
Neural Mechanisms of Cancer Cachexia


Abstract
:Simple Summary
Abstract

1. Introduction
2. Central Inflammation: Lessons from IL-1β
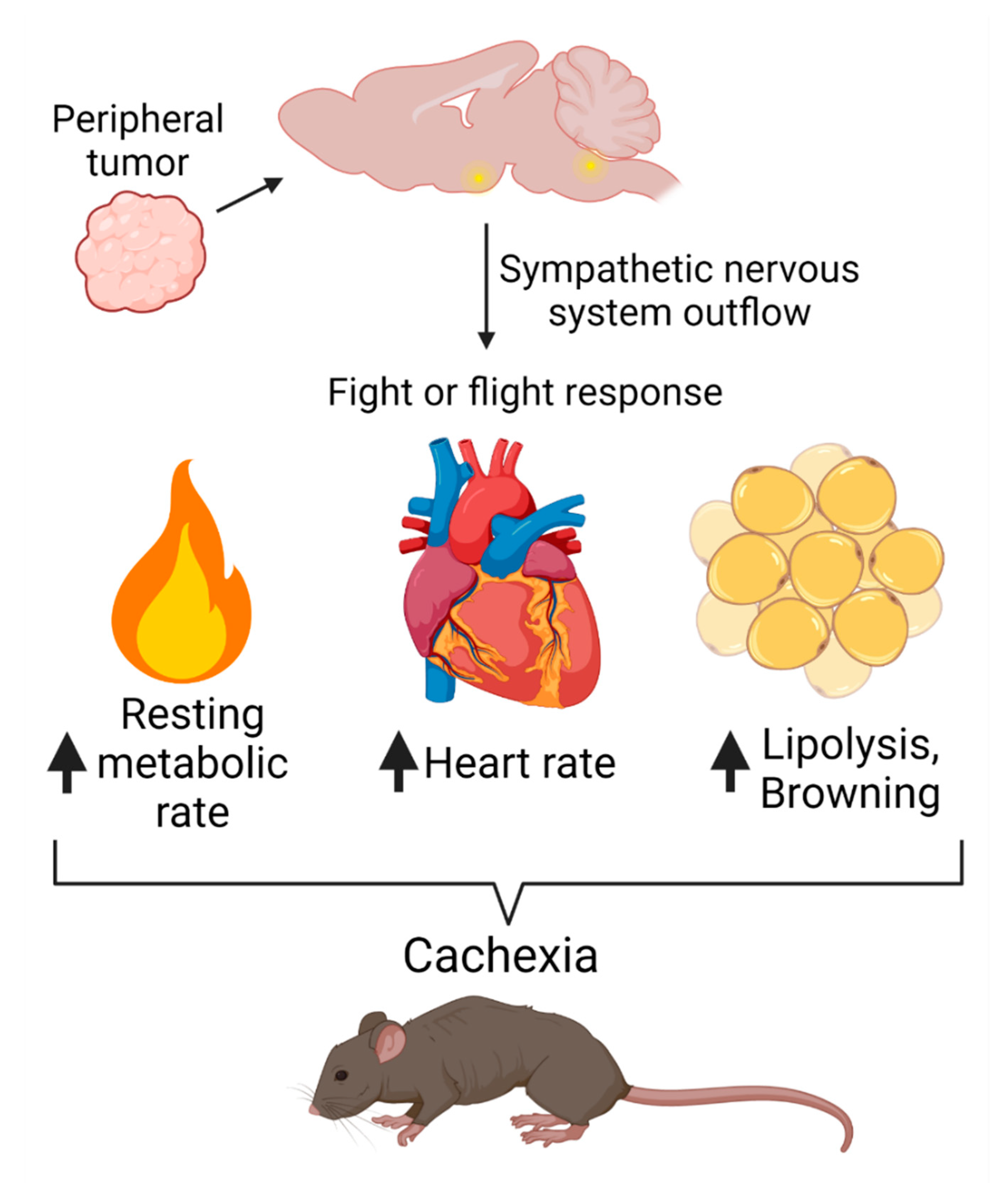
3. Sympathetic Nervous System Engagement
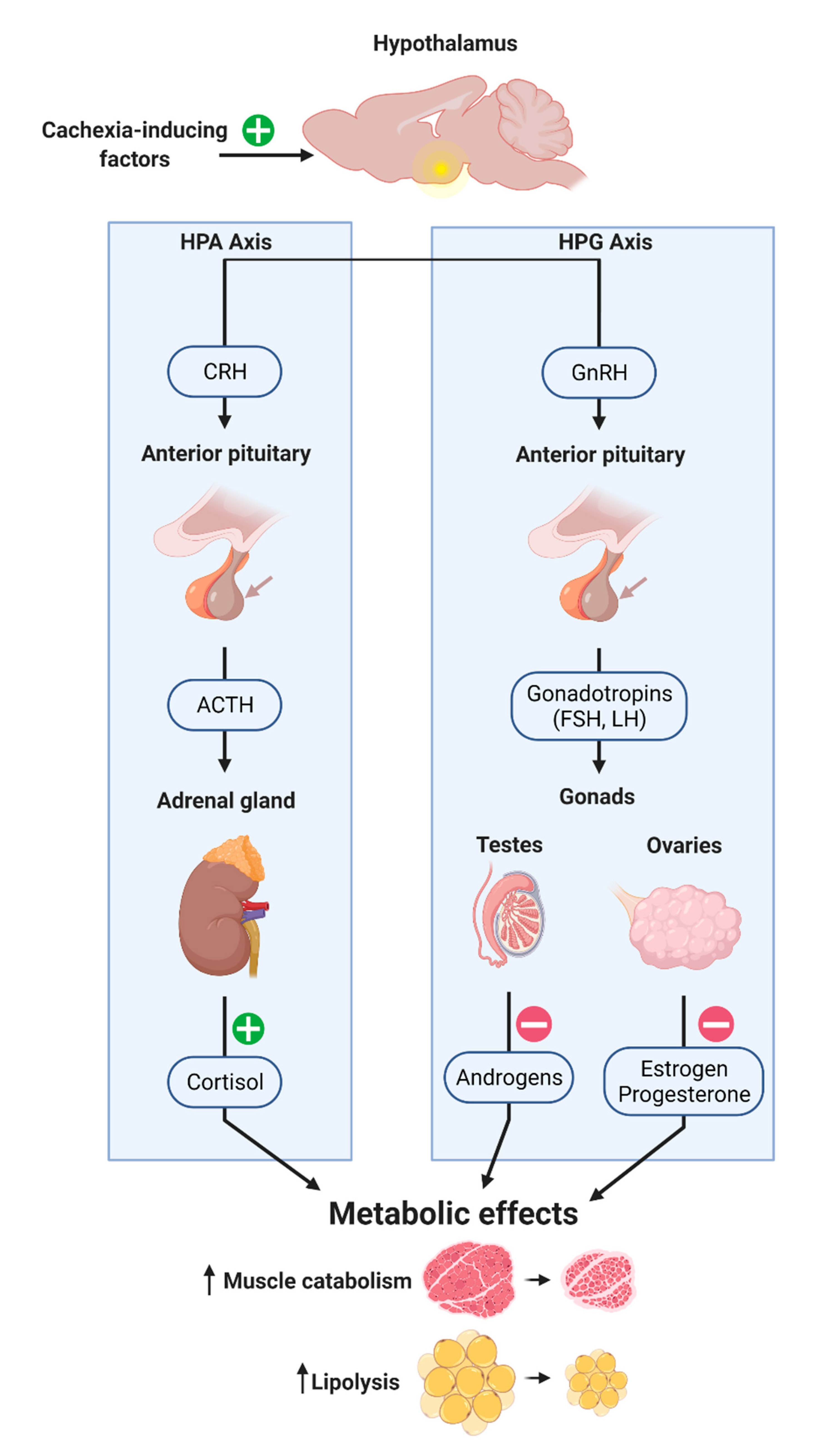
4. Neuroendocrine Modulation
5. Recently Identified CNS Mediators of Cachexia: GDF15, LCN2 and INSL3
5.1. GDF15
5.2. LCN2
5.3. INSL3
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
References
- Olson, B.; Marks, D.L.; Grossberg, A.J. Diverging metabolic programmes and behaviours during states of starvation, protein malnutrition, and cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 1429–1446. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Scarlett, J.M.; Marks, D.L. Hypothalamic mechanisms in cachexia. Physiol. Behav. 2010, 100, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.J.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J.G.; Krasnow, S.M.; Marks, D.L. Interleukin-1beta signaling in fenestrated capillaries is sufficient to trigger sickness responses in mice. J. Neuroinflamm. 2017, 14, 219. [Google Scholar] [CrossRef] [Green Version]
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2016, 54, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, M.; Molfese, D.L.; Viswanath, H.; Curtis, K.; Jones, A.; Hayes, T.G.; Marcelli, M.; Mediwala, S.; Baldwin, P.; Garcia, J.M.; et al. The habenula as a novel link between the homeostatic and hedonic pathways in cancer-associated weight loss: A pilot study. J. Cachexia Sarcopenia Muscle 2018, 9, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Buenafe, A.C.; Olson, B.; Michaelis, K.A.; Torres, E.R.; Jeng, S.; et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. eLife 2020, 9, e54095. [Google Scholar] [CrossRef]
- Seelaender, M.; Laviano, A.; Busquets, S.; Püschel, G.P.; Margaria, T.; Batista, M.L., Jr. Inflammation in Cachexia. Mediat. Inflamm. 2015, 2015, 536954. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Robertson, G. Cancer cachexia: Malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 2013, 24, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Stemmler, B.; López-Soriano, F.J.; Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 2019, 15, 9–20. [Google Scholar] [CrossRef]
- Scott, H.R.; McMillan, D.C.; Crilly, A.; McArdle, C.S.; Milroy, R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br. J. Cancer 1996, 73, 1560–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, J.; Tachibana, M.; Ueno, M.; Miyajima, A.; Baba, S.; Murai, M. Association between tumor necrosis factor in serum and cachexia in patients with prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 1743–1748. [Google Scholar]
- Derman, B.A.; Macklis, J.N.; Azeem, M.S.; Sayidine, S.; Basu, S.; Batus, M.; Esmail, F.; Borgia, J.A.; Bonomi, P.; Fidler, M.J. Relationships between longitudinal neutrophil to lymphocyte ratios, body weight changes, and overall survival in patients with non-small cell lung cancer. BMC Cancer 2017, 17, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borish, L.C.; Steinke, J.W. 2. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111, S460–S475. [Google Scholar] [CrossRef]
- Steinke, J.W.; Borish, L. 3. Cytokines and chemokines. J. Allergy Clin. Immunol. 2006, 117, S441–S445. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11. [Google Scholar] [CrossRef]
- Seruga, B.; Zhang, H.; Bernstein, L.J.; Tannock, I.F. Cytokines and their relationship to the symptoms and outcome of cancer. Nat. Rev. Cancer 2008, 8, 887–899. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; Lόpez-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef]
