
The Multifaceted Role of TGF-β in Gastrointestinal Tumors

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
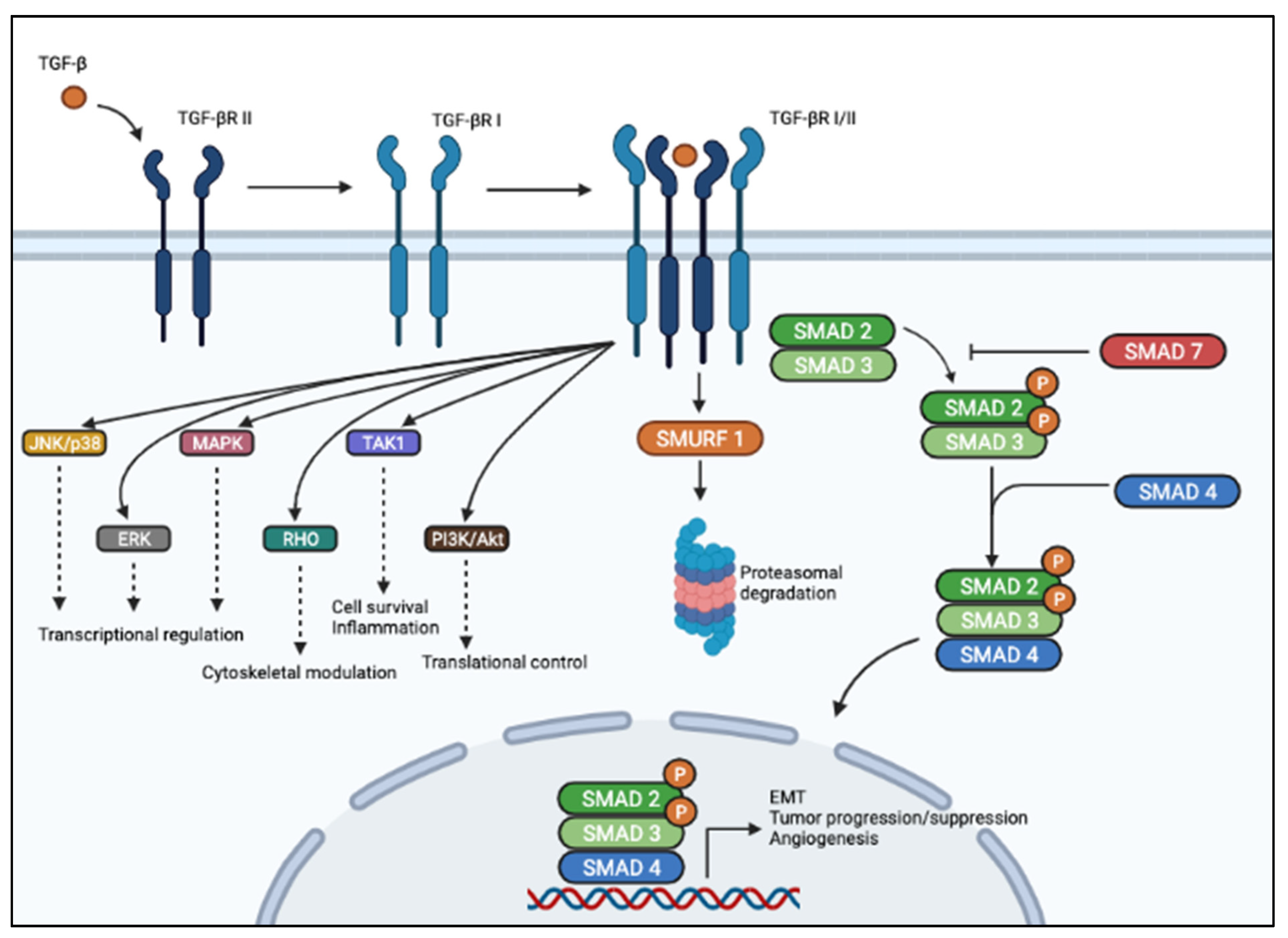
2. TGF-β Secretion and Activation
3. The TGF-β Signaling Pathway
4. TGF-β Signaling in Cancer
5. TGF- in Gastrointestinal Tumors
5.1. Pancreatic Cancer
5.2. Colorectal Cancer
5.3. Gastric Cancer
5.4. Hepatocellular Carcinoma
5.5. Esophageal Cancer
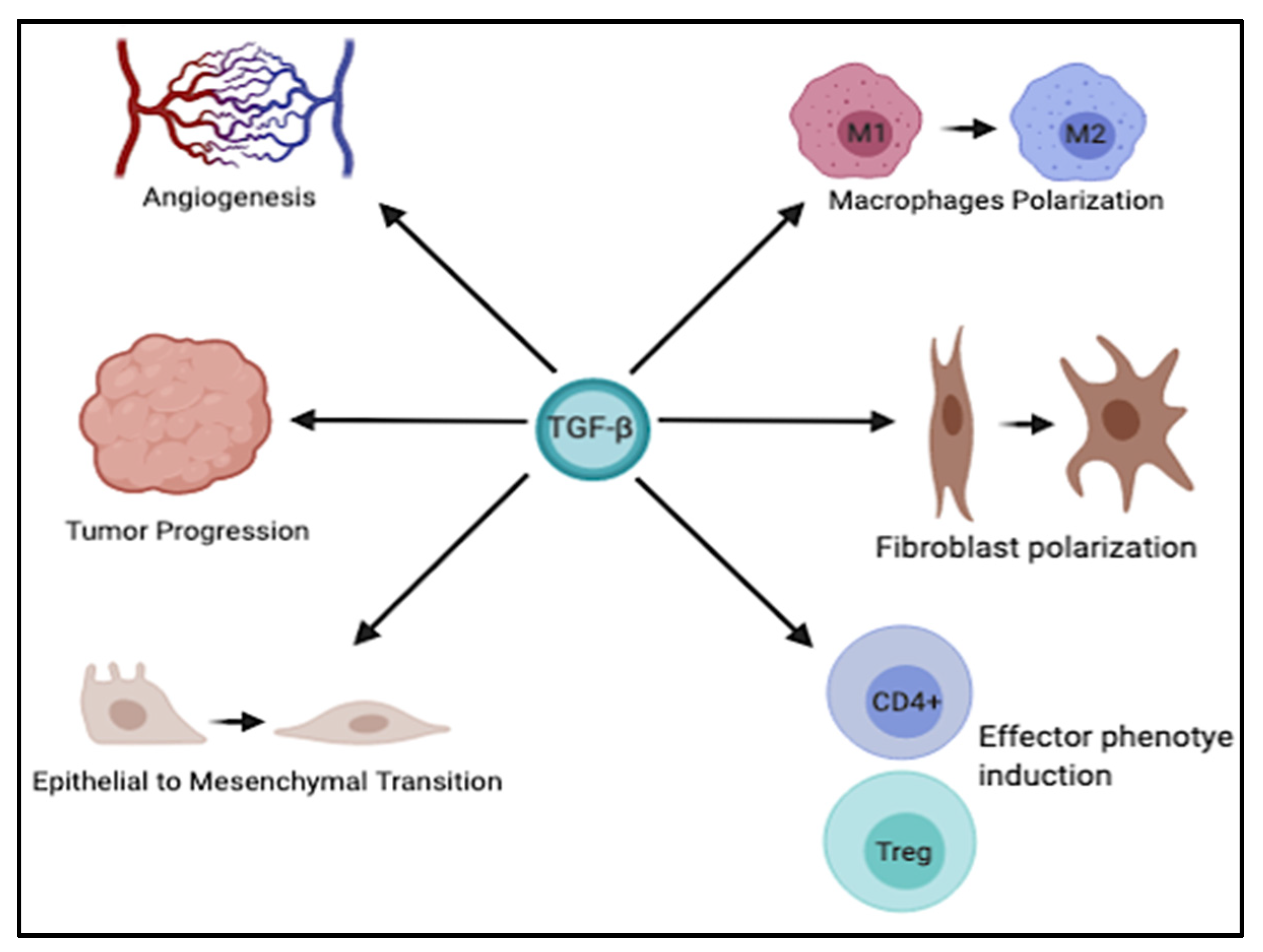
6. TGF-β and Tumor Microenvironment
6.1. Regulation of Macrophage Plasticity
6.2. Regulation of Neutrophil Plasticity
6.3. Induction of T Regulatory Phenotype
6.4. Crosstalk with Cancer Associated Fibroblasts
7. TGF-β Targeted Therapies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| α-SMA | a-Smooth Muscle Actin |
| ALK1–7 | Activin Receptor-Like Kinase 1–7 |
| APC | Antigen-Presenting Cells |
| ASO | Antisense Oligonucleotides |
| bHLH | Basic/Helix-Loop-Helix |
| BMPs | Bone Morphogenetic Proteins |
| C/EBPb | CCAAT/Enhancer-Binding Protein beta |
| CAFs | Cancer-Associated Fibroblasts |
| CDK | Cyclin-Dependent Kinase |
| CMSs | Consensus Molecular Subtypes |
| CRC | Colorectal Cancer |
| CTGF | Connective Tissue Growth Factor |
| CTLA4 | Cytotoxic T Lymphocyte Antigen 4 |
| CXXC5 | CXXC-type zinc finger protein 5 |
| DAXX | Death-Associated protein 6 |
| EC | Esophageal Cancer |
| ECM | Extracellular Matrix |
| ELF | Embryonic Liver Fodrin |
| EMT | Epithelial to Mesenchymal Transition |
| ESCC | Esophageal Squamous Cell Carcinoma |
| ERK | Extracellular Signal-Regulated Kinase |
| FAP | Fibroblast Activation Protein |
| G3BP1 | GTPase-Activating Protein SH3 Domain-Binding Protein 1 |
| GC | Gastric Cancer |
| GI | Gastrointestinal |
| GP73 | Golgi Protein 73 |
| GSK-3alpha | Glycogen Synthase Kinase-3alpha |
| HCC | Hepatocellular Carcinoma |
| HDAC | Histone Deacetylase |
| iCAF | Inflammatory CAF |
| IGFBP7 | Insulin-like Growth Factor-Binding Protein 7 |
| IRAK-M | Interleukin-1 Receptor Associated Kinase-M |
| JNK | c-Jun N-terminal Kinases |
| LAP | Latency-Associated Peptide |
| LIMK | LIM domain Kinase |
| LLP | Large Latent Complex |
| LRG | Leucine-Rich alpha-2 Glycoprotein |
| LTBP | Latent TGF-β Binding Protein |
| MALAT1 | Metastasis-Associated Lung Adenocarcinoma Transcript 1 |
| MAPK | Mitogen-Activated Protein Kinases |
| MTD | Maximum Tolerated Dose |
| MDSCs | Myeloid-Derived Suppressor Cells |
| MeCP2 | Methyl-CpG-Binding Protein 2 |
| MEL1 | MDS1/EVI 1 Like gene |
| MHC | Major Histocompatibility Complex |
| MMPs | Matrix Metalloproteinases |
| MMR | Mismatch Repair |
| myCAF | Myofibroblastic CAF |
| NF-κB | Nuclear Factor-κB |
| PC | Pancreatic Cancer |
| PD-1 | Programmed Cell Death 1 |
| PDACs | Pancreatic Ductal Adenocarcinomas |
| PI3K | Phosphoinositide 3-Kinase |
| PXR | Xenobiotic Nuclear Receptor |
| ROCK | Rho-associated Protein Kinase |
| R-SMAD | Receptor Regulated SMAD |
| RGD | Arginine-Glycine Aspartate |
| RHO | Rhodopsin |
| RORGT | RAR-related Orphan Receptor Gamma |
| RUNX3 | Runx-related Transcription Factor 3 |
| SALL4 | Spalt Like Transcription Factor 4 |
| SLC | Small Latent Complex |
| SMAD | Acronym obtained by merging Caenorhabditis elegans Sma genes and the Drosophila Mad, Mothers against decapentaplegic |
| SMURF | SMAD Ubiquitination Regulatory Factor |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAK1 | TGF-β-activated kinase-1 |
| TAMs | Tumor-Associated Macrophages |
| TANs | Tumor-Associated Neutrophils |
| TβRI-III | Type I-III Receptors |
| TCR | T-cell Receptor |
| TGF-βR | Transforming growth factor-beta receptor |
| TGF-β | Transforming growth factor-beta |
| TLR | Toll-like Receptor |
| TME | Tumor Microenvironment |
| TREG | T Cells to Regulatory T cell |
| TRM | Resident Memory T cell |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFC | Vascular Endothelial Growth Factor C |
| XIAP | X-linked Inhibitor of Apoptosis Protein |
| ZEB | Zinc finger E-Box-Binding Homeobox |
References
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Korc, M. Role of growth factors in pancreatic cancer. Surg. Oncol. Clin. N. Am. 1998, 7, 25–41. [Google Scholar] [CrossRef]
- Horiguchi, K.; Shirakihara, T.; Nakano, A.; Imamura, T.; Miyazono, K.; Saitoh, M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J. Biol. Chem. 2009, 284, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Oft, M.; Akhurst, R.J.; Balmain, A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat. Cell Biol. 2002, 4, 487–494. [Google Scholar] [CrossRef]
