
Adipocytes Promote Breast Cancer Cell Survival and Migration through Autophagy Activation

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Chemicals
2.2. Differentiation of OP9 and Production of Conditioned Medium
2.3. Co-Culture System
2.4. Oil Red O Staining
2.5. RNA Extraction, RNA Sequencing and RT-qPCR
2.6. Western Blot
2.7. siRNA and Plasmid Transfections
2.8. Intracellular Degradation
2.9. Lysosome pH and LAMP1 Staining
2.10. Oxygen Consumption Measurement
2.11. Lipid Analysis Using High Performance Thin Layer Chromatography (HPTLC) and Gas Chromatography (GC)
2.12. Viability Assays
2.13. Migration and Invasion Assays
2.14. Statistical Analysis
3. Results
3.1. Co-Culture Model for Adipocyte and Breast Cancer Cell Interactions
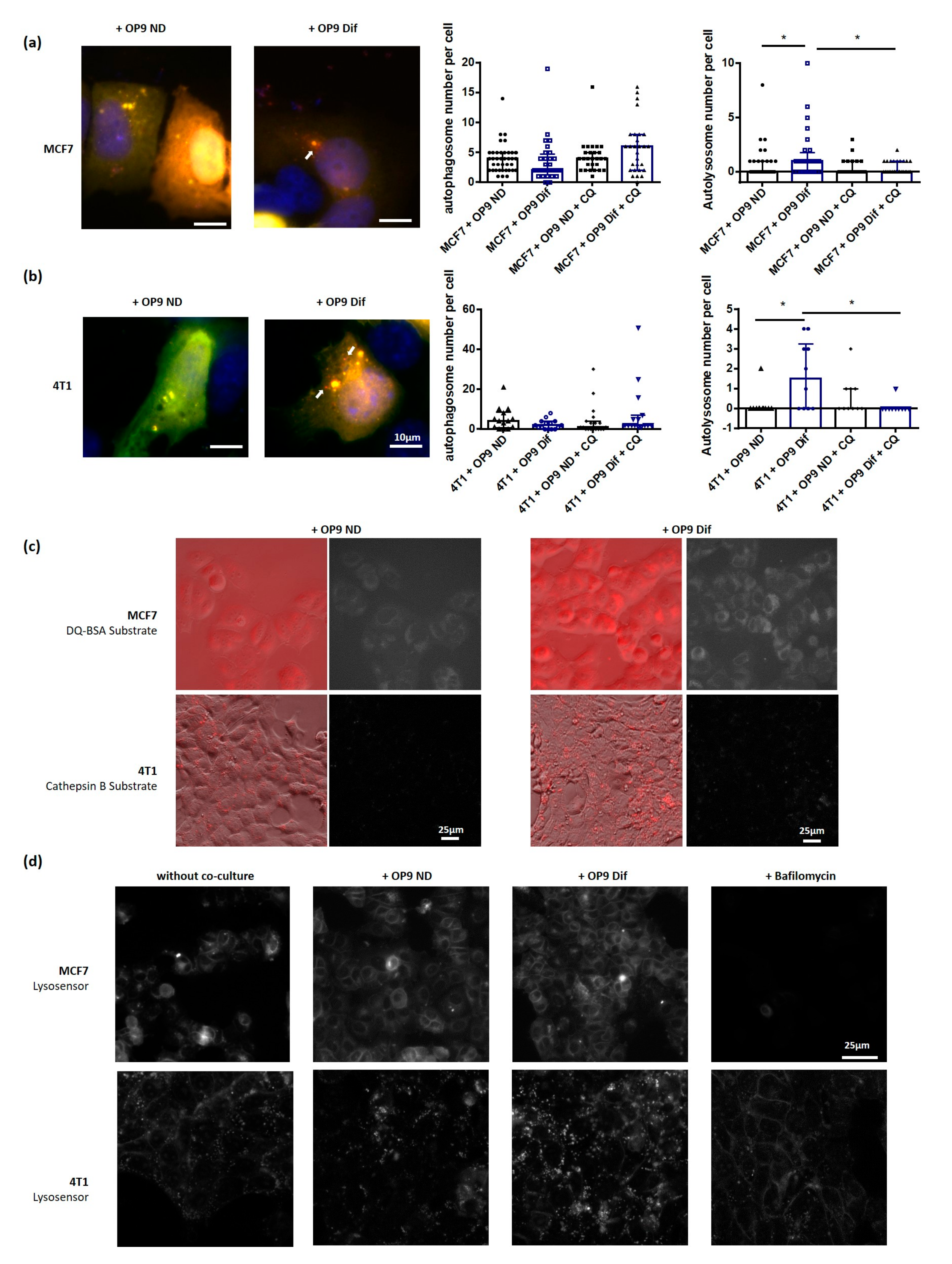
3.2. Adipocytes Activate Autophagy in Breast Cancer Cells
3.3. Role of Fatty Acids in the Promotion of Autophagy by Adipocytes
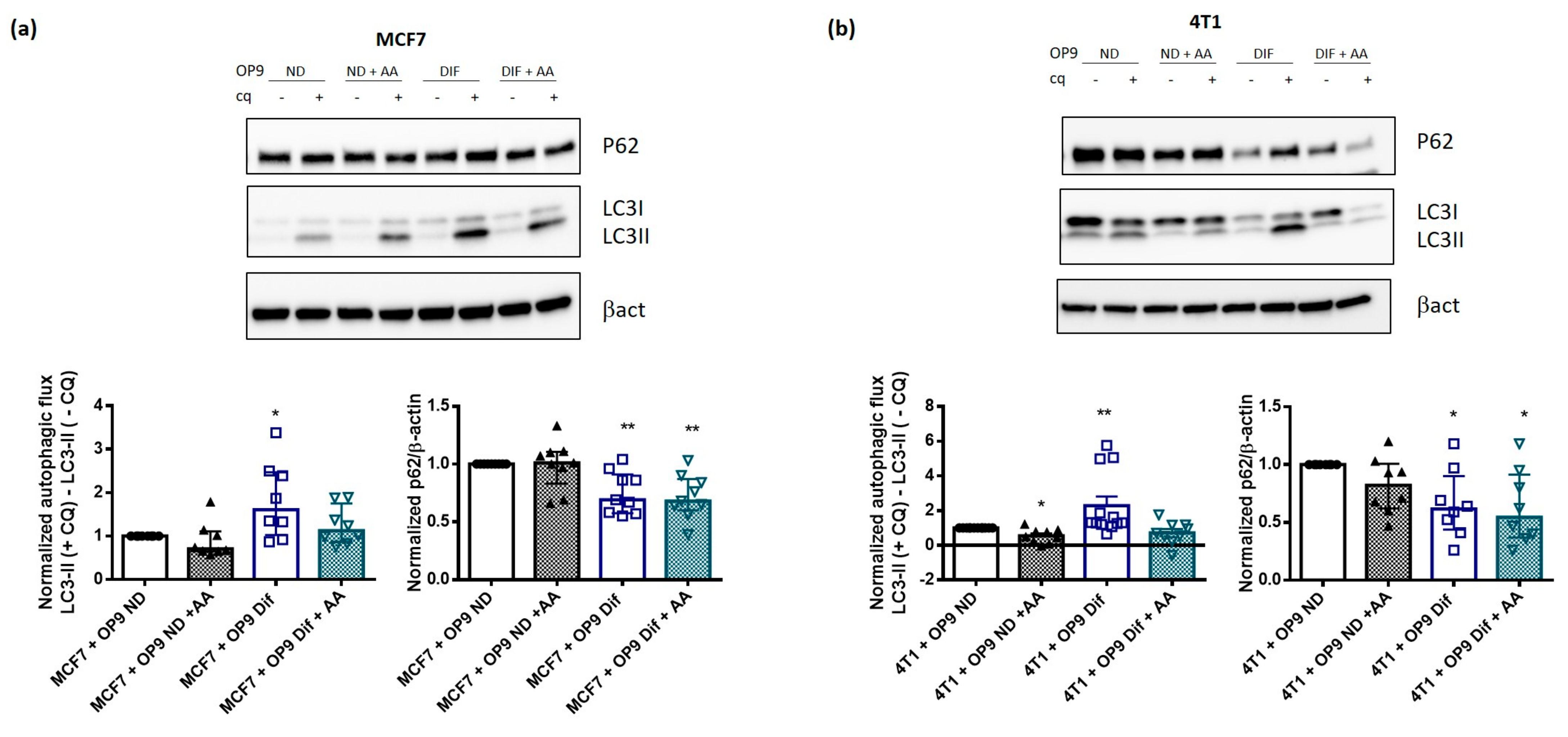
3.4. AA Prevents Autophagy Activation by Adipocytes
3.5. Autophagy Induction by Adipocytes Promotes Cancer Cell Survival and Migration
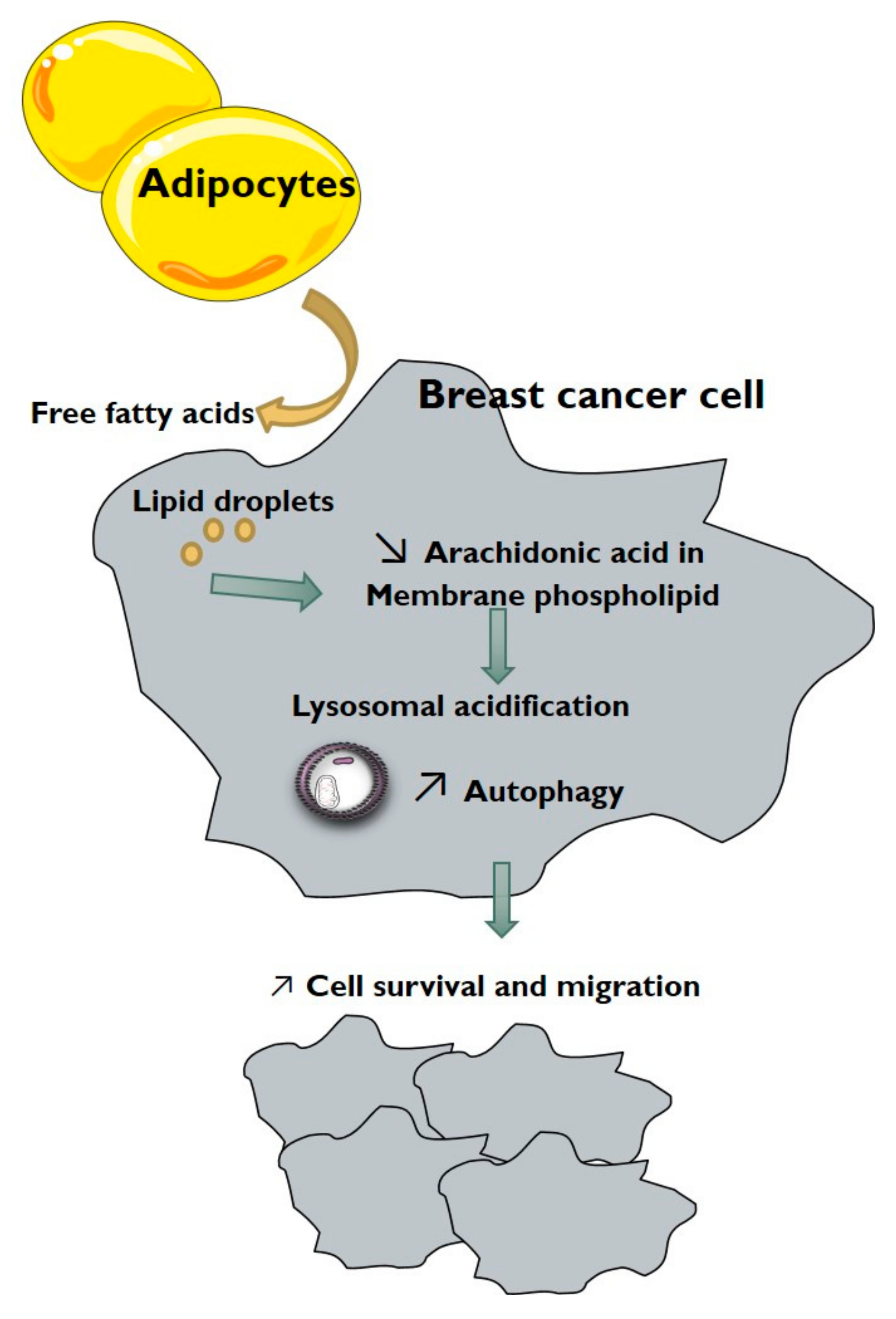
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer Metabolism in Space and Time: Beyond the Warburg Effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The Diversity and Breadth of Cancer Cell Fatty Acid Metabolism. Cancer Metab. 2021, 9, 1–28. [Google Scholar] [CrossRef]
- McKillop, I.H.; Girardi, C.A.; Thompson, K.J. Role of Fatty Acid Binding Proteins (FABPs) in Cancer Development and Progression. Cell Signal. 2019, 62, 109336. [Google Scholar] [CrossRef]
- Deng, Y.; Scherer, P.E. Adipokines as Novel Biomarkers and Regulators of the Metabolic Syndrome. Ann. N. Y. Acad. Sci. 2010, 1212, E1–E19. [Google Scholar] [CrossRef]