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef]
- Olson, B.; Norgard, M.A.; Levasseur, P.R.; Zhu, X.; Marks, D.L. Physiologic and molecular characterization of a novel murine model of metastatic head and neck cancer cachexia. J. Cachexia Sarcopenia Muscle 2021. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.J. Endotoxin-induced activation of cerebral catecholamine and serotonin metabolism: Comparison with interleukin-1. J. Pharmacol. Exp. Ther. 1992, 261, 964–969. [Google Scholar] [PubMed]
- Zalcman, S.; Green-Johnson, J.M.; Murray, L.; Nance, D.M.; Dyck, D.; Anisman, H.; Greenberg, A.H. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res. 1994, 643, 40–49. [Google Scholar] [CrossRef]
- Müller, N.; Ackenheil, M. Psychoneuroimmunology and the cytokine action in the CNS: Implications for psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 1998, 22, 1–33. [Google Scholar] [CrossRef]
- Tancredi, V.; Zona, C.; Velotti, F.; Eusebi, F.; Santoni, A. Interleukin-2 suppresses established long-term potentiation and inhibits its induction in the rat hippocampus. Brain Res. 1990, 525, 149–151. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Seto, D.; Quirion, R. Modulation of hippocampal acetylcholine release: A potent central action of interleukin-2. J. Neurosci. Off. J. Soc. Neurosci. 1993, 13, 3368–3374. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.D.; Krieg, R.J.; Povlishock, J.T. Differential central nervous system responses following single and multiple recombinant interleukin-2 infusions. J. Neuroimmunol. 1990, 28, 249–260. [Google Scholar] [CrossRef]
- Qiu, Z.; Sweeney, D.D.; Netzeband, J.G.; Gruol, D.L. Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 10445–10456. [Google Scholar] [CrossRef]
- Krady, J.K.; Lin, H.W.; Liberto, C.M.; Basu, A.; Kremlev, S.G.; Levison, S.W. Ciliary neurotrophic factor and interleukin-6 differentially activate microglia. J. Neurosci. Res. 2008, 86, 1538–1547. [Google Scholar] [CrossRef]
- Arruda, A.P.; Milanski, M.; Romanatto, T.; Solon, C.; Coope, A.; Alberici, L.C.; Festuccia, W.T.; Hirabara, S.M.; Ropelle, E.; Curi, R.; et al. Hypothalamic actions of tumor necrosis factor alpha provide the thermogenic core for the wastage syndrome in cachexia. Endocrinology 2010, 151, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, R.; Kamilaris, T.C.; Calogero, A.E.; Johnson, E.O.; Gomez, M.T.; Gold, P.W.; Chrousos, G.P. Interactions between tumor necrosis factor-alpha, hypothalamic corticotropin-releasing hormone, and adrenocorticotropin secretion in the rat. Endocrinology 1990, 126, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Romanatto, T.; Cesquini, M.; Amaral, M.E.; Roman, E.A.; Moraes, J.C.; Torsoni, M.A.; Cruz-Neto, A.P.; Velloso, L.A. TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient—Effects on leptin and insulin signaling pathways. Peptides 2007, 28, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Wang, B.; Kodali, M.C.; Chen, C.; Kim, E.; Patters, B.J.; Lan, L.; Kumar, S.; Wang, X.; Yue, J.; et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflamm. 2018, 15, 8. [Google Scholar] [CrossRef]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, V.R.; Micheletti, T.O.; Pimentel, G.D.; Katashima, C.K.; Lenhare, L.; Morari, J.; Mendes, M.C.; Razolli, D.S.; Rocha, G.Z.; de Souza, C.T.; et al. Hypothalamic S1P/S1PR1 axis controls energy homeostasis. Nat. Commun. 