- Tang, B.; Vu, M.; Booker, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Chen, X.Q.; Li, P. The Role of TGF-beta and Its Receptors in Gastrointestinal Cancers. Transl. Oncol. 2019, 12, 475–484. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Roberts, A.B.; Kim, S.J.; Noma, T.; Glick, A.B.; Lafyatis, R.; Lechleider, R.; Jakowlew, S.B.; Geiser, A.; O’Reilly, M.A.; Danielpour, D.; et al. Multiple forms of TGF-beta: Distinct promoters and differential expression. Ciba Found. Symp. 1991, 157, 7–15; discussion 15–28. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B. Molecular and cell biology of TGF-beta. Miner. Electrolyte Metab. 1998, 24, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Makanji, Y.; Chen, J.; Wilce, M.C.; Chan, K.L.; Robertson, D.M.; Harrison, C.A. Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-beta1 complex. J. Biol. Chem. 2010, 285, 17029–17037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef]
- Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-beta binding proteins: Extracellular matrix association and roles in TGF-beta activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, D.B. Latent transforming growth factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, V.; Rifkin, D.B. LTBPs, more than just an escort service. J. Cell Biochem. 2012, 113, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, L.M.; Winokur, T.S.; Hollands, R.S.; Christopherson, K.; Levinson, A.D.; Sporn, M.B. Recombinant latent transforming growth factor beta 1 has a longer plasma half-life in rats than active transforming growth factor beta 1, and a different tissue distribution. J. Clin. Investig. 1990, 86, 1976–1984. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [Green Version]
- Franzen, P.; ten Dijke, P.; Ichijo, H.; Yamashita, H.; Schulz, P.; Heldin, C.H.; Miyazono, K. Cloning of a TGF beta type I receptor that forms a heteromeric complex with the TGF beta type II receptor. Cell 1993, 75, 681–692. [Google Scholar] [CrossRef]
- Yamashita, H.; ten Dijke, P.; Franzen, P.; Miyazono, K.; Heldin, C.H. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-beta. J. Biol. Chem. 1994, 269, 20172–20178. [Google Scholar] [CrossRef]
- Attisano, L.; Carcamo, J.; Ventura, F.; Weis, F.M.; Massague, J.; Wrana, J.L. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993, 75, 671–680. [Google Scholar] [CrossRef]
- Liu, F.; Ventura, F.; Doody, J.; Massague, J. Human type II receptor for bone morphogenic proteins (BMPs): Extension of the two-kinase receptor model to the BMPs. Mol. Cell Biol. 1995, 15, 3479–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenzweig, B.L.; Imamura, T.; Okadome, T.; Cox, G.N.; Yamashita, H.; ten Dijke, P.; Heldin, C.H.; Miyazono, K. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Attisano, L.; Wrana, J.L.; Montalvo, E.; Massague, J. Activation of signalling by the activin receptor complex. Mol. Cell Biol. 1996, 16, 1066–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatza, C.E.; Oh, S.Y.; Blobe, G.C. Roles for the type III TGF-beta receptor in human cancer. Cell Signal. 2010, 22, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Santoro, R.; Carbone, C.; Piro, G.; Chiao, P.J.; Melisi, D. TAK-ing aim at chemoresistance: The emerging role of MAP3K7 as a target for cancer therapy. Drug Resist. Updat. 2017, 33–35, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M., Jr.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef]
- Hayashida, T.; Decaestecker, M.; Schnaper, H.W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J. 2003, 17, 1576–1578. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [Green Version]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Liang, M.; Feng, X.H. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J. Biol. Chem. 2000, 275, 36818–36822. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Xu, X.; Li, L.; Ning, H.; Rong, Y.; Shang, Y.; Wang, Y.; Fu, X.Y.; Chang, Z. CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 2005, 280, 20842–20850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moren, A.; Imamura, T.; Miyazono, K.; Heldin, C.H.; Moustakas, A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 2005, 280, 22115–22123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrakis, K.J.; Andrew, R.L.; Lee, K.L.; Petropoulou, C.; Dixon, J.E.; Navaratnam, N.; Norris, D.P.; Episkopou, V. Arkadia enhances Nodal/TGF-beta signaling by coupling phospho-Smad2/3 activity and turnover. PLoS Biol. 2007, 5, e67. [Google Scholar] [CrossRef] [PubMed]
- Kit Leng Lui, S.; Iyengar, P.V.; Jaynes, P.; Isa, Z.; Pang, B.; Tan, T.Z.; Eichhorn, P.J.A. USP26 regulates TGF-beta signaling by deubiquitinating and stabilizing SMAD7. EMBO Rep. 2017, 18, 797–808. [Google Scholar] [CrossRef]