- Proenca, A.R.; Sertie, R.A.; Oliveira, A.C.; Campana, A.B.; Caminhotto, R.O.; Chimin, P.; Lima, F.B. New Concepts in White Adipose Tissue Physiology. Braz. J. Med. Biol. Res. 2014, 47, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, J.-F.; Brisson, L. Interaction between Adipose Tissue and Cancer Cells: Role for Cancer Progression. Cancer Metastasis Rev. 2021, 40, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and Adverse Breast Cancer Risk and Outcome: Mechanistic Insights and Strategies for Intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary Adipocytes Stimulate Breast Cancer Invasion through Metabolic Remodeling of Tumor Cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- He, J.Y.; Wei, X.H.; Li, S.J.; Liu, Y.; Hu, H.L.; Li, Z.Z.; Kuang, X.H.; Wang, L.; Shi, X.; Yuan, S.T.; et al. Adipocyte-Derived IL-6 and Leptin Promote Breast Cancer Metastasis via Upregulation of Lysyl Hydroxylase-2 Expression. Cell Commun. Signal. 2018, 16, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Bougnoux, P.; Giraudeau, B.; Couet, C. Diet, Cancer, and the Lipidome. Cancer Epidemiol. Prev. Biomark. 2006, 15, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Ouldamer, L.; Goupille, C.; Vilde, A.; Arbion, F.; Body, G.; Chevalier, S.; Cottier, J.P.; Bougnoux, P. N-3 Polyunsaturated Fatty Acids of Marine Origin and Multifocality in Human Breast Cancer. PLoS ONE 2016, 11, e0147148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, S.; Nakamura, S.; Yoshimori, T. Phospholipids in Autophagosome Formation and Fusion. J. Mol. Biol. 2016, 428, 4819–4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A Dual Role for Autophagy in a Murine Model of Lung Cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [Green Version]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B Expression Is a Common Feature of Solid Tumors and Associated with Proliferation, Metastasis, and Poor Outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High Expression of LC3B Is Associated with Progression and Poor Outcome in Triple-Negative Breast Cancer. Med. Oncol. 2013, 30, 475. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and Chemotherapy Resistance: A Promising Therapeutic Target for Cancer Treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.