2014, 5, 4859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Dwarkasing, J.T.; Boekschoten, M.V.; Argilès, J.M.; van Dijk, M.; Busquets, S.; Penna, F.; Toledo, M.; Laviano, A.; Witkamp, R.F.; van Norren, K. Differences in food intake of tumour-bearing cachectic mice are associated with hypothalamic serotonin signalling. J. Cachexia Sarcopenia Muscle 2015, 6, 84–94. [Google Scholar] [CrossRef]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Borner, T.; Liberini, C.G.; Lutz, T.A.; Riediger, T. Brainstem GLP-1 signalling contributes to cancer anorexia-cachexia syndrome in the rat. Neuropharmacology 2018, 131, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Sonti, G.; Ilyin, S.E.; Plata-Salamán, C.R. Anorexia induced by cytokine interactions at pathophysiological concentrations. Am. J. Physiol. 1996, 270, R1394–R1402. [Google Scholar] [CrossRef]
- Plata-Salamán, C.R. Anorexia induced by activators of the signal transducer gp 130. Neuroreport 1996, 7, 841–844. [Google Scholar] [CrossRef]
- Dinarello, C.A. The proinflammatory cytokines interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J. Infect. Dis. 1991, 163, 1177–1184. [Google Scholar] [CrossRef]
- Turrin, N.P.; Plata-Salamán, C.R. Cytokine-cytokine interactions and the brain. Brain Res. Bull. 2000, 51, 3–9. [Google Scholar] [CrossRef]
- Carlson, C.D.; Bai, Y.; Jonakait, G.M.; Hart, R.P. Interleukin-1 beta increases leukemia inhibitory factor mRNA levels through transient stimulation of transcription rate. Glia 1996, 18, 141–151. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Zhu, X.; Burfeind, K.G.; Krasnow, S.M.; Levasseur, P.R.; Morgan, T.K.; Marks, D.L. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 824–838. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Zhu, X.; Levasseur, P.R.; Michaelis, K.A.; Norgard, M.A.; Marks, D.L. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain Behav. Immun. 2018, 73, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nemeth, D.P.; McKim, D.B.; Zhu, L.; DiSabato, D.J.; Berdysz, O.; Gorantla, G.; Oliver, B.; Witcher, K.G.; Wang, Y.; et al. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity 2019, 50, 312–333. [Google Scholar] [CrossRef] [Green Version]
- Krasnow, S.M.; Knoll, J.G.; Verghese, S.C.; Levasseur, P.R.; Marks, D.L. Amplification and propagation of interleukin-1beta signaling by murine brain endothelial and glial cells. J. Neuroinflamm. 2017, 14, 133. [Google Scholar] [CrossRef]
- Zhu, X.; Burfeind, K.G.; Michaelis, K.A.; Braun, T.P.; Olson, B.; Pelz, K.R.; Morgan, T.K.; Marks, D.L. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Ruud, J.; Backhed, F.; Engblom, D.; Blomqvist, A. Deletion of the gene encoding MyD88 protects from anorexia in a mouse tumor model. Brain Behav. Immun. 2010, 24, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Vichaya, E.G.; Christian, D.L.; Molkentine, J.M.; Vermeer, D.W.; Gross, P.S.; Vermeer, P.D.; Lee, J.H.; Dantzer, R. Tumor-Associated Fatigue in Cancer Patients Develops Independently of IL1 Signaling. Cancer Res. 2018, 78, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, A.; Dakhil, S.R.; Nguyen, P.L.; Sloan, J.A.; Kugler, J.W.; Rowland, K.M., Jr.; Soori, G.S.; Wender, D.B.; Fitch, T.R.; Novotny, P.J.; et al. A placebo-controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: Results from N00C1 from the North Central Cancer Treatment Group. Cancer 2007, 110, 1396–1403. [Google Scholar] [CrossRef]