- Tang, W.B.; Ling, G.H.; Sun, L.; Liu, F.Y. Smad anchor for receptor activation (SARA) in TGF-beta signaling. Front. Biosci. 2010, 2, 857–860. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Katuri, V.; Dillner, A.; Mishra, B.; Deng, C.X.; Mishra, L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 2003, 299, 574–577. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, C.N.; Gnanapragasam, V.; Byrne, R.L.; Collins, A.T.; Neal, D.E. Transforming growth factor-beta1 up-regulates p15, p21 and p27 and blocks cell cycling in G1 in human prostate epithelium. J. Endocrinol. 1999, 160, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Fiore, A.P.; Osaki, L.H.; Gama, P. Transforming growth factor beta1 increases p27 levels via synthesis and degradation mechanisms in the hyperproliferative gastric epithelium in rats. PLoS ONE 2014, 9, e101965. [Google Scholar] [CrossRef] [Green Version]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.D.; Choi, J.Y.; Lee, S.J.; Kim, J.S.; Min, B.R.; Lee, Y.I.; Kang, Y.K. TGF-beta-induced cell-cycle arrest through the p21(WAF1/CIP1)-G1 cyclin/Cdks-p130 pathway in gastric-carcinoma cells. Int. J. Cancer 1999, 83, 512–517. [Google Scholar] [CrossRef]
- Kang, S.H.; Bang, Y.J.; Jong, H.S.; Seo, J.Y.; Kim, N.K.; Kim, S.J. Rapid induction of p21WAF1 but delayed down-regulation of Cdc25A in the TGF-beta-induced cell cycle arrest of gastric carcinoma cells. Br. J. Cancer 1999, 80, 1144–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Ammanamanchi, S.; Ko, T.C.; Brattain, M.G. Transforming growth factor beta 1 increases the stability of p21/WAF1/CIP1 protein and inhibits CDK2 kinase activity in human colon carcinoma FET cells. Cancer Res. 2003, 63, 3340–3346. [Google Scholar]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Cui, J.; Qin, Q.; Zhang, J.; Liu, L.; Deng, S.; Wu, C.; Yang, M.; Li, S.; Wang, C. Mechanical stiffness of liver tissues in relation to integrin beta1 expression may influence the development of hepatic cirrhosis and hepatocellular carcinoma. J. Surg. Oncol. 2010, 102, 482–489. [Google Scholar] [CrossRef]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbi, A.; Attisano, L.; Bernabeu, C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535. [Google Scholar] [CrossRef] [Green Version]
- Pak, K.H.; Park, K.C.; Cheong, J.H. VEGF-C induced by TGF- beta1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis. BMC Cancer 2019, 19, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pen, A.; Moreno, M.J.; Durocher, Y.; Deb-Rinker, P.; Stanimirovic, D.B. Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGF-beta signaling. Oncogene 2008, 27, 6834–6844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Ma, J.; Han, J.D.; Wang, N.; Chen, Y.G. Distinct regulation of gene expression in human endothelial cells by TGF-beta and its receptors. Microvasc. Res. 2006, 71, 12–19. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Chen, Y.; He, T.L.; Wang, C.; Guo, S.W.; Hu, H.; Ni, C.M.; Jin, G.; Zhang, Y.J. Menin Coordinates C/EBPbeta-Mediated TGF-beta Signaling for Epithelial-Mesenchymal Transition and Growth Inhibition in Pancreatic Cancer. Mol. Ther. Nucleic Acids 2019, 18, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, J.; He, J.; Liu, Y.; Feng, W.; Zhou, H.; Zhou, M.; Wei, H.; Lu, Y.; Peng, W.; et al. Methyl-CpG-binding protein 2 drives the Furin/TGF-beta1/Smad axis to promote epithelial-mesenchymal transition in pancreatic cancer cells. Oncogenesis 2020, 9, 76. [Google Scholar] [CrossRef]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar]
- Duda, D.G.; Sunamura, M.; Lefter, L.P.; Furukawa, T.; Yokoyama, T.; Yatsuoka, T.; Abe, T.; Inoue, H.; Motoi, F.; Egawa, S.; et al. Restoration of SMAD4 by gene therapy reverses the invasive phenotype in pancreatic adenocarcinoma cells. Oncogene 2003, 22, 6857–6864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.; Kong, Y.; Huang, H.; Wang, C.; Zhang, H. TGFbeta signaling in pancreatic ductal adenocarcinoma. Tumour Biol. 2015, 36, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; DeCant, B.; Mascarinas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef] [Green Version]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 Expression Inhibits YAP and TAZ Oncogenic Functions in Pancreatic Cancer. Mol. Cancer Ther. 2020, 19, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Hao, Y.; Zhang, X.; Ward, S.; Houldsworth, J.; Polydorides, A.D.; Harpaz, N. Clinicopathological characterization of SMAD4-mutated intestinal adenocarcinomas: A case-control study. PLoS ONE 2019, 14, e0212142. [Google Scholar] [CrossRef] [Green Version]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Grady, W.M.; Willis, J.E.; Trobridge, P.; Romero-Gallo, J.; Munoz, N.; Olechnowicz, J.; Ferguson, K.; Gautam, S.; Markowitz, S.D. Proliferation and Cdk4 expression in microsatellite unstable colon cancers with TGFBR2 mutations. Int. J. Cancer 2006, 118, 600–608. [Google Scholar] [CrossRef]