-F.; Brisson, L. Autophagy and Mitophagy in Cancer Metabolic Remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic Acid Induces Autophagy through P53/AMPK/MTOR Signaling and Promotes Apoptosis in Human Cancer Cells Harboring Wild-Type P53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential Roles of Unsaturated and Saturated Fatty Acids on Autophagy and Apoptosis in Hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Giulitti, F.; Petrungaro, S.; Mandatori, S.; Tomaipitinca, L.; de Franchis, V.; D’Amore, A.; Filippini, A.; Gaudio, E.; Ziparo, E.; Giampietri, C. Anti-Tumor Effect of Oleic Acid in Hepatocellular Carcinoma Cell Lines via Autophagy Reduction. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Park, C.; Choi, K.; Bickel, P.E. OP9 Mouse Stromal Cells Rapidly Differentiate into Adipocytes: Characterization of a Useful New Model of Adipogenesis. J. Lipid Res. 2006, 47, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Peyta, L.; Jarnouen, K.; Pinault, M.; Coulouarn, C.; Guimaraes, C.; Goupille, C.; de Barros, J.P.; Chevalier, S.; Dumas, J.F.; Maillot, F.; et al. Regulation of Hepatic Cardiolipin Metabolism by TNFalpha: Implication in Cancer Cachexia. Biochim. Biophys. Acta 2015, 1851, 1490–1500. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Pinault, M.; Guimaraes, C.; Ben Hassen, C.; Gutierrez-Pajares, J.L.; Chevalier, S.; Goupille, C.; Bernard-Savary, P.; Frank, P.G. Development of a Novel High-Performance Thin Layer Chromatography-Based Method for the Simultaneous Quantification of Clinically Relevant Lipids from Cells and Tissue Extracts. Lipids 2020, 55, 403–412. [Google Scholar] [CrossRef]
- Pinault, M.; Guimaraes, C.; Dumas, J.-F.; Servais, S.; Chevalier, S.; Besson, P.; Goupille, C. A 1D High Performance Thin Layer Chromatography Method Validated to Quantify Phospholipids Including Cardiolipin and Monolysocardiolipin from Biological Samples. Eur. J. Lipid Sci. Technol. 2020, 122, 1900240. [Google Scholar] [CrossRef]
