D.; Wehrle, J.; Follo, M.; Philipp, U.; Hussung, S.; Pfeifer, D.; et al. Circulating Tumor DNA Allows Early Treatment Monitoring in BRAF- and NRAS-Mutant Malignant Melanoma. JCO Precis. Oncol. 2020, 4, 20–31. [Google Scholar] [CrossRef]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [Green Version]
- Sanmamed, M.F.; Fernandez-Landazuri, S.; Rodriguez, C.; Zarate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martin-Algarra, S.; Gonzalez, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Forthun, R.B.; Hovland, R.; Schuster, C.; Puntervoll, H.; Brodal, H.P.; Namlos, H.M.; Aasheim, L.B.; Meza-Zepeda, L.A.; Gjertsen, B.T.; Knappskog, S.; et al. ctDNA detected by ddPCR reveals changes in tumour load in metastatic malignant melanoma treated with bevacizumab. Sci. Rep. 2019, 9, 17471. [Google Scholar] [CrossRef]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Ziman, M.; et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, S.P.; Luber, B.; Makell, M.; Brothers, P.; Santmyer, J.; Schollenberger, M.D.; Quinn, H.; Edelstein, D.L.; Jones, F.S.; Bleich, K.B.; et al. From validity to clinical utility: The influence of circulating tumor DNA on melanoma patient management in a real-world setting. Mol. Oncol. 2018, 12, 1661–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.A.; Tadepalli, J.S.; Shao, Y.; Zhang, Y.; Weiss, S.; Robinson, E.; Spittle, C.; Furtado, M.; Shelton, D.N.; Karlin-Neumann, G.; et al. Sensitivity of plasma BRAFmutant and NRASmutant cell-free DNA assays to detect metastatic melanoma in patients with low RECIST scores and non-RECIST disease progression. Mol. Oncol. 2016, 10, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Mayo de Las Casas, C.; Jordana Ariza, N.; Manzano, J.L.; Molina-Vila, M.A.; Soriano, V.; Puertolas, T.; Balada, A.; Soria, A.; Majem, M.; et al. Early evolution of BRAFV600 status in the blood of melanoma patients correlates with clinical outcome and identifies patients refractory to therapy. Melanoma Res. 2018, 28, 195–203. [Google Scholar] [CrossRef]
- Varaljai, R.; Wistuba-Hamprecht, K.; Seremet, T.; Diaz, J.M.S.; Nsengimana, J.; Sucker, A.; Griewank, K.; Placke, J.M.; Horn, P.A.; von Neuhoff, N.; et al. Application of Circulating Cell-Free Tumor DNA Profiles for Therapeutic Monitoring and Outcome Prediction in Genetically Heterogeneous Metastatic Melanoma. JCO Precis. Oncol. 2019, 3, PO.18.00229. [Google Scholar] [CrossRef] [Green Version]
- Forschner, A.; Weißgraeber, S.; Hadaschik, D.; Schulze, M.; Kopp, M.; Kelkenberg, S.; Sinnberg, T.; Garbe, C.; Biskup, S.; Battke, F. Circulating Tumor DNA Correlates with Outcome in Metastatic Melanoma Treated by BRAF and MEK Inhibitors—Results of a Prospective Biomarker Study. OncoTargets Ther. 2020, 13, 5017–5032. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.-J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Van Wilpe, S.; Koornstra, R.; Den Brok, M.; De Groot, J.W.; Blank, C.; De Vries, J.; Gerritsen, W.; Mehra, N. Lactate dehydrogenase: A marker of diminished antitumor immunity. Oncoimmunology 2020, 9, 1731942. [Google Scholar] [CrossRef] [Green Version]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosrati, A.; Tsai, K.K.; Goldinger, S.M.; Tumeh, P.; Grimes, B.; Loo, K.; Algazi, A.P.; Nguyen-Kim, T.D.L.; Levesque, M.; Dummer, R.; et al. Evaluation of clinicopathological factors in PD-1 response: Derivation and validation of a prediction scale for response to PD-1 monotherapy. Br. J. Cancer 2017, 116, 1141–1147. [Google Scholar] [CrossRef] [Green Version]