- Hill, A.G.; Jacobson, L.; Gonzalez, J.; Rounds, J.; Majzoub, J.A.; Wilmore, D.W. Chronic central nervous system exposure to interleukin-1 beta causes catabolism in the rat. Am. J. Physiol. 1996, 271, R1142–R1148. [Google Scholar] [CrossRef]
- Schafers, M.; Lee, D.H.; Brors, D.; Yaksh, T.L.; Sorkin, L.S. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J. Neurosci. 2003, 23, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Van Riemsdijk, I.C.; Baan, C.C.; Loonen, E.H.; Knoop, C.J.; Navarro Betonico, G.; Niesters, H.G.; Zietse, R.; Weimar, W. T cells activate the tumor necrosis factor-alpha system during hemodialysis, resulting in tachyphylaxis. Kidney Int. 2001, 59, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Brouckaert, P.; Fiers, W. Induction of tolerance allows separation of lethal and antitumor activities of tumor necrosis factor in mice. Cancer Res. 1991, 51, 2366–2372. [Google Scholar]
- Tsoli, M.; Moore, M.; Burg, D.; Painter, A.; Taylor, R.; Lockie, S.H.; Turner, N.; Warren, A.; Cooney, G.; Oldfield, B.; et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. 2012, 72, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; Flak, J.; Jankord, R. Chronic stress plasticity in the hypothalamic paraventricular nucleus. Prog. Brain Res. 2008, 170, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef]
- Brooks, S.L.; Neville, A.M.; Rothwell, N.J.; Stock, M.J.; Wilson, S. Sympathetic activation of brown-adipose-tissue thermogenesis in cachexia. Biosci. Rep. 1981, 1, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Bing, C.; Brown, M.; King, P.; Collins, P.; Tisdale, M.J.; Williams, G. Increased gene expression of brown fat uncoupling protein (UCP)1 and skeletal muscle UCP2 and UCP3 in MAC16-induced cancer cachexia. Cancer Res. 2000, 60, 2405–2410. [Google Scholar]
- Hundsberger, T.; Omlin, A.; Haegele-Link, S.; Vehoff, J.; Strasser, F. Autonomic dysfunction in cancer cachexia coincides with large fiber polyneuropathy. J. Pain Symptom Manag. 2014, 48, 611–618.e1. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Sequeria, A.; Manderson, C.; Maddocks, M.; Wasley, D.; Wilcock, A. Exploring autonomic nervous system dysfunction in patients with cancer cachexia: A pilot study. Auton. Neurosci. Basic Clin. 2012, 166, 93–95. [Google Scholar] [CrossRef]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef]
- Jiang, Z.; Rajamanickam, S.; Justice, N.J. Local Corticotropin-Releasing Factor Signaling in the Hypothalamic Paraventricular Nucleus. J. Neurosci. 2018, 38, 1874–1890. [Google Scholar] [CrossRef]
- Kim, M.S.; Yan, J.; Wu, W.; Zhang, G.; Zhang, Y.; Cai, D. Rapid linkage of innate immunological signals to adaptive immunity by the brain-fat axis. Nat. Immunol. 2015, 16, 525–533. [Google Scholar] [CrossRef]
- Stanley, S.; Pinto, S.; Segal, J.; Pérez, C.A.; Viale, A.; DeFalco, J.; Cai, X.; Heisler, L.K.; Friedman, J.M. Identification of neuronal subpopulations that project from hypothalamus to both liver and adipose tissue polysynaptically. Proc. Natl. Acad. Sci. USA 2010, 107, 7024–7029. [Google Scholar] [CrossRef] [Green Version]
- Habecker, B.A.; Sachs, H.H.; Rohrer, H.; Zigmond, R.E. The dependence on gp130 cytokines of axotomy induced neuropeptide expression in adult sympathetic neurons. Dev. Neurobiol. 2009, 69, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Luan, H.H.; Wang, A.; Hilliard, B.K.; Carvalho, F.; Rosen, C.E.; Ahasic, A.M.; Herzog, E.L.; Kang, I.; Pisani, M.A.; Yu, S.; et al. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 2019, 178, 1231–1244.e11. [Google Scholar] [CrossRef]
