
The Emerging Role of Suppressors of Cytokine Signaling (SOCS) in the Development and Progression of Leukemia


Abstract
:Simple Summary
Abstract
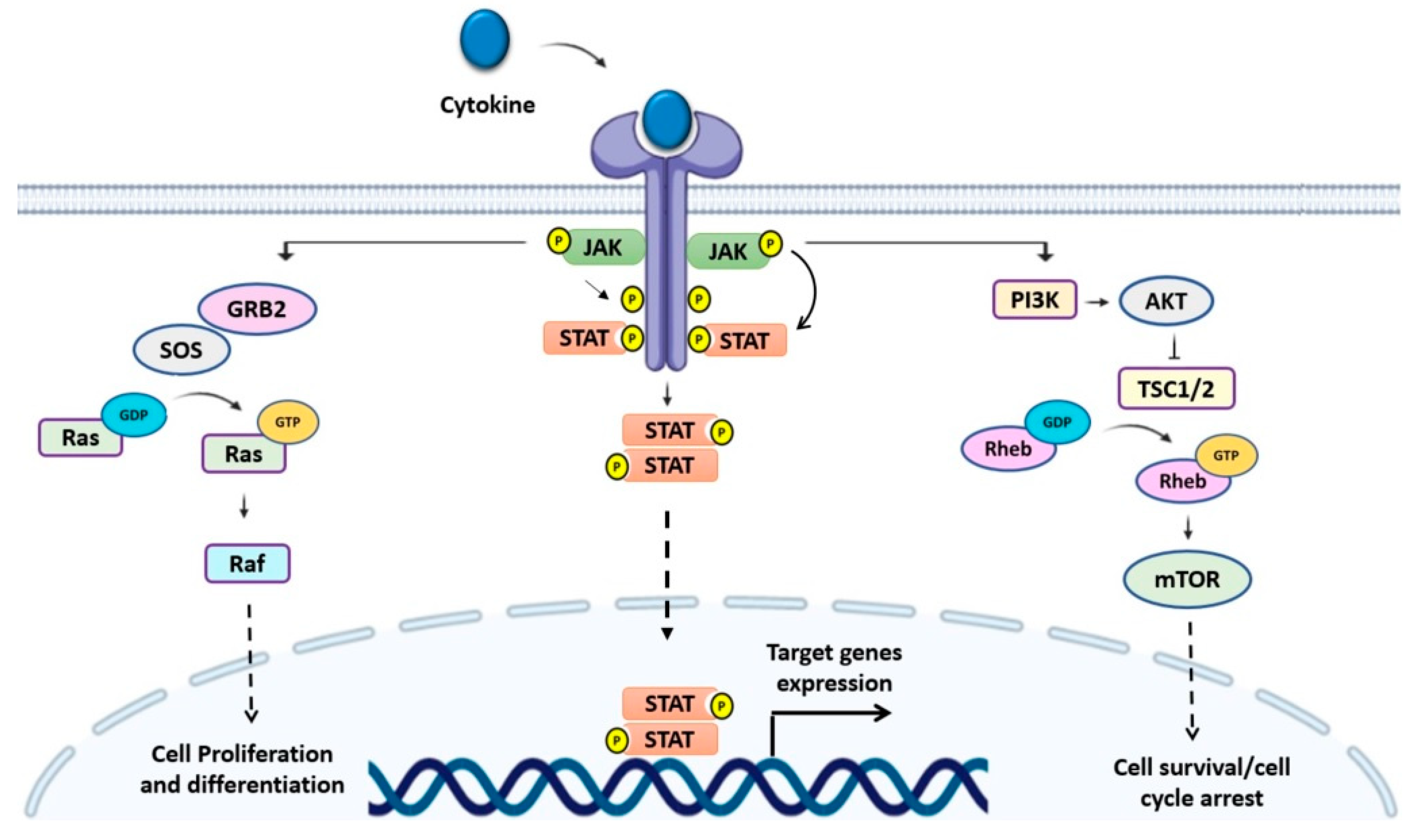
1. Introduction
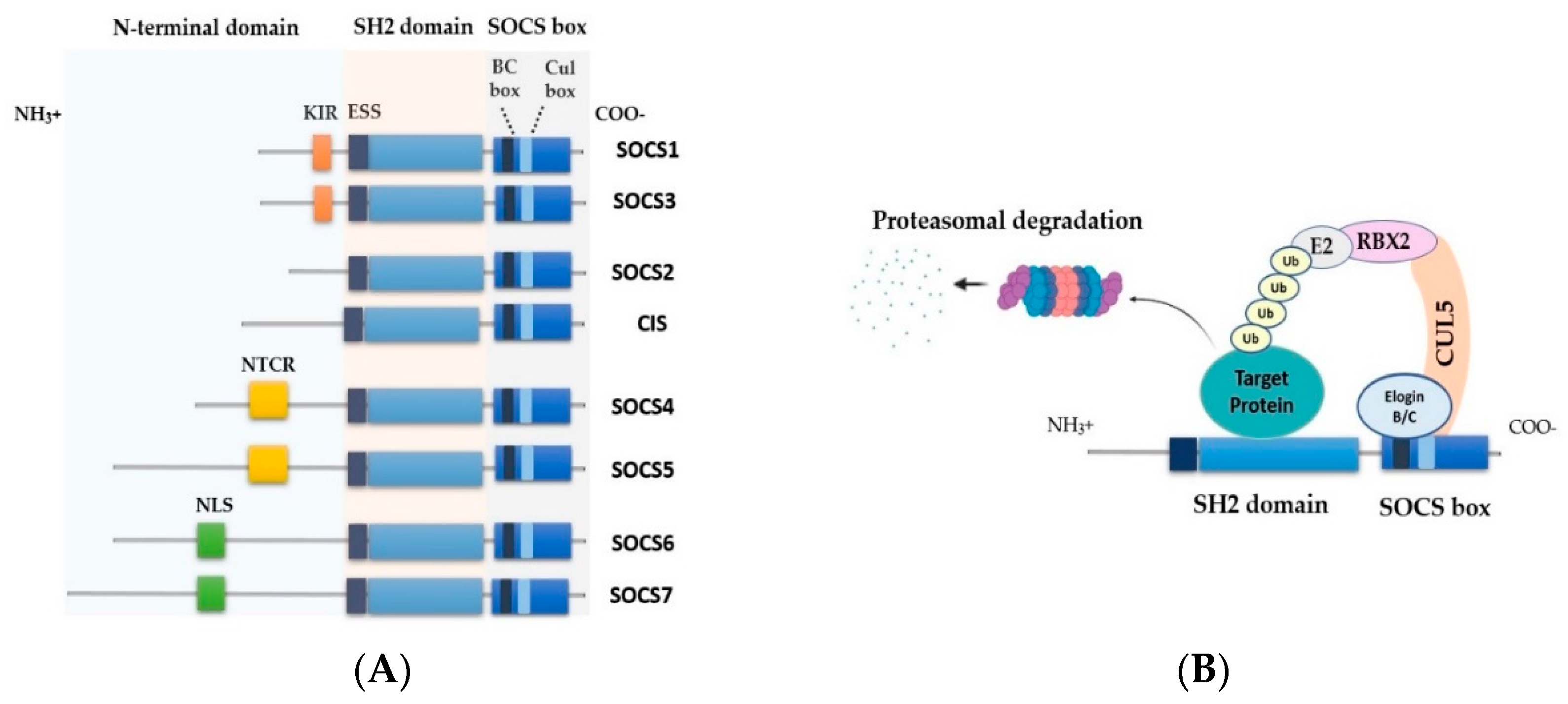
2. SOCS Family Proteins
2.1. SOCS Structure and Function
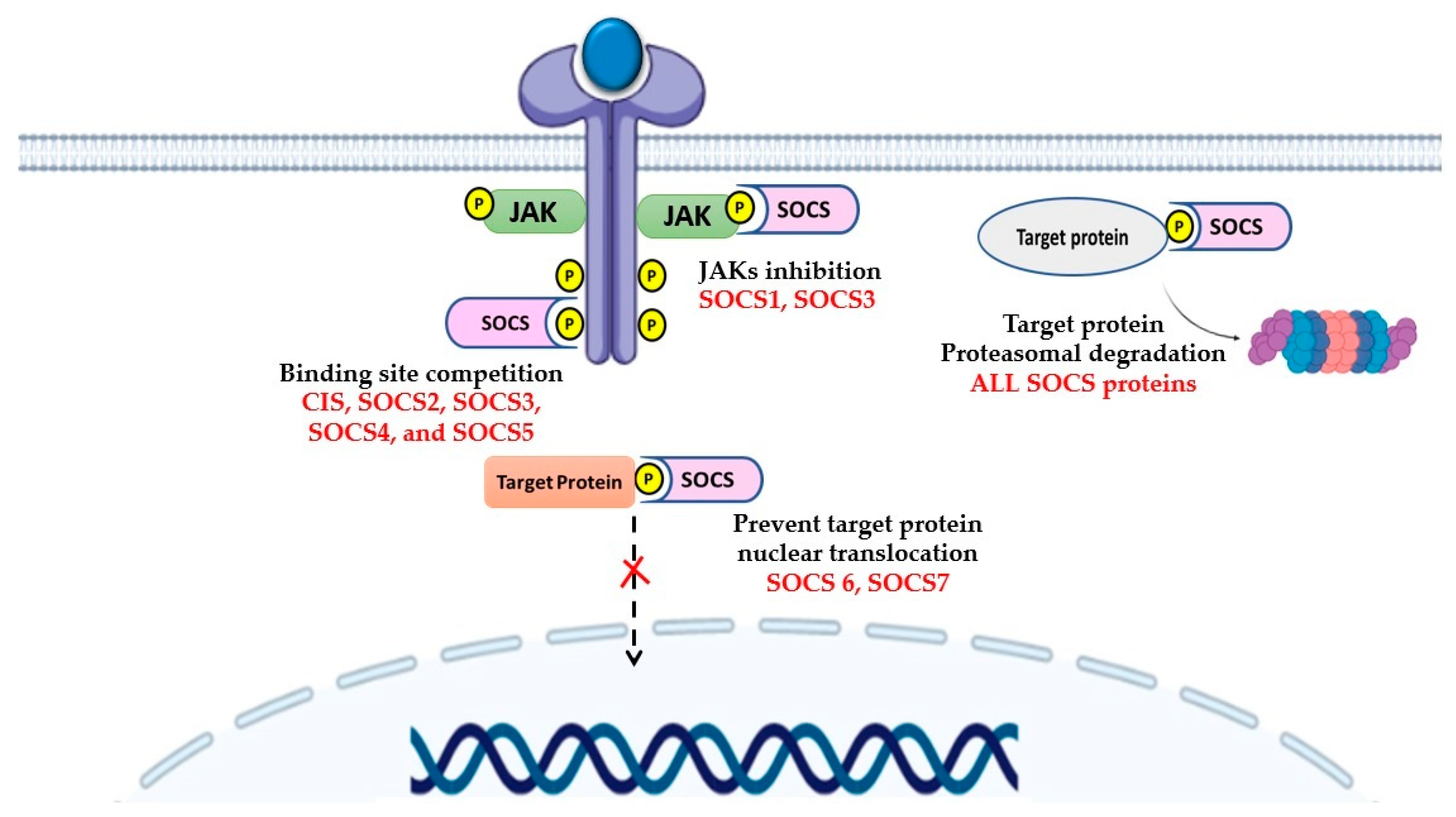
2.2. Control of Signaling by SOCS
3. SOCS Proteins in Leukemia
3.1. SOCS1 and SOCS3
3.2. CIS and SOCS2
3.3. SOCS4 and SOCS5
3.4. SOCS6 and SOCS7
4. Regulation of SOCS Expression and Functions
4.1. Epigenetic Dysregulation of SOCS Genes
4.2. MicroRNA Regulation of SOCS Genes
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Etten, R.A. Aberrant Cytokine Signaling in Leukemia. Oncogene 2007, 26, 6738–6749. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhang, Z.; Qu, X.; Zhu, X.; Zhao, L.; Wei, R.; Guo, Q.; Sun, L.; Yin, X.; Zhang, Y.; et al. Roles of STAT3 in Leukemia (Review). Int. J. Oncol. 2018, 53, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, S.; Desai, J.S.; Adam, A.; Mishra, P.R. JAK/STAT as a Novel Target for Treatment of Leukemia. Int. J. Pharm. Pharm. Sci. 2014, 6, 1–7. [Google Scholar]
- Mirantes, C.; Passegue, E.; Pietras, E.M. Pro-inflammatory cytokines: Emerging Players Regulating HSC Function in Normal and Diseased Hematopoiesis. Exp. Cell Res. 2014, 329, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. The JAK-STAT Signaling Pathway: Input and Output Integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.; Pearlman, E. Targeting JAK/STAT Signaling Pathway in Inflammatory Diseases. Curr. Signal. Transduct. Ther. 2009, 4, 201–221. [Google Scholar] [CrossRef]
- Ram, P.T.; Iyengar, R. G Protein Coupled Receptor Signaling through the Src and Stat3 Pathway: Role in Proliferation and Transformation. Oncogene 2001, 20, 1601–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in Oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, W.W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Sig-naling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wormald, S.; Hilton, D. Inhibitors of Cytokine Signal Transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Weniger, M.; Melzner, I.; Menz, C.K.; Wegener, S.; Bucur, A.J.; Dorsch, K.; Mattfeldt, T.; E Barth, T.F.; Möller, P. Mutations of the Tumor Suppressor Gene SOCS-1 in Classical Hodgkin Lymphoma are Frequent and Associated with Nuclear Phospho-STAT5 Accumulation. Oncogene 2006, 25, 2679–2684. [Google Scholar] [CrossRef] [Green Version]
- Mottok, A.; Renné, C.; Seifert, M.; Oppermann, E.; Bechstein, W.; Hansmann, M.-L.; Küppers, R.; Bräuninger, A. Inactivating SOCS1 Mutations are Caused by Aberrant Somatic Hypermutation and Restricted to a Subset of B-Cell Lymphoma Entities. Blood 2009, 114, 4503–4506. [Google Scholar] [CrossRef]
- Melzner, I.; Bucur, A.J.; Brüderlein, S.; Dorsch, K.; Hasel, C.; Barth, T.F.E.; Leithäuser, F.; Möller, P. Biallelic Mutation of SOCS-1 Impairs JAK2 Degradation and Sustains Phospho-JAK2 Action in the MedB-1 Mediastinal Lymphoma Line. Blood 2005, 105, 2535–2542. [Google Scholar] [CrossRef]