- Bucheit, A.D.; Syklawer, E.; Jakob, J.A.; Bassett, R.L., Jr.; Curry, J.L.; Gershenwald, J.E.; Kim, K.B.; Hwu, P.; Lazar, A.J.; Davies, M.A. Clinical characteristics and outcomes with specific BRAF and NRAS mutations in patients with metastatic melanoma. Cancer 2013, 119, 3821–3829. [Google Scholar] [CrossRef] [PubMed]
- Calbet-Llopart, N.; Potrony, M.; Tell-Marti, G.; Carrera, C.; Barreiro, A.; Aguilera, P.; Podlipnik, S.; Puig, S.; Malvehy, J.; Puig-Butille, J.A. Detection of cell-free circulating BRAF(V) (600E) by droplet digital polymerase chain reaction in patients with and without melanoma under dermatological surveillance. Br. J. Dermatol. 2020, 182, 382–389. [Google Scholar] [CrossRef]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Total Patients, n (%) | 53 (100%) |
|---|---|
| Sex, n (%) | |
| Female | 19 (36%) |
| Male | 34 (64%) |
| Age | |
| Median years (range) | 61 (28–78) |
| ECOG, n (%) | |
| 0 | 34 (64%) |
| 1 | 16 (30%) |
| 2 | 3 (6%) |
| Initiated treatment, n (%) | |
| Immunotherapy | 28 (53%) |
| BRAF/MEK inhibitor | 25 (47%) |
| LDH (U/L) | |
| Median (range) | 357 (261–1560) |
| S100 (ng/mL) | |
| Median (range) | 1.43 (0.06–10.97) |
| Metastasis location, n (%) | |
| Skin | 9 (17%) |
| Lymph node | 34 (64%) |
| Lung | 23 (43%) |
| Kidney | 21 (40%) |
| Liver | 19 (36%) |
| Follow-up | |
| Median months (range) | 12.3 (0–38.1) |
| Progression-Free Survival | |||||||
|---|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariate Analysis | ||||||
| Variable | HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Liver metastasis | Present vs. absent | 3.65 | 1.73–7.71 | <0.001 | 1.38 | 0.36–5.3 | 0.643 |
| ctDNA baseline | log10 (mBRAF per mL plasma) | 1.68 | 1.07–2.64 | 0.02 | 1.26 | 0.67–2.4 | 0.473 |
| S100 dynamics | Above vs. below ULN at 12-18 weeks | 5.5 | 2.21–13.71 | <0.001 | 0.95 | 0.23–4.0 | 0.94 |
| ctDNA dynamics | Positive vs. negative at 12-18 weeks | 12.57 | 4.30–36.76 | <0.001 | 18.75 | 3.55–98.9 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolmeijer, S.H.; Koornstra, R.H.T.; de Groot, J.W.B.; Geerlings, M.J.; van Rens, D.H.; Boers-Sonderen, M.J.; Schalken, J.A.; Gerritsen, W.R.; Ligtenberg, M.J.L.; Mehra, N. Plasma BRAF Mutation Detection for the Diagnostic and Monitoring Trajectory of Patients with LDH-High Stage IV Melanoma. Cancers 2021, 13, 3913. https://doi.org/10.3390/cancers13153913
Tolmeijer SH, Koornstra RHT, de Groot JWB, Geerlings MJ, van Rens DH, Boers-Sonderen MJ, Schalken JA, Gerritsen WR, Ligtenberg MJL, Mehra N. Plasma BRAF Mutation Detection for the Diagnostic and Monitoring Trajectory of Patients with LDH-High Stage IV Melanoma. Cancers. 2021; 13(15):3913. https://doi.org/10.3390/cancers13153913
Chicago/Turabian StyleTolmeijer, Sofie H., Rutger H. T. Koornstra, Jan Willem B. de Groot, Maartje J. Geerlings, Dirk H. van Rens, Marye J. Boers-Sonderen, Jack A. Schalken, Winald R. Gerritsen, Marjolijn J. L. Ligtenberg, and Niven Mehra. 2021. "Plasma BRAF Mutation Detection for the Diagnostic and Monitoring Trajectory of Patients with LDH-High Stage IV Melanoma" Cancers 13, no. 15: 3913. https://doi.org/10.3390/cancers13153913