- Lerner, L.; Hayes, T.G.; Tao, N.; Krieger, B.; Feng, B.; Wu, Z.; Nicoletti, R.; Chiu, M.I.; Gyuris, J.; Garcia, J.M. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachexia Sarcopenia Muscle 2015, 6, 317–324. [Google Scholar] [CrossRef]
- Lerner, L.; Tao, J.; Liu, Q.; Nicoletti, R.; Feng, B.; Krieger, B.; Mazsa, E.; Siddiquee, Z.; Wang, R.; Huang, L.; et al. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 2016, 7, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Suriben, R.; Chen, M.; Higbee, J.; Oeffinger, J.; Ventura, R.; Li, B.; Mondal, K.; Gao, Z.; Ayupova, D.; Taskar, P.; et al. Antibody-mediated inhibition of GDF15-GFRAL activity reverses cancer cachexia in mice. Nat. Med. 2020, 26, 1264–1270. [Google Scholar] [CrossRef]
- Mendes, M.C.; Pimentel, G.D.; Costa, F.O.; Carvalheira, J.B. Molecular and neuroendocrine mechanisms of cancer cachexia. J. Endocrinol. 2015, 226, R29–R43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laviano, A.; Inui, A.; Marks, D.L.; Meguid, M.M.; Pichard, C.; Rossi Fanelli, F.; Seelaender, M. Neural control of the anorexia-cachexia syndrome. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1000–E1008. [Google Scholar] [CrossRef] [PubMed]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Michaelis, K.A.; Olson, B.; Marks, D.L. Microglia in the hypothalamus respond to tumor-derived factors and are protective against cachexia during pancreatic cancer. Glia 2020, 68, 1479–1494. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Grossberg, A.J.; Krasnow, S.M.; Levasseur, P.R.; Szumowski, M.; Zhu, X.X.; Maxson, J.E.; Knoll, J.G.; Barnes, A.P.; Marks, D.L. Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J. 2013, 27, 3572–3582. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Szumowski, M.; Levasseur, P.R.; Grossberg, A.J.; Zhu, X.; Agarwal, A.; Marks, D.L. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014, 9, e106489. [Google Scholar] [CrossRef] [Green Version]
- Waddell, D.S.; Baehr, L.M.; van den Brandt, J.; Johnsen, S.A.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E785–E797. [Google Scholar] [CrossRef]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Burney, B.O.; Garcia, J.M. Hypogonadism in male cancer patients. J. Cachexia Sarcopenia Muscle 2012, 3, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, R. The assessment and management of cancer cachexia: Hypogonadism and hypermetabolism among supportive and palliative care patients. Curr. Opin. Support. Palliat. Care 2014, 8, 279–285. [Google Scholar] [CrossRef]
- Garcia, J.M.; Li, H.; Mann, D.; Epner, D.; Hayes, T.G.; Marcelli, M.; Cunningham, G.R. Hypogonadism in male patients with cancer. Cancer 2006, 106, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, A.; Vassilopoulou-Sellin, R.; Palmer, J.L.; Kaur, G.; Bruera, E. Symptomatic hypogonadism in male survivors of cancer with chronic exposure to opioids. Cancer 2004, 100, 851–858. [Google Scholar] [CrossRef]
- Skipworth, R.J.; Moses, A.G.; Sangster, K.; Sturgeon, C.M.; Voss, A.C.; Fallon, M.T.; Anderson, R.A.; Ross, J.A.; Fearon, K.C. Interaction of gonadal status with systemic inflammation and opioid use in determining nutritional status and prognosis in advanced pancreatic cancer. Support. Care Cancer Off. J. Multinatl. Assoc. Support. Care Cancer 2011, 19, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Dev, R.; Hui, D.; Del Fabbro, E.; Delgado-Guay, M.O.; Sobti, N.; Dalal, S.; Bruera, E. Association between hypogonadism, symptom burden, and survival in male patients with advanced cancer. Cancer 2014, 120, 1586–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chang, C.C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjær, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Borner, T.; Shaulson, E.D.; Ghidewon, M.Y.; Barnett, A.B.; Horn, C.C.; Doyle, R.P.; Grill, H.J.; Hayes, M.R.; De Jonghe, B.C. GDF15 Induces Anorexia through Nausea and Emesis. Cell Metab. 2020, 31, 351–362.e5. [Google Scholar] [CrossRef]