- Yang, M.; Chen, H.; Zhou, L.; Huang, X.; Su, F.; Wang, P. Identification of SOCS Family Members with Prognostic Values in Human Ovarian Cancer. Am. J. Transl. Res. 2020, 12, 1824–1838. [Google Scholar]
- Qiu, X.; Zheng, J.; Guo, X.; Gao, X.; Liu, H.; Tu, Y.; Zhang, Y. Reduced Expression of SOCS2 and SOCS6 in Hepatocellular Carcinoma Correlates with Aggressive Tumor Progression and Poor Prognosis. Mol. Cell. Biochem. 2013, 378, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Tsay, W.; Tang, J.-L.; Shen, H.-L.; Lin, S.-W.; Huang, S.-Y.; Yao, M.; Chen, Y.-C.; Shen, M.-C.; Wang, C.-H.; et al. SOCS1 Methylation in Patients with Newly Diagnosed Acute Myeloid Leukemia. Genes Chromosom. Cancer 2003, 37, 300–305. [Google Scholar] [CrossRef]
- Raccurt, M.; Tam, S.P.; Lau, P.; Mertani, H.C.; Lambert, A.; Garcia-Caballero, T.; Li, H.; Brown, R.J.; A McGuckin, M.; Morel, G.; et al. Suppressor of Cytokine Signalling Gene Expression is Elevated in Breast Carcinoma. Br. J. Cancer 2003, 89, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, B.; Carapeti-Marootian, M.; Hochhaus, A.; Weiβer, A.; Goldman, J.M.; Melo, J.V. Overexpression of SOCS-2 in Advanced Stages of Chronic Myeloid Leukemia: Possible Inadequacy of a Negative Feedback Mechanism. Blood 2002, 99, 1766–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasi, W.; Jiang, W.G.; Sharma, A.; Mokbel, K. Higher Expression Levels of SOCS 1,3,4,7 are Associated with Earlier Tumour Stage and better Clinical Outcome in Human Breast Cancer. BMC Cancer 2010, 10, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Zhang, H.-W.; Lu, M.-H.; He, X.-H.; Li, Y.; Gu, H.; Liu, M.-F.; Wang, E.-D. MicroRNA-155 Functions as an OncomiR in Breast Cancer by Targeting the Suppressor of Cytokine Signaling 1 Gene. Cancer Res. 2010, 70, 3119–3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.; Naudin, C.; Letourneur, M.; Polrot, M.; Renoir, J.-M.; Lazar, V.; Dessen, P.; Roche, S.; Bertoglio, J.; Pierre, J. Suppressor of Cytokine Signaling 1 Modulates Invasion and Metastatic Potential of Colorectal Cancer Cells. Mol. Oncol. 2014, 8, 942–955. [Google Scholar] [CrossRef]
- Galm, O.; Yoshikawa, H.; Esteller, M.; Osieka, R.; Herman, J.G. SOCS-1, A Negative Regulator of Cytokine Signaling, is Fre-quently Silenced by Methylation in Multiple Myeloma. Blood 2003, 101, 2784–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevrier, M.; Bobbala, D.; Villalobos-Hernandez, A.; Khan, G.M.; Ramanathan, S.; Saucier, C.; Ferbeyre, G.; Geha, S.; Ilangumaran, S. Expression of SOCS1 and the Downstream Targets of Its Putative Tumor Suppressor Functions in Prostate Cancer. BMC Cancer 2017, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Gregory, P.; Fernandes, R.C.; Denis, I.; Wang, Q.; Townley, S.L.; Zhao, S.G.; Hanson, A.R.; Pickering, M.A.; Armstrong, H.; et al. MicroRNA-194 Promotes Prostate Cancer Metastasis by Inhibiting SOCS2. Cancer Res. 2016, 77, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Pierconti, F.; Martini, M.; Pinto, F.; Cenci, T.; Capodimonti, S.; Calarco, A.; Bassi, P.F.; Larocca, L.M. Epigenetic Silencing of SOCS3 Identifies a Subset of Prostate Cancer with an Aggressive