- Zhang, B.; Halder, S.K.; Kashikar, N.D.; Cho, Y.J.; Datta, A.; Gorden, D.L.; Datta, P.K. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 2010, 138, 969–980.e3. [Google Scholar] [CrossRef] [Green Version]
- Gatza, C.E.; Holtzhausen, A.; Kirkbride, K.C.; Morton, A.; Gatza, M.L.; Datto, M.B.; Blobe, G.C. Type III TGF-beta receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia 2011, 13, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Ai, X.; Wu, Y.; Zhang, W.; Zhang, Z.; Jin, G.; Zhao, J.; Yu, J.; Lin, Y.; Zhang, W.; Liang, H.; et al. Targeting the ERK pathway reduces liver metastasis of Smad4-inactivated colorectal cancer. Cancer Biol. Ther. 2013, 14, 1059–1067. [Google Scholar] [CrossRef] [Green Version]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208.e113. [Google Scholar] [CrossRef]
- Hu, W.Q.; Wang, L.W.; Yuan, J.P.; Yan, S.G.; Li, J.D.; Zhao, H.L.; Peng, C.W.; Yang, G.F.; Li, Y. High expression of transform growth factor beta 1 in gastric cancer confers worse outcome: Results of a cohort study on 184 patients. Hepatogastroenterology 2014, 61, 245–250. [Google Scholar]
- Xiong, R.; Gao, J.L.; Yin, T. G3BP1 activates the TGF-beta/Smad signaling pathway to promote gastric cancer. Onco Targets Ther. 2019, 12, 7149–7156. [Google Scholar] [CrossRef] [Green Version]
- Leng, A.; Liu, T.; He, Y.; Li, Q.; Zhang, G. Smad4/Smad7 balance: A role of tumorigenesis in gastric cancer. Exp. Mol. Pathol. 2009, 87, 48–53. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, P.; Shao, M.; Zang, X.; Zhang, J.; Mao, F.; Qian, H.; Xu, W. SALL4 activates TGF-beta/SMAD signaling pathway to induce EMT and promote gastric cancer metastasis. Cancer Manag. Res. 2018, 10, 4459–4470. [Google Scholar] [CrossRef] [Green Version]
- Takahata, M.; Inoue, Y.; Tsuda, H.; Imoto, I.; Koinuma, D.; Hayashi, M.; Ichikura, T.; Yamori, T.; Nagasaki, K.; Yoshida, M.; et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J. Biol. Chem. 2009, 284, 3334–3344. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liu, T.; Wu, J.C.; Luo, S.Z.; Chen, R.; Lu, L.G.; Xu, M.Y. STAT3 aggravates TGF-beta1-induced hepatic epithelial-to-mesenchymal transition and migration. Biomed. Pharmacother. 2018, 98, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Li, Z.; Zhang, R.; Jia, C.; Yang, Z.; Zhao, H.; Ya, S.; Mao, R.; Ailijiang, T.; et al. GP73 promotes epithelial-mesenchymal transition and invasion partly by activating TGF-beta1/Smad2 signaling in hepatocellular carcinoma. Carcinogenesis 2018, 39, 900–910. [Google Scholar] [CrossRef]
- Mazzocca, A.; Fransvea, E.; Lavezzari, G.; Antonaci, S.; Giannelli, G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology 2009, 50, 1140–1151. [Google Scholar] [CrossRef]
- Bhagyaraj, E.; Ahuja, N.; Kumar, S.; Tiwari, D.; Gupta, S.; Nanduri, R.; Gupta, P. TGF-beta induced chemoresistance in liver cancer is modulated by xenobiotic nuclear receptor PXR. Cell Cycle 2019, 18, 3589–3602. [Google Scholar] [CrossRef]
- Yan, X.; Wu, J.; Jiang, Q.; Cheng, H.; Han, J.J.; Chen, Y.G. CXXC5 suppresses hepatocellular carcinoma by promoting TGF-beta-induced cell cycle arrest and apoptosis. J. Mol. Cell Biol. 2018, 10, 48–59. [Google Scholar] [CrossRef]
- Wang, B.; Xu, X.; Yang, Z.; Zhang, L.; Liu, Y.; Ma, A.; Xu, G.; Tang, M.; Jing, T.; Wu, L.; et al. POH1 contributes to hyperactivation of TGF-beta signaling and facilitates hepatocellular carcinoma metastasis through deubiquitinating TGF-beta receptors and caveolin-1. EBioMedicine 2019, 41, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Lu, X.; Wang, M.; Zhao, X.; Xue, L. X-linked inhibitor of apoptosis protein accelerates migration by inducing epithelial-mesenchymal transition through TGF-beta signaling pathway in esophageal cancer cells. Cell Biosci. 2019, 9, 76. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, S.; Chen, Y.; Liu, T.; Han, X.; Zhang, X.; Shen, T.; Lu, X. TGF-beta1-Induced Upregulation of MALAT1 Promotes Kazakh’s Esophageal Squamous Cell Carcinoma Invasion by EMT. J. Cancer 2020, 11, 6892–6901. [Google Scholar] [CrossRef]
- Wang, J.; Wu, M.; Zheng, D.; Zhang, H.; Lv, Y.; Zhang, L.; Tan, H.S.; Zhou, H.; Lao, Y.Z.; Xu, H.X. Garcinol inhibits esophageal cancer metastasis by suppressing the p300 and TGF-beta1 signaling pathways. Acta Pharmacol. Sin. 2020, 41, 82–92. [Google Scholar] [CrossRef]
- Parikh, P.Y.; Lillemoe, K.D. Surgical management of pancreatic cancer--distal pancreatectomy. Semin. Oncol. 2015, 42, 110–122. [Google Scholar] [CrossRef]