- Kjeldsen, L.; Cowland, J.B.; Borregaard, N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta 2000, 1482, 272–283. [Google Scholar] [CrossRef]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- Mosialou, I.; Shikhel, S.; Liu, J.M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 543, 385–390. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, H.; Deis, J.A.; Mashek, M.G.; Zhao, M.; Ariyakumar, D.; Armien, A.G.; Bernlohr, D.A.; Mashek, D.G.; Chen, X. Lipocalin 2 regulates brown fat activation via a nonadrenergic activation mechanism. J. Biol. Chem. 2014, 289, 22063–22077. [Google Scholar] [CrossRef] [Green Version]
- Meyers, K.; López, M.; Ho, J.; Wills, S.; Rayalam, S.; Taval, S. Lipocalin-2 deficiency may predispose to the progression of spontaneous age-related adiposity in mice. Sci. Rep. 2020, 10, 14589. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Butler, J.T.; Buenafe, A.; Burfeind, K.G.; Michaelis, K.A.; Pelz, K.R.; Mendez, H.; et al. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat. Commun. 2021, 12, 2057. [Google Scholar] [CrossRef]
- Esteban-Lopez, M.; Agoulnik, A.I. Diverse functions of insulin-like 3 peptide. J. Endocrinol. 2020, 247, R1–R12. [Google Scholar] [CrossRef]
- Ferlin, A.; Perilli, L.; Gianesello, L.; Taglialavoro, G.; Foresta, C. Profiling insulin like factor 3 (INSL3) signaling in human osteoblasts. PLoS ONE 2011, 6, e29733. [Google Scholar] [CrossRef]
- Ferlin, A.; De Toni, L.; Agoulnik, A.I.; Lunardon, G.; Armani, A.; Bortolanza, S.; Blaauw, B.; Sandri, M.; Foresta, C. Protective Role of Testicular Hormone INSL3 From Atrophy and Weakness in Skeletal Muscle. Front. Endocrinol. 2018, 9, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeom, E.; Shin, H.; Yoo, W.; Jun, E.; Kim, S.; Hong, S.H.; Kwon, D.W.; Ryu, T.H.; Suh, J.M.; Kim, S.C.; et al. Tumour-derived Dilp8/INSL3 induces cancer anorexia by regulating feeding neuropeptides via Lgr3/8 in the brain. Nat. Cell Biol. 2021, 23, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Song, W.; Droujinine, I.A.; Hu, Y.; Asara, J.M.; Perrimon, N. Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev. Cell 2015, 33, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Clarevega, A.; Bilder, D. Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 2015, 33, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Biag, J.; Huang, Y.; Gou, L.; Hintiryan, H.; Askarinam, A.; Hahn, J.D.; Toga, A.W.; Dong, H.W. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: A study of immunostaining and multiple fluorescent tract tracing. J. Comp. Neurol. 2012, 520, 6–33. [Google Scholar] [CrossRef] [Green Version]
- Currow, D.; Temel, J.S.; Abernethy, A.; Milanowski, J.; Friend, J.; Fearon, K.C. ROMANA 3: A phase 3 safety extension study of anamorelin in advanced non-small-cell lung cancer (NSCLC) patients with cachexia. Ann. Oncol. 2017, 28, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Brisbois, T.; De Kock, I.; Watanabe, S.; Mirhosseini, M.; Lamoureux, D.; Chasen, M.; MacDonald, N.; Baracos, V.; Wismer, W. Delta-9-tetrahydrocannabinol may palliate altered chemosensory perception in cancer patients: Results of a randomized, double-blind, placebo-controlled pilot trial. Ann. Oncol. 2011, 22, 2086–2093. [Google Scholar] [CrossRef]