Behavior. Prostate 2010, 71, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Shen, D.; He, L.; Wang, H.; Liu, C.; Zhang, W. Prognostic Significance of SOCS3 and Its Biological Function in Col-orectal Cancer. Gene 2017, 627, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Chen, S.; Chen, C.; Xiao, B.; Li, F.; Wang, Y.; Tao, Z. Interleukin-23 Facilitates Thyroid Cancer Cell Migration and Invasion by Inhibiting SOCS4 Expression via MicroRNA-25. PLoS ONE 2015, 10, e0139456. [Google Scholar] [CrossRef] [PubMed]
- Song, W.B.; Chen, Y.L.; Zhu, G.D.; Xie, H.J.; Yang, Z.S.; Li, L. Exosome-Mediated miR-9-5p Promotes Proliferation and Mi-gration of Renal Cancer Cells both In Vitro and In Vivo by Targeting SOCS4. Biochem. Biophys. Res. Commun. 2020, 529, 1216–1224. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Q.; Chen, W.; Wu, T.; Liu, S.; Li, X.; Luo, B.; Zhang, T.; Yan, G.; Lu, H.; et al. MicroRNA-301a Promotes Pancreatic Cancer Invasion and Metastasis through the JAK/STAT3 Signaling Pathway by Targeting SOCS5. Carcinogenesis 2019, 41, 502–514. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, S.; Chua, M.-S.; Li, H.; Luo, D.; Wang, S.; Zhang, S.; Han, B.; Sun, C. SOCS5 Inhibition Induces Autophagy to Impair Metastasis in Hepatocellular Carcinoma Cells via the PI3K/Akt/mTOR Pathway. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shen, R.; Wang, Y.; Wang, C.-X.; Yin, M.; Liu, H.-L.; Chen, J.-P.; Han, J.-Q.; Wang, W.-B. MiRNA-155 Mediates TAM Resistance by Modulating SOCS6-STAT3 Signalling Pathway in Breast Cancer. Am. J. Transl. Res. 2015, 7, 2115–2126. [Google Scholar]
- Lai, R.-H.; Hsiao, Y.-W.; Wang, M.-J.; Lin, H.-Y.; Wu, C.-W.; Chi, C.-W.; Li, A.F.-Y.; Jou, Y.-S.; Chen, J.-Y. SOCS6, Down-Regulated in Gastric Cancer, Inhibits Cell Proliferation and Colony Formation. Cancer Lett. 2010, 288, 75–85. [Google Scholar] [CrossRef]
- Noguchi, S.; Yamada, N.; Kumazaki, M.; Yasui, Y.; Iwasaki, J.; Naito, S. socs7, A Target Gene of microRNA-145, Reg-ulates Interferon-β Induction Through STAT3 Nuclear Translocation in Bladder. Cancer Cells 2013, 4, e482. [Google Scholar]
- Sasi, W.; Ye, L.; Jiang, W.G.; Mokbel, K.; Sharma, A. Observations on The effects of Suppressor of Cytokine Signaling 7 (SOCS7) Knockdown in Breast Cancer Cells: Their In Vitro response to Insulin Like Growth Factor I (IGF-I). Clin. Transl. Oncol. 2013, 16, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, K.; Cheng, A.; Wang, M.; Cui, M.; Huang, J.; Zhu, D.; Chen, S.; Liu, M.; Zhao, X.; et al. SOCS Proteins Participate in the Regulation of Innate Immune Response Caused by Viruses. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS Proteins, Cytokine Signalling and Immune Regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Trengove, M.C.; Ward, A.C. SOCS Proteins in Development and Disease. Am. J. Clin. Exp. Immunol. 2013, 2, 1–29. [Google Scholar]