- Goupille, C.; Frank, P.G.; Arbion, F.; Jourdan, M.-L.; Guimaraes, C.; Pinault, M.; Body, G.; Chevalier, S.; Bougnoux, P.; Ouldamer, L. Low Levels of Omega-3 Long-Chain Polyunsaturated Fatty Acids Are Associated with Bone Metastasis Formation in Premenopausal Women with Breast Cancer: A Retrospective Study. Nutrients 2020, 12, 3832. [Google Scholar] [CrossRef]
- Dória, M.L.; Cotrim, C.Z.; Simões, C.; Macedo, B.; Domingues, P.; Domingues, M.R.; Helguero, L.A. Lipidomic Analysis of Phospholipids from Human Mammary Epithelial and Breast Cancer Cell Lines. J. Cell. Physiol. 2013, 228, 457–468. [Google Scholar] [CrossRef]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Marino, G.; Cianfanelli, V.; Ben-Younes, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated Fatty Acids Induce Non-Canonical Autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yi, Y.-P.; Zhang, G.-J. Effects of Arachidonic Acid on the Lysosomal Ion Permeability and Osmotic Stability. J. Bioenerg. Biomembr. 2006, 38, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte Lipolysis Links Obesity to Breast Cancer Growth: Adipocyte-Derived Fatty Acids Drive Breast Cancer Cell Proliferation and Migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes Activate Mitochondrial Fatty Acid Oxidation and Autophagy to Promote Tumor Growth in Colon Cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Thomas, M.H.; Pelleieux, S.; Vitale, N.; Olivier, J.L. Dietary Arachidonic Acid as a Risk Factor for Age-Associated Neurodegenerative Diseases: Potential Mechanisms. Biochimie 2016, 130, 168–177. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy Suppresses Progression of K-Ras-Induced Lung Tumors to Oncocytomas and Maintains Lipid Homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy Provides Metabolic Substrates to Maintain Energy Charge and Nucleotide Pools in Ras-Driven Lung Cancer Cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BrafV600E-Driven