- Melisi, D.; Calvetti, L.; Frizziero, M.; Tortora, G. Pancreatic cancer: Systemic combination therapies for a heterogeneous disease. Curr. Pharm. Des. 2014, 20, 6660–6669. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Yang, H.; Liu, J.; Zheng, X.; Feng, J.; Li, X.; Li, W. Prognostic Value of SMAD4 in Pancreatic Cancer: A Meta-Analysis. Transl. Oncol. 2016, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Vincent, D.F.; Yan, K.P.; Treilleux, I.; Gay, F.; Arfi, V.; Kaniewski, B.; Marie, J.C.; Lepinasse, F.; Martel, S.; Goddard-Leon, S.; et al. Inactivation of TIF1gamma cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 2009, 5, e1000575. [Google Scholar] [CrossRef]
- Leung, L.; Radulovich, N.; Zhu, C.Q.; Wang, D.; To, C.; Ibrahimov, E.; Tsao, M.S. Loss of canonical Smad4 signaling promotes KRAS driven malignant transformation of human pancreatic duct epithelial cells and metastasis. PLoS ONE 2013, 8, e84366. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liang, Y.; Yin, Q.; Liu, S.; Wang, Q.; Tang, Y.; Cao, C. Clinical and prognostic significance of serum transforming growth factor-beta1 levels in patients with pancreatic ductal adenocarcinoma. Braz. J. Med. Biol. Res. 2016, 49. [Google Scholar] [CrossRef]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-beta signaling pathway and prognosis of pancreatic cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef]
- Otsuru, T.; Kobayashi, S.; Wada, H.; Takahashi, T.; Gotoh, K.; Iwagami, Y.; Yamada, D.; Noda, T.; Asaoka, T.; Serada, S.; et al. Epithelial-mesenchymal transition via transforming growth factor beta in pancreatic cancer is potentiated by the inflammatory glycoprotein leucine-rich alpha-2 glycoprotein. Cancer Sci. 2019, 110, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Merz, V.; Zecchetto, C.; Santoro, R.; Simionato, F.; Sabbadini, F.; Mangiameli, D.; Piro, G.; Cavaliere, A.; Deiana, M.; Valenti, M.T.; et al. Plasma IL8 Is a Biomarker for TAK1 Activation and Predicts Resistance to Nanoliposomal Irinotecan in Patients with Gemcitabine-Refractory Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4661–4669. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Fessler, E.; Medema, J.P. Colorectal Cancer Subtypes: Developmental Origin and Microenvironmental Regulation. Trends Cancer 2016, 2, 505–518. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef]
- Grady, W.M.; Markowitz, S.D. Genetic and epigenetic alterations in colon cancer. Annu. Rev. Genom. Hum. Genet. 2002, 3, 101–128. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Bellam, N.; Pasche, B. Tgf-beta signaling alterations and colon cancer. Cancer Treat. Res. 2010, 155, 85–103. [Google Scholar] [CrossRef]
- Morris, S.M.; Davison, J.; Carter, K.T.; O’Leary, R.M.; Trobridge, P.; Knoblaugh, S.E.; Myeroff, L.L.; Markowitz, S.D.; Brett, B.T.; Scheetz, T.E.; et al. Transposon mutagenesis identifies candidate genes that cooperate with loss of transforming growth factor-beta signaling in mouse intestinal neoplasms. Int. J. Cancer 2017, 140, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Roman, C.; Saha, D.; Beauchamp, R. TGF-beta and colorectal carcinogenesis. Microsc. Res. Tech. 2001, 52, 450–457. [Google Scholar] [CrossRef]
- Pinheiro, M.; Pinto, C.; Peixoto, A.; Veiga, I.; Lopes, P.; Henrique, R.; Baldaia, H.; Carneiro, F.; Seruca, R.; Tomlinson, I.; et al. Target gene mutational pattern in Lynch syndrome colorectal carcinomas according to tumour location and germline mutation. Br. J. Cancer 2015, 113, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaksson-Mettavainio, M.; Palmqvist, R.; Forssell, J.; Stenling, R.; Oberg, A. SMAD4/DPC4 expression and prognosis in human colorectal cancer. Anticancer Res. 2006, 26, 507–510. [Google Scholar] [PubMed]
- Perekatt, A.O.; Shah, P.P.; Cheung, S.; Jariwala, N.; Wu, A.; Gandhi, V.; Kumar, N.; Feng, Q.; Patel, N.; Chen, L.; et al. SMAD4 Suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res. 2018, 78, 4878–4890. [Google Scholar] [CrossRef] [Green Version]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Wu, Y.; Yang, J.; Yang, D.; Fang, X. Progress in the treatment of advanced gastric cancer. Tumour Biol. 2017, 39, 1010428317714626. [Google Scholar] [CrossRef] [Green Version]
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, G.J.A.; Maciejewski, R.; Polkowski, W.P. Gastric cancer: Epidemiology, prevention, classification, and treatment. Cancer Manag. Res. 2018, 10, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.P.; Yu, J.; Miehlke, S.; Fei, G.; Lendeckel, U.; Ridwelski, K.; Stolte, M.; Bayerdorffer, E.; Malfertheiner, P. Expression of transforming growth factor beta-1 in gastric cancer and in the gastric mucosa of first-degree relatives of patients with gastric cancer. Br. J. Cancer 2000, 82, 1795–1800. [Google Scholar] [CrossRef] [Green Version]