- Ahmed, C.M.I.; Larkin, J.; Johnson, H.M. SOCS1 Mimetics and Antagonists: A Complementary Approach to Positive and Negative Regulation of Immune Function. Front. Immunol. 2015, 6, 183. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.-P.; Chandrashekaran, I.; Low, A.; Speed, T.P.; Nicholson, S.E.; Norton, R.S. The N-Terminal Domains of SOCS Proteins: A Conserved Region in the Disordered N-Termini of SOCS4 and 5. Proteins: Struct. Funct. Bioinform. 2011, 80, 946–957. [Google Scholar] [CrossRef]
- Hwang, M.N.; Min, C.H.; Kim, H.S.; Lee, H.; Yoon, K.A.; Park, S.Y.; Lee, E.S.; Yoon, S. The Nuclear Localization of SOCS6 Requires the N-Terminal Region and Negatively Regulates Stat3 Protein Levels. Biochem Biophys Res. Commun. 2007, 360, 333–338. [Google Scholar] [CrossRef]
- Kremer, B.E.; Adang, L.A.; Macara, I.G. Septins Regulate Actin Organization and Cell-Cycle Arrest through Nuclear Accumulation of NCK Mediated by SOCS7. Cell 2007, 130, 837–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of Cytokine Signaling and Immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef]
- Bullock, A.N.; Rodriguez, M.C.; Debreczeni, J.; Songyang, Z.; Knapp, S. Structure of the SOCS4-ElonginB/C Complex Reveals a Distinct SOCS Box Interface and the Molecular Basis for SOCS-Dependent EGFR Degradation. Structure 2007, 15, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Linossi, E.M.; Calleja, D.J.; Nicholson, S.E. Understanding SOCS Protein Specificity. Growth Factors 2018, 36, 104–117. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D. SOCS Proteins: Negative Regulators of Cytokine Signaling. STEM Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Zhang, J.-G.; Farley, A.; Nicholson, S.E.; Willson, T.A.; Zugaro, L.M.; Simpson, R.; Moritz, R.L.; Cary, D.; Richardson, R.; Hausmann, G.; et al. The Conserved SOCS box Motif in Suppressors of Cytokine Signaling Binds to Elongins B and C and May Couple bound Proteins to Proteasomal Degradation. Proc. Natl. Acad. Sci. USA 1999, 96, 2071–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.; Nakayama, K.I. VHL-box and SOCS-box Domains Determine Binding Specificity for Cul2-Rbx1 and Cul5-Rbx2 Modules of Ubiquitin Ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohroki, J.; Nishiyama, T.; Nakamura, T.; Masuho, Y. ASB Proteins Interact with Cullin5 and Rbx2 to form E3 Ubiquitin Ligase Complexes. FEBS Lett. 2005, 579, 6796–6802. [Google Scholar] [CrossRef] [Green Version]
- Gianfelici, V.; Chiaretti, S.; Demeyer, S.; Di Giacomo, F.; Messina, M.; La Starza, R.; Peragine, N.; Paoloni, F.; Geerdens, E.; Pierini, V. RNA Sequencing Unravels the Genetics of Refractory/Relapsed T-cell Acute Lymphoblastic Leukemia. Prognostic and Therapeutic Implications. Haematology 2016, 101, 941–950. [Google Scholar] [CrossRef]
- Lin, T.S.; Mahajan, S.; A Frank, D. STAT Signaling in The Pathogenesis and Treatment of Leukemias. Oncogene 2000, 19, 2496–2504. [Google Scholar] [CrossRef] [Green Version]