Lung Tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of Autophagy in the Tumor Microenvironment and Cancer Metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte Extracellular Vesicles Carry Enzymes and Fatty Acids That Stimulate Mitochondrial Metabolism and Remodeling in Tumor Cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Zaoui, M.; Morel, M.; Ferrand, N.; Fellahi, S.; Bastard, J.-P.; Lamazière, A.; Larsen, A.K.; Béréziat, V.; Atlan, M.; Sabbah, M. Breast-Associated Adipocytes Secretome Induce Fatty Acid Uptake and Invasiveness in Breast Cancer Cells via CD36 Independently of Body Mass Index, Menopausal Status and Mammary Density. Cancers 2019, 11, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-Induced CD36 Expression Drives Ovarian Cancer Progression and Metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K.; et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab. 2018, 28, 689–705.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Li, Y.; Xing, L.; Tan, Y.; Sun, J.; Zeng, B.; Xiang, T.; Tan, J.; Ren, G.; Wang, Y. Utilization of Adipocyte-Derived Lipids and Enhanced Intracellular Trafficking of Fatty Acids Contribute to Breast Cancer Progression. Cell Commun. Signal. 2018, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4t1 Control | 4t1 Condi | MCF7 Control | MCF7 Condi | ||
|---|---|---|---|---|---|
| Saturated | |||||
| Myristic Acid | 14:0 | 2.34[2.03–3.44] | 2.5[2.17–3.05] | 2.92[2.24–3.39] | 3.15[2.62–3.46] |
| Pentadecanoic Acid | 15:0 | 0.33[0.27–0.53] | 0.23[0.18–0.32] | 0.27[0.1–0.32] | 0.23[0.17–0.27] |
| Palmitic Acid | 16:0 | 18.49[15.61–21.84] | 18.95[15.54–20.18] | 18.17[17.45–20.57] | 17.96[17.05–20.42] |
| Margaric Acid | 17:0 | 0.42[0.3–0.54] | 0.28[0.14–0.45] | 0.25[0.18–0.4] | 0.2[0.11–0.37] |
| Stearic Acid | 18:0 | 12.27[11.31–12.91] | 12.35[11.24–12.81] | 8.88[8.31–11.15] | 8.69[7.81–9.98] |
| Arachidic Acid | 20:0 | 0.25[0.14–0.5] | 0.27[0.19–0.48] | 0.22[0.11–0.31] | 0.22[0.19–0.26] |