- Ohue, M.; Tomita, N.; Monden, T.; Miyoshi, Y.; Ohnishi, T.; Izawa, H.; Kawabata, Y.; Sasaki, M.; Sekimoto, M.; Nishisho, I.; et al. Mutations of the transforming growth factor beta type II receptor gene and microsatellite instability in gastric cancer. Int. J. Cancer 1996, 68, 203–206. [Google Scholar] [CrossRef]
- Kang, S.H.; Bang, Y.J.; Im, Y.H.; Yang, H.K.; Lee, D.A.; Lee, H.Y.; Lee, H.S.; Kim, N.K.; Kim, S.J. Transcriptional repression of the transforming growth factor-beta type I receptor gene by DNA methylation results in the development of TGF-beta resistance in human gastric cancer. Oncogene 1999, 18, 7280–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, X.Z.; Yang, J.O.; Lee, K.Y.; Ito, K.; Sakakura, C.; Li, Q.L.; Kim, H.R.; Cha, E.J.; Lee, Y.H.; Kaneda, A.; et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor {beta}-activated SMAD. Mol. Cell Biol. 2005, 25, 8097–8107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef]
- Yoo, J.; Ghiassi, M.; Jirmanova, L.; Balliet, A.G.; Hoffman, B.; Fornace, A.J., Jr.; Liebermann, D.A.; Bottinger, E.P.; Roberts, A.B. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 2003, 278, 43001–43007. [Google Scholar] [CrossRef] [Green Version]
- Perlman, R.; Schiemann, W.P.; Brooks, M.W.; Lodish, H.F.; Weinberg, R.A. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 2001, 3, 708–714. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Yoshimura, T.; Matsuhashi, S.; Eguchi, Y.; Yasutake, T.; Hisatomi, A.; Sakai, T.; Yamamoto, K. Transforming growth factor-beta 1-induced apoptosis is blocked by beta 1-integrin-mediated mitogen-activated protein kinase activation in human hepatoma cells. Cancer Sci. 2004, 95, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Burch, M.L.; Zheng, W.; Little, P.J. Smad linker region phosphorylation in the regulation of extracellular matrix synthesis. Cell. Mol. Life Sci. 2011, 68, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Short, M.W.; Burgers, K.G.; Fry, V.T. Esophageal Cancer. Am. Fam. Physician 2017, 95, 22–28. [Google Scholar]
- Li, Y.; Yang, H.X.; Luo, R.Z.; Zhang, Y.; Li, M.; Wang, X.; Jia, W.H. High expression of p300 has an unfavorable impact on survival in resectable esophageal squamous cell carcinoma. Ann. Thorac. Surg. 2011, 91, 1531–1538. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Ding, S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10, 590941. [Google Scholar] [CrossRef]
- Gambardella, V.; Castillo, J.; Tarazona, N.; Gimeno-Valiente, F.; Martinez-Ciarpaglini, C.; Cabeza-Segura, M.; Rosello, S.; Roda, D.; Huerta, M.; Cervantes, A.; et al. The role of tumor-associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat. Rev. 2020, 86, 102015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef]
- Huang, Y.; Ge, W.; Zhou, J.; Gao, B.; Qian, X.; Wang, W. The Role of Tumor Associated Macrophages in Hepatocellular Carcinoma. J. Cancer 2021, 12, 1284–1294. [Google Scholar] [CrossRef]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Wahl, S.M.; Allen, J.B.; Weeks, B.S.; Wong, H.L.; Klotman, P.E. Transforming growth factor beta enhances integrin expression and type IV collagenase secretion in human monocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 4577–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Kim, J.G.; Moon, M.Y.; Jeon, C.Y.; Won, H.Y.; Kim, H.J.; Jeon, Y.J.; Seo, J.Y.; Kim, J.I.; Kim, J.; et al. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood 2006, 108, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Standiford, T.J.; Kuick, R.; Bhan, U.; Chen, J.; Newstead, M.; Keshamouni, V.G. TGF-beta-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 2011, 30, 2475–2484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Lim, S.; Li, A.G.; Lee, C.; Lee, Y.S.; Lee, E.K.; Park, S.H.; Wang, X.J.; Kim, S.J. Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat. Immunol. 2007, 8, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Xia, L.; Li, J.; Ni, S.; Song, H.; Wu, X. Tumor-Associated Macrophages Derived TGF-betaInduced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells through Smad2,3-4/Snail Signaling Pathway. Cancer Res. Treat. 2019, 51, 252–266. [Google Scholar] [CrossRef]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-beta secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle 2018, 17, 2766–2778. [Google Scholar] [CrossRef] [Green Version]