- Girardi, T.; Vereecke, S.; O Sulima, S.; Khan, Y.; Fancello, L.; Briggs, J.W.; Schwab, C.; De Beeck, J.O.; Verbeeck, J.; Royaert, J. The T-cell Leukemia-Associated Ribosomal RPL10 R98S Mutation Enhances JAK-STAT Signaling. Leukemia 2017, 32, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Gouilleux-Gruart, V.; Gouilleux, F.; Desaint, C.; Claisse, J.F.; Capiod, J.C.; Delobel, J. STAT-Related Transcription Factors are Con-stitutively Activated in Peripheral Blood Cells from Acute Leukemia Patients. Blood 1996, 87, 1692–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikezoe, T.; Kojima, S.; Furihata, M.; Yang, J.; Nishioka, C.; Takeuchi, A.; Isaka, M.; Koeffler, H.P.; Yokoyama, A. Expression of p-JAK2 Predicts Clinical Outcome and is a Potential Molecular Target of Acute Myelogenous Leukemia. Int. J. Cancer 2011, 129, 2512–2521. [Google Scholar] [CrossRef]
- Benekli, M.; Xia, Z.; Donohue, K.A.; Ford, L.A.; Pixley, L.A.; Baer, M.R.; Baumann, H.; Wetzler, M. Constitutive Activity of Signal Transducer and Activator of Transcription 3 Protein in Acute Myeloid Leukemia Blasts is Associated with Short Disease-Free Survival. Blood 2002, 99, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; White, S.M.; Schaefer, T.S.; Ball, E.D.; Dyer, K.F.; Tweardy, D.J. Granulocyte Colony-Stimulating Factor Activation of Stat3 Alpha and Stat3 Beta in Immature Normal and Leukemic Human Myeloid Cells. Blood 1996, 88. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.; Li, L.; Ho, Y.; Lin, A.; Stein, A.; Forman, S.; Perrotti, D.; Jove, R.; Bhatia, R. Role of Altered Growth Factor Receptor-Mediated JAK2 Signaling in Growth and Maintenance of Human Acute Myeloid Leukemia Stem Cells. Blood 2014, 123, 2826–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Bempt, M.V.; Dagklis, A.; et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov. 2018, 8, 616–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncero, A.M.; Nieva, P.L.; Cobos-Fernández, M.A.; Villa-Morales, M.C.; Gonzalez-Sanchez, L.; López-Lorenzo, J.L.; Llamas, P.; Ayuso, C.; Rodriguezpinilla, S.M.; Arriba, M.C.; et al. Contribution of JAK2 Mutations to T-Cell Lymphoblastic Lymphoma Development. Leukemia 2016, 30, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Flex, E.; Petrangeli, V.; Stella, L.; Chiaretti, S.; Hornakova, T.; Knoops, L.; Ariola, C.; Fodale, V.; Clappier, E.; Paoloni, F.; et al. Somatically Acquired JAK1 Mutations in Adult Acute Lymphoblastic Leukemia. J. Exp. Med. 2008, 205, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic Mutations and Germline Sequence Variants in The Expressed Tyrosine Kinase Genes of Patients with de Novo Acute Myeloid Leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef] [Green Version]
- Degryse, S.; Bornschein, S.; De Bock, C.E.; Leroy, E.; Bempt, M.V.; Demeyer, S.; Jacobs, K.; Geerdens, E.; Gielen, O.; Soulier, J.; et al. Mutant JAK3 Signaling is Increased by Loss of Wild-Type JAK3 or by Acquisition of Secondary JAK3 Mutations in T-ALL. Blood 2018, 131, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; da Silva, M.C.; Paganin, M.; Tritapoe, J.; A Hixon, J.; Silveira, A.; Cardoso, B.; et al. Oncogenic IL7R Gain-of-Function Mutations in Childhood T-Cell Acute Lymphoblastic Leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Mangolini, M.; De Boer, J.; Walf-Vorderwülbecke, V.; Pieters, R.; Boer, M.L.D.; Williams, O. STAT3 Mediates Oncogenic Addiction to TEL-AML1 in t(12;21) Acute Lymphoblastic Leukemia. Blood 2013, 122, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianfelici, V.; Messina, M.; Paoloni, F.; Peragine, N.; Lauretti, A.; Fedullo, A.L.; Di Giacomo, F.; Vignetti, M.; Vitale, A.; Guarini, A.; et al. IL7R Overexpression in Adult Acute Lymphoblastic Leukemia is Associated to JAK/STAT Pathway Mutations and Identifies Patients who Could Benefit from Targeted Therapies. Leuk. Lymphoma 2018, 60, 829–832. [Google Scholar] [CrossRef]