| Behenic Acid | 22:0 | 0.52[0.23–0.64] | 0.55[0.38–1.58] | 0.41[0.1–0.78] | 0.38[0.14–0.5] |
| Tricosanoic Acid | 23:0 | 0.28[0.13–0.47] | 0.2[0.02–0.41] | 0.09[0.04–0.2] | 0.1[0.02–0.22] |
| Lignoceric Acid | 24:0 | 0.43[0.09–0.8] | 0.38[0.2–0.81] | 0.57[0.2–0.85] | 0.31[0.24–0.52] |
| Total SFA | 35.27[39.84–31.85] | 35.85[38.34–31.74] | 32.11[35.82–30.65] | 31.11[34.48–30.22] | |
| Monounsaturated | |||||
| Myristoleic Acid | 14:1 | 0.03[0.01–0.04] | 0.03[0.02–0.06] | 0.06[0.04–0.25] | 0.09[0.04–0.2] |
| Pentadecenoic Acid | 15:1 | 0.3[0.06–0.51] | 0.21[0.05–0.29] | 0.18[0.02–0.26] | 0.3[0.02–0.4] |
| cis-7-Hexadecenoic Acid | 16:1n-9 | 2.43[2.14–3.05] | 2.65[2.39–3.38] | 1.06[0.91–5.48] | 0.99[0.85–5.86] |
| Palmitoleic Acid | 16:1n-7 | 4.12[2.8–5.19] | 5.12[4.37–7.15] | 12.65[7.33–15.16] | 14.04[6.99–14.83] |
| Heptadecenoic Acid | 17:1 | 0.41[0.08–0.53] | 0.29[0.07–0.4] | 0.5[0.42–0.61] | 0.48[0.34–0.52] |
| Oleic Acid | 18:1 | 26.57[24.78–28.44] | 23.89[20.43–31.5] | 36.2[28.64–38.86] | 37.62[35.63–39.86] |
| Gondoic Acid | 20:1 | 0.89[0.54–1.26] | 0.88[0.56–1.43] | 0.4[0.31–0.7] | 0.62[0.53–0.78] |
| Erucic Acid | 22:1 | 0.29[0.13–0.45] | 0.3[0.13–0.84] | 0.26[0.12–0.41] | 0.19[0.12–0.5] |
| Nervonic Acid | 24:1 | 0.92[0.63–1.01] | 0.64[0.16–1.18] | 0.12[0.41–0.55] | 0.12[0.5–0.57] |
| Total MUFA | 33.19[38.23–30.81] | 31.15[42.48–28.11] | 51.45[56.95–43.21] | 55.26[56.59–52.06] *** | |
| Polyunsaturated | |||||
| Linoleic Acid | 18:2n-6 | 1.53[1.17–1.86] | 1.09[0.88–1.51] ** | 1.51[1.1–1.8] | 1.11[0.89–2.03] |
| Gamma Linolenic Acid | 18:3n-6 | 0.1[0.03–0.2] | 0.08[0.02–0.13] | 0.04[0.02–0.07] | 0.06[0.04–0.11] |
| Eicosadienoic Acid | 20:2n-6 | 2.28[1.72–3.33] | 2.24[1.77–3.26] | 0.82[0.31–1.2] | 0.7[0.34–1.16] |
| Dihomo-γ-linolenic Acid | 20:3n-6 | 0.96[0.91–1.17] | 0.81[0.67–0.91] | 0.23[0.09–0.26] | 0.16[0.06–0.25] |
| Arachidonic Acid | 20:4n-6 | 3.8[3.24–4.09] | 2.93[2.62–3.38] *** | 2.04[1.41–2.66] | 1.63[1.28–2.23] ** |