- Xiong, C.; Zhu, Y.; Xue, M.; Jiang, Y.; Zhong, Y.; Jiang, L.; Shi, M.; Chen, H. Tumor-associated macrophages promote pancreatic ductal adenocarcinoma progression by inducing epithelial-to-mesenchymal transition. Aging 2021, 13, 3386–3404. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Reibman, J.; Meixler, S.; Lee, T.C.; Gold, L.I.; Cronstein, B.N.; Haines, K.A.; Kolasinski, S.L.; Weissmann, G. Transforming growth factor beta 1, a potent chemoattractant for human neutrophils, bypasses classic signal-transduction pathways. Proc. Natl. Acad. Sci. USA 1991, 88, 6805–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Pang, Y.; Gara, S.K.; Achyut, B.R.; Heger, C.; Goldsmith, P.K.; Lonning, S.; Yang, L. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-beta in breast cancer progression. Int. J. Cancer 2012, 131, 2584–2595. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Franciszkiewicz, K.; Le Floc’h, A.; Jalil, A.; Vigant, F.; Robert, T.; Vergnon, I.; Mackiewicz, A.; Benihoud, K.; Validire, P.; Chouaib, S.; et al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. 2009, 69, 6249–6255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, N.; Kuranaga, Y.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Akao, Y. Colorectal cancer cell-derived extracellular vesicles induce phenotypic alteration of T cells into tumor-growth supporting cells with transforming growth factor-beta1-mediated suppression. Oncotarget 2016, 7, 27033–27043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. 2018, 1417, 104–115. [Google Scholar] [CrossRef]
- Moo-Young, T.A.; Larson, J.W.; Belt, B.A.; Tan, M.C.; Hawkins, W.G.; Eberlein, T.J.; Goedegebuure, P.S.; Linehan, D.C. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J. Immunother. 2009, 32, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C. TH17 cells in development: An updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 2008, 8, 337–348. [Google Scholar] [CrossRef]
- Kinoshita, M.; Kobayashi, S.; Gotoh, K.; Kubo, M.; Hayashi, K.; Iwagami, Y.; Yamada, D.; Akita, H.; Noda, T.; Asaoka, T.; et al. Heterogeneity of Treg/Th17 According to Cancer Progression and Modification in Biliary Tract Cancers via Self-Producing Cytokines. Dig. Dis. Sci. 2020, 65, 2937–2948. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Mo, Q.; Dong, Y.; Wang, G.; Ji, A. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. Int. J. Clin. Exp. Pathol. 2015, 8, 5702–5708. [Google Scholar]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [Green Version]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal. Cell Pathol. 2010, 33, 61–79. [Google Scholar] [CrossRef]
- Fukino, K.; Shen, L.; Matsumoto, S.; Morrison, C.D.; Mutter, G.L.; Eng, C. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004, 64, 7231–7236. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.W.; Lind Nielsen, H.; Gudjonsson, T.; Villadsen, R.; Ronnov-Jessen, L.; Bissell, M.J. The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res. 2001, 3, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Yoon, H.; Tang, C.M.; Banerjee, S.; Delgado, A.L.; Yebra, M.; Davis, J.; Sicklick, J.K. TGF-beta1-mediated transition of resident fibroblasts to cancer-associated fibroblasts promotes cancer metastasis in gastrointestinal stromal tumor. Oncogenesis 2021, 10, 13. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Hawinkels, L.J.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Zubeldia, I.; Dotor, J.; Redrado, M.; Bleau, A.M.; Manrique, I.; de Aberasturi, A.L.; Villalba, M.; Calvo, A. Co-migration of colon cancer cells and CAFs induced by TGFbeta(1) enhances liver metastasis. Cell Tissue Res. 2015, 359, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.; Harryvan, T.J.; Groenewoud, A.; van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Snaar-Jagalska, B.E.; et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, M.F.; Mead, A.; Ali, R.R.; Alexander, R.A.; Murray, S.; Chen, C.; York-Defalco, C.; Dean, N.M.; Schultz, G.S.; Khaw, P.T. Novel antisense oligonucleotides targeting TGF-beta inhibit in vivo scarring and improve surgical outcome. Gene Ther. 2003, 10, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, D.R.; Bielefeldt-Ohmann, H.; Himbeck, R.P.; Jarnicki, A.G.; Marzo, A.L.; Robinson, B.W. Transforming growth factor-beta: Antisense RNA-mediated inhibition affects anchorage-independent growth, tumorigenicity and tumor-infiltrating T-cells in malignant mesothelioma. Growth Factors 1994, 11, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Yang, T.; Janulis, L.; Goodwin, S.; Kundu, S.D.; Karpus, W.J.; Lee, C. Down-regulation of TGF-beta1 production restores immunogenicity in prostate cancer cells. Br. J. Cancer 2000, 83, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Kemaladewi, D.U.; Pasteuning, S.; van der Meulen, J.W.; van Heiningen, S.H.; van Ommen, G.J.; Ten Dijke, P.; Aartsma-Rus, A.; t Hoen, P.A.; Hoogaars, W.M. Targeting TGF-beta Signaling by Antisense Oligonucleotide-mediated Knockdown of TGF-beta Type I Receptor. Mol. Ther. Nucleic Acids 2014, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, O.J.; Qazi, S.; Hwang, L.; Ng, K.; Trieu, V. Impact of targeting transforming growth factor beta-2 with antisense OT-101 on the cytokine and chemokine profile in patients with advanced pancreatic cancer. Onco Targets Ther. 2018, 11, 2779–2796. [Google Scholar] [CrossRef] [Green Version]
- Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Therapeutic Targeting of TGFbeta Ligands in Glioblastoma Using Novel Antisense Oligonucleotides Reduces the Growth of Experimental Gliomas. Clin. Cancer Res. 2019, 25, 7189–7201. [Google Scholar] [CrossRef] [Green Version]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Lonning, S.; Mannick, J.; McPherson, J.M. Antibody targeting of TGF-beta in cancer patients. Curr. Pharm. Biotechnol. 2011, 12, 2176–2189. [Google Scholar] [CrossRef]
- Bedinger, D.; Lao, L.; Khan, S.; Lee, S.; Takeuchi, T.; Mirza, A.M. Development and characterization of human monoclonal antibodies that neutralize multiple TGFbeta isoforms. MAbs 2016, 8, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy, V.; Ge, R.; Grazioli, A.; Xie, W.; Banach-Petrosky, W.; Kang, Y.; Lonning, S.; McPherson, J.; Yingling, J.M.; Biswas, S.; et al. Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 2010, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, N.J.; Li, L.; Kapoun, A.M.; Medicherla, S.; Reddy, M.; Li, G.; O’Young, G.; Quon, D.; Henson, M.; Damm, D.L.; et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol. Pharmacol. 2007, 72, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.P.; Hahn, T.; Ramanathapuram, L.; Bradley-Dunlop, D.; Mahadevan, D.; Mercado-Pimentel, M.E.; Runyan, R.B.; Besselsen, D.G.; Zhang, X.; Cheung, H.K.; et al. An orally active small molecule TGF-beta receptor I antagonist inhibits the growth of metastatic murine breast cancer. Anticancer Res. 2009, 29, 2099–2109. [Google Scholar] [PubMed]
- Shinto, O.; Yashiro, M.; Kawajiri, H.; Shimizu, K.; Shimizu, T.; Miwa, A.; Hirakawa, K. Inhibitory effect of a TGFbeta receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous gastric cancer cells. Br. J. Cancer 2010, 102, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Lee, H.J.; Park, S.J.; Park, H.J.; Lee, K.; Sheen, Y.Y.; et al. Discovery of N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2 -yl)methyl)-2-fluoroaniline (EW-7197): A highly potent, selective, and orally bioavailable inhibitor of TGF-beta type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J. Med. Chem. 2014, 57, 4213–4238. [Google Scholar] [CrossRef]
- Son, J.Y.; Park, S.Y.; Kim, S.J.; Lee, S.J.; Park, S.A.; Kim, M.J.; Kim, S.W.; Kim, D.K.; Nam, J.S.; Sheen, Y.Y. EW-7197, a novel ALK-5 kinase inhibitor, potently inhibits breast to lung metastasis. Mol. Cancer Ther. 2014, 13, 1704–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueorguieva, I.; Tabernero, J.; Melisi, D.; Macarulla, T.; Merz, V.; Waterhouse, T.H.; Miles, C.; Lahn, M.M.; Cleverly, A.; Benhadji, K.A. Population pharmacokinetics and exposure-overall survival analysis of the transforming growth factor-beta inhibitor galunisertib in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 84, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.; Wild, C.P. World Cancer Report 2014; International Agency for Research on Cancer (IARC): Lyon, France, 2014. [Google Scholar]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; de Gramont, A. Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef] [Green Version]
- Hwang, L.; Ng, K.; Wang, W.; Trieu, V. Abstract 3742: Treatment with trabedersen, an anti-TGF-beta 2 antisense, primed tumors to subsequent chemotherapies. Cancer Res. 2016, 76, 3742. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Role of TGF-β signaling in GI Tumors | |
|---|---|
| Pancreatic Cancer | |
| Colorectal Cancer |
|
| Gastric Cancer | |
| Hepatocellular Carcinoma | |
| Esophageal Cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabbadini, F.; Bertolini, M.; De Matteis, S.; Mangiameli, D.; Contarelli, S.; Pietrobono, S.; Melisi, D. The Multifaceted Role of TGF-β in Gastrointestinal Tumors. Cancers 2021, 13, 3960. https://doi.org/10.3390/cancers13163960
Sabbadini F, Bertolini M, De Matteis S, Mangiameli D, Contarelli S, Pietrobono S, Melisi D. The Multifaceted Role of TGF-β in Gastrointestinal Tumors. Cancers. 2021; 13(16):3960. https://doi.org/10.3390/cancers13163960
Chicago/Turabian StyleSabbadini, Fabio, Monica Bertolini, Serena De Matteis, Domenico Mangiameli, Serena Contarelli, Silvia Pietrobono, and Davide Melisi. 2021. "The Multifaceted Role of TGF-β in Gastrointestinal Tumors" Cancers 13, no. 16: 3960. https://doi.org/10.3390/cancers13163960
APA StyleSabbadini, F., Bertolini, M., De Matteis, S., Mangiameli, D., Contarelli, S., Pietrobono, S., & Melisi, D. (2021). The Multifaceted Role of TGF-β in Gastrointestinal Tumors. Cancers, 13(16), 3960. https://doi.org/10.3390/cancers13163960