- Govaerts, I.; Jacobs, K.; Vandepoel, R.; Cools, J. JAK/STAT Pathway Mutations in T-ALL, Including the STAT5B N642H Mutation, are Sensitive to JAK1/JAK3 Inhibitors. HemaSphere 2019, 3. [Google Scholar] [CrossRef]
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT Pathway Inhibition in Murine Xenograft Models of Early T-Cell Precursor (ETP) Acute Lymphoblastic Leukemia. Blood 2015, 125, 1759–1767. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Griner, L.A.M.; Ju, W.; Duveau, D.Y.; Guha, R.; Petrus, M.N. Selective Targeting of JAK/STAT Signaling is Potentiated by Bcl-xL Blockade in IL-2–Dependent Adult T-Cell Leukemia. Proc. Natl. Acad. Sci. USA 2015, 112, 12480–12485. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Constantinescu, S. JAK/STAT Signaling in Hematological Malignancies. Oncogene 2012, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Vitali, C.; Bassani, C.; Chiodoni, C.; Fellini, E.; Guarnotta, C.; Miotti, S. SOCS2 Controls Proliferation and Stemness of Hema-topoietic Cells under Stress Conditions and Its Deregulation Marks Unfavorable Acute Leukemias. Cancer Res. 2015, 75, 2387–2399. [Google Scholar] [CrossRef] [Green Version]
- Abdelrasoul, H.; Vadakumchery, A.; Werner, M.; Lenk, L.; Khadour, A.; Young, M.; El Ayoubi, O.; Vogiatzi, F.; Krämer, M.; Schmid, V.; et al. Synergism between IL7R and CXCR4 Drives BCR-ABL Induced Transformation in Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Liu, K.; Wu, Z.; Chu, J.; Yang, L.; Wang, N. Promoter Methylation and Expression of SOCS3 Affect The Clinical Outcome of Pediatric Acute Lymphoblastic Leukemia by JAK/STAT Pathway. Biomed. Pharmacother. 2019, 115, 108913. [Google Scholar] [CrossRef]
- Sharma, N.D.; Nickl, C.K.; Kang, H.; Ornatowski, W.; Brown, R.; Ness, S.; Loh, M.L.; Mullighan, C.G.; Winter, S.S.; Hunger, S.P.; et al. Epigenetic Silencing of SOCS 5 Potentiates JAK-STAT Signaling and Progression of T-Cell Acute Lymphoblastic Leukemia. Cancer Sci. 2019, 110, 1931–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zheng, Y.; Gao, J.; Zhu, G.; Gao, K.; Zhang, W.; Shi, F.; Zhang, Q. Expression of SHP-1 and SOCS6 in Patients with Acute Leukemia and their Clinical Implication. OncoTargets Ther. 2017, 10, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.-Y.; Kim, H.; Madden, K.; Loftus, J.P.; Chen, G.M.; Allen, D.H.; Zhang, R.; Xu, J.; Chen, C.