- Bar-Sela, G.; Zalman, D.; Semenysty, V.; Ballan, E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: Pilot study. Integr. Cancer Ther. 2019, 18, 1534735419881498. [Google Scholar] [CrossRef]
- Turcott, J.G.; Núñez, M.d.R.G.; Flores-Estrada, D.; Oñate-Ocaña, L.F.; Zatarain-Barrón, Z.L.; Barrón, F.; Arrieta, O. The effect of nabilone on appetite, nutritional status, and quality of life in lung cancer patients: A randomized, double-blind clinical trial. Support. Care Cancer 2018, 26, 3029–3038. [Google Scholar] [CrossRef]
- Hiura, Y.; Takiguchi, S.; Yamamoto, K.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; Nakajima, K.; Miyata, H.; Fujiwara, Y.; Mori, M. Effects of ghrelin administration during chemotherapy with advanced esophageal cancer patients: A prospective, randomized, placebo-controlled phase 2 study. Cancer 2012, 118, 4785–4794. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.R.; López-Briz, E.; Sanchis, R.C.; Perales, J.L.G.; Bort-Martí, S. Megestrol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Madeddu, C.; Macciò, A.; Panzone, F.; Tanca, F.M.; Mantovani, G. Medroxyprogesterone acetate in the management of cancer cachexia. Expert Opin. Pharmacother. 2009, 10, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Lai, V.; George, J.; Richey, L.; Kim, H.J.; Cannon, T.; Shores, C.; Couch, M. Results of a pilot study of the effects of celecoxib on cancer cachexia in patients with cancer of the head, neck, and gastrointestinal tract. Head Neck J. Sci. Spec. Head Neck 2008, 30, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, G.; Macciò, A.; Madeddu, C.; Serpe, R.; Antoni, G.; Massa, E.; Dessì, M.; Panzone, F. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J. Mol. Med. 2010, 88, 85–92. [Google Scholar] [CrossRef]
- Mehrzad, V.; Afshar, R.; Akbari, M. Pentoxifylline treatment in patients with cancer cachexia: A double-blind, randomized, placebo-controlled clinical trial. Adv. Biomed. Res. 2016, 5, 60. [Google Scholar]
- Goldberg, R.M.; Loprinzi, C.L.; Mailliard, J.A.; O’Fallon, J.R.; Krook, J.E.; Ghosh, C.; Hestorff, R.D.; Chong, S.F.; Reuter, N.F.; Shanahan, T.G. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 1995, 13, 2856–2859. [Google Scholar] [CrossRef]
- Gordon, J.; Trebble, T.; Ellis, R.; Duncan, H.; Johns, T.; Goggin, P. Thalidomide in the treatment of cancer cachexia: A randomised placebo controlled trial. Gut 2005, 54, 540–545. [Google Scholar] [CrossRef]
- Mantovani, G., III. Randomised phase III clinical trial of 5 different arms of treatment on 332 patients with cancer cachexia. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 292–301. [Google Scholar] [PubMed]
- Yennurajalingam, S.; Willey, J.S.; Palmer, J.L.; Allo, J.; Fabbro, E.D.; Cohen, E.N.; Tin, S.; Reuben, J.M.; Bruera, E. The role of thalidomide and placebo for the treatment of cancer-related anorexia-cachexia symptoms: Results of a double-blind placebo-controlled randomized study. J. Palliat. Med. 2012, 15, 1059–1064. [Google Scholar] [CrossRef]
- Jatoi, A.; Ritter, H.L.; Dueck, A.; Nguyen, P.L.; Nikcevich, D.A.; Luyun, R.F.; Mattar, B.I.; Loprinzi, C.L. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer 2010, 68, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor-Induced Factor | Effect on CNS Function | References |
|---|---|---|
| Il-1b | Modulates neurotransmitter secretion; decreases gluamatergic transmission | [23,24,25] |
| Il-2 | Mediates cognitive decline by hippocampal neurodegeneration, decreases hippocampal acetylcholine secretion and demyelination | [26,27,28] |
| Il-6 | NMDA receptor neurotoxicity; microglial activation | [29,30] |
| TNF-alpha | Modulates anorexia; increases thermogenesis and respiratory quotient | [31,32,33] |
| Neutrophils | CNS infiltration via CCR2–CCL2 axis to induce anorexia | [7] |
| Extracellular vesicles | Axonogenesis, microRNA signaling to induce inflammation | [34,35,36] |
| Sphingosin-1-phosphate | Promotes anorexia and energy expenditure via persistent activation of hypothalamic STAT3 | [37,38] |
| Serotonin | Inhibits hypothalamic neuropeptide Y secretion | [39] |
| Macrophage inhibitory cytokine-1 | Decreases appetite by interacting with TGF-B type II receptor in the hypothalamus; decreases neuropeptide Y expression and increases POMC expression in arcuate | [40] |
| Glucagon-like peptide-1 | Meditates food intake and body weight by acting on GLP-1R in the brainstem | [41] |
| CNS-Targeting Drug/Therapy | Mechanism of Action | Clinical Trial Outcomes | References |
|---|---|---|---|
| Anamorelin HCl | Ghrelin receptor agonist | Improved appetite, food intake, body weight and lean mass. No significant improvement in handgrip strength | [106,107] |
| THC | CB1/CB2 receptors agonist | Improved appetite, food intake, fatigue reversal | [108,109] |
| Nabilone | CB1/CB2 receptors agonist Synthetic analog of THC | Improved appetite, food intake, decrease in insomnia, pain | [110] |
| Ghrelin | Orexigenic hormone | Improved appetite, food intake, reduced iatrogenic burden of chemotherapy (nausea, vomiting) | [111] |
| Megestrol acetate | Progesterone receptor agonist Synthetic analog of progesterone | Improved appetite, food intake, weight gain. Downregulation of proinflammatory cytokines. | [112] |
| Medroxyprogesterone acetate | Progesterone receptor agonist Synthetic analog of progesterone | Improved appetite, food intake, weight gain. Downregulation of IL-6, IL-1, TNFα | [113] |
| Celecoxib | COX-2 inhibitor | Weight gain, BMI increase. Downregulation of proinflammatory cytokines. Improvement in handgrip strength, performance | [114,115] |
| Pentoxifylline/Oxpentifylline | Hemorheological agent/Dimethylxanthine derivative | No significant change in appetite, food intake. Potential negative effects on QoL | [116,117] |
| Thalidomide | Glutamic acid derivative | Weight gain. Downregulation of proinflammatory cytokines | [118,119,120] |
| Infliximab | IgG1k monoclonal antibody | No significant change in weight. Potential negative effects on QoL | [121] |
| Etanercept | TNFα receptor blocker | No significant change in weight | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olson, B.; Diba, P.; Korzun, T.; Marks, D.L. Neural Mechanisms of Cancer Cachexia. Cancers 2021, 13, 3990. https://doi.org/10.3390/cancers13163990
Olson B, Diba P, Korzun T, Marks DL. Neural Mechanisms of Cancer Cachexia. Cancers. 2021; 13(16):3990. https://doi.org/10.3390/cancers13163990
Chicago/Turabian StyleOlson, Brennan, Parham Diba, Tetiana Korzun, and Daniel L. Marks. 2021. "Neural Mechanisms of Cancer Cachexia" Cancers 13, no. 16: 3990. https://doi.org/10.3390/cancers13163990
APA StyleOlson, B., Diba, P., Korzun, T., & Marks, D. L. (2021). Neural Mechanisms of Cancer Cachexia. Cancers, 13(16), 3990. https://doi.org/10.3390/cancers13163990