| Docosadienoic Acid | 22:2n-6 | 0.11[0.02–0.32] | 0.14[0.02–0.18] | 0.11[0.01–0.38] | 0.09[0.04–0.21] |
| Docosatetraenoic Acid | 22:4n-6 | 0.17[0.08–0.51] | 0.08[0.05–0.4] | 0.08[0.03–0.23] | 0.06[0.03–0.11] |
| Total n-6 PUFA | 9.12[10.11–8.19] | 7.64[8.46–6.42] | 4.67[5.74–4.01] | 4.25[4.76–3.41] | |
| Alpha linolenic Acid | 18:3n-3 | 0.09[0.05–0.28] | 0.06[0.05–0.17] | 0.05[0.03–0.11] | 0.06[0.02–0.12] |
| Eicosatrienoic Acid | 20:3n-3 | 0.1[0.03–0.13] | 0.09[0.04–0.15] | 0.04[0.01–0.09] | 0.03[0.02–0.06] |
| Eicosapentaenoic Acid | 20:5n-3 | 1.08[0.61–1.97] | 0.99[0.65–1.6] | 0.36[0.21–0.77] | 0.23[0.11–0.4] |
| Docosapentaenoic Acid | 22:5n-3 | 3.38[1.89–3.79] | 2.24[1.73–3.18] *** | 0.8[0.27–1.25] | 0.53[0.27–0.93] ** |
| Docosahexaenoic Acid | 22:6n-3 | 3.11[2.1–3.98] | 2.41[1.89–2.97] *** | 1.64[1.18–2.23] | 1.38[1.08–1.89] * |
| Total n-3 PUFA | 7.77[10.13–4.68] | 5.91[7.69–4.98] | 3.01[4.07–2.12] | 2.18[3.16–1.53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellanger, D.; Dziagwa, C.; Guimaraes, C.; Pinault, M.; Dumas, J.-F.; Brisson, L. Adipocytes Promote Breast Cancer Cell Survival and Migration through Autophagy Activation. Cancers 2021, 13, 3917. https://doi.org/10.3390/cancers13153917
Bellanger D, Dziagwa C, Guimaraes C, Pinault M, Dumas J-F, Brisson L. Adipocytes Promote Breast Cancer Cell Survival and Migration through Autophagy Activation. Cancers. 2021; 13(15):3917. https://doi.org/10.3390/cancers13153917
Chicago/Turabian StyleBellanger, Dorine, Cléa Dziagwa, Cyrille Guimaraes, Michelle Pinault, Jean-François Dumas, and Lucie Brisson. 2021. "Adipocytes Promote Breast Cancer Cell Survival and Migration through Autophagy Activation" Cancers 13, no. 15: 3917. https://doi.org/10.3390/cancers13153917
APA StyleBellanger, D., Dziagwa, C., Guimaraes, C., Pinault, M., Dumas, J.-F., & Brisson, L. (2021). Adipocytes Promote Breast Cancer Cell Survival and Migration through Autophagy Activation. Cancers, 13(15), 3917. https://doi.org/10.3390/cancers13153917