-H.; Hu, Y.; et al. Network Analysis Reveals Synergistic Genetic Dependencies for Rational Combination Therapy in Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Chen, T.L.; Gupta, N.; Lehman, A.; Ruppert, A.S.; Yu, L.; Oakes, C.C.; Claus, R.; Plass, C.; Maddocks, K.J.; Andritsos, L.; et al. Hsp90 Inhibition Increases SOCS3 Transcript and Regulates Migration and Cell Death in Chronic Lymphocytic Leukemia. Oncotarget 2016, 7, 28684–28696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toniolo, P.A.; Liu, S.; Yeh, J.E.; Ye, D.Q.; Barbuto, J.A.M.; Frank, D.A. Deregulation of SOCS5 Suppresses Dendritic Cell Function in Chronic Lymphocytic Leukemia. Oncotarget 2016, 7, 46301–46314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yang, L.; Liu, X.; Zhan, Y.; Pan, Y.; Wang, X.; Luo, J. Association between Methylation of Tumor Suppressor Gene SOCS1 and Acute Myeloid Leukemia. Oncol. Rep. 2018, 40, 1008–1016. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Ries, R.E.; Gudgeon, C.J.; Harrington, K.H.; Alonzo, T.A.; Gerbing, R.B. High Expression of Suppressor of Cytokine Signaling-2 Predicts Poor Outcome in Pediatric Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Leuk. Lymphoma 2014, 55, 2817–2821. [Google Scholar] [CrossRef] [Green Version]
- Jacobia, A.; Thiemea, S.; Lehmann, R.; Ugarte, F.; Malech, H.; Koch, S.; Thiede, C.; Müller, K.; Bornhäuser, M.; Ryser, M. Impact of CXCR4 Inhibition on FLT3-ITD−Positive Human AML Blasts. Exp. Hematol. 2010, 38, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell. 2020, 27, 224–237.e6. [Google Scholar] [CrossRef]
- Al-Jamal, H.; Jusoh, S.A.M.; Yong, A.C.; Asan, J.M.; Hassan, R.; Johan, M.F. Silencing of Suppressor of Cytokine Signaling-3 due to Methylation Results in Phosphorylation of STAT3 in Imatinib Resistant BCR-ABL Positive Chronic Myeloid Leukemia Cells. Asian Pac. J. Cancer Prev. 2014, 15, 4555–4561. [Google Scholar] [CrossRef] [Green Version]
- Behzad, M.M.; Shahrabi, S.; Jaseb, K.; Bertacchini, J.; Ketabchi, N.; Saki, N. Aberrant DNA Methylation in Chronic Myeloid Leukemia: Cell Fate Control, Prognosis, and Therapeutic Response. Biochem. Genet. 2018, 56, 149–175. [Google Scholar] [CrossRef]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Castillejo, J.A.; Cervantes, F.; Barrios, M.; Colomer, D. The Suppressor of Cytokine Signaling-1 is Constitutively Expressed in Chronic Myeloid Leukemia and Correlates with Poor Cytogenetic Response to Interfer-on-Alpha. Haematologica 2004, 89, 42–48. [Google Scholar]
- Zhu, Z.; Lu, X.; Jiang, L.; Sun, X.; Zhou, H.; Jia, Z.; Zhang, X.; Ma, L. STAT3 Signaling Pathway is Involved in Decitabine Induced Biological Phenotype Regulation of Acute Myeloid Leukemia Cells. Am. J. Transl. Res. 2015, 7, 1896–1907. [Google Scholar] [PubMed]
- Teramo, A.; Gattazzo, C.; Passeri, F.; Lico, A.; Tasca, G.; Cabrelle, A.; Martini, V.; Frezzato, F.; Trimarco, V.; Ave, E.; et al. Intrinsic and Extrinsic Mechanisms Contribute to Maintain the JAK/STAT Pathway Aberrantly Activated in T-Type Large Granular Lymphocyte Leukemia. Blood 2013, 121, 3843–3854. [Google Scholar] [CrossRef] [Green Version]
- Rottapel, R.; Ilangumaran, S.; Neale, C.; La Rose, J.; Ho, J.M.-Y.; Nguyen, M.H.-H.; Barber, D.; Dubreuil, P.; De Sepulveda, P. The Tumor Suppressor Activity of SOCS-1. Oncogene 2002, 21, 4351–4362. [Google Scholar] [CrossRef] [Green Version]
- Frantsve, J.; Schwaller, J.; Sternberg, D.W.; Kutok, J.; Gilliland, D.G. Socs-1 Inhibits TEL-JAK2-Mediated Transformation of Hemato-Poietic Cells through Inhibition of JAK2 Kinase Activity and Induction of Proteasome-Mediated Degradation. Mol. Cell. Biol. 2001, 21, 3547–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, D.; Ezoe, S.; Fujimoto, M.; Kimura, A.; Saito, Y.; Nagai, H.; Tachibana, I.; Matsumura, I.; Tanaka, T.; Kanegane, H.; et al. Suppressor of Cytokine Signalling-1 Gene Silencing in Acute Myeloid Leukaemia and Human Haematopoietic Cell Lines. Br. J. Haematol. 2004, 126, 726–735. [Google Scholar] [CrossRef]
- Chen, S.-S.; Wu, W.-Z.; Zhang, Y.-P.; Huang, W.-J. Gene Polymorphisms of SOCS1 and SOCS2 and Acute Lymphoblastic Leukemia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5564–5572. [Google Scholar]
- Ng, O.H.; Ure, U.; Ar, M.C.; Akyerli, C.; Soysal, T.; Ferhanoğlu, B.; Özçelik, T.; Ozbek, U. TheSOCS-1 Gene Methylation in Chronic Myeloid Leukemia Patients. Am. J. Hematol. 2007, 82, 729–730. [Google Scholar] [CrossRef]
- Liu, T.C.; Lin, S.F.; Chang, J.G.; Yang, M.Y.; Hung, S.Y.; Chang, C.S. Epigenetic Alteration of the SOCS1 Gene in Chronic Myeloid leu-Kaemia. Br. J. Haematol. 2003, 123, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Guillem, V.; Amat, P.; Cervantes, F.; Alvarez-Larrán, A.; Cervera, J.; Maffioli, M.; Bellosillo, B.; Collado, M.; Marugán, I.; Martínez-Ruiz, F.; et al. Functional Polymorphisms in SOCS1 and PTPN22 Genes Correlate with The Response to Imatinib Treatment in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. Leuk. Res. 2012, 36, 174–181. [Google Scholar] [CrossRef]
- Le, Y.; Zhu, B.M.; Harley, B.; Park, S.Y.; Kobayashi, T.; Manis, J.P. SOCS3 Protein Developmentally Regulates the Chemokine Receptor CXCR4-FAK Signaling Pathway during B Lymphopoiesis. Immunity 2007, 27, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Fierro, F.A.; Brenner, S.; Oelschlaegel, U.; Jacobi, A.; Knoth, H.; Ehninger, G. Combining SDF-1/CXCR4 Antagonism and Chemo Therapy in Relapsed Acute Myeloid Leukemia. Leukemia 2009, 23, 393–396. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ohkubo, T.; Kiguchi, T.; A Jenkins, N.; Gilbert, D.J.; Copeland, N.G.; Hara, T.; Miyajima, A. A Novel Cytokine-Inducible Gene CIS Encodes an SH2-Containing Protein that Binds to Tyrosine-Phosphorylated Interleukin 3 and Erythropoietin Receptors. EMBO J. 1995, 14, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, S.; Xu, X.; Sundstedt, A.; Paulsson, K.M.; Anderson, P.; Karlsson, S.; Sjögren, H.-O.; Wang, P. Cytokine-Induced Src Homology 2 Protein (Cis) Promotes T Cell Receptor–Mediated Proliferation and Prolongs Survival of Activated T Cells. J. Exp. Med. 2000, 191, 985–994. [Google Scholar] [CrossRef]
- Matsumoto, A.; Masuhara, M.; Mitsui, K.; Yokouchi, M.; Ohtsubo, M.; Misawa, H.; Miyajima, A.; Yoshimura, A. CIS, A Cytokine Inducible SH2 Protein, is a Target of the JAK-STAT5 Pathway and Modulates STAT5 Activation. Blood 1997, 89. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Wu, X.; Wu, D.; Jiang, R.-L.; Castillo, E.F.; Chock, C.J.; Zhou, Q.; Liu, M.; Dong, C.; Yang, X.O. Treg Expression of CIS Suppresses Allergic Airway Inflammation through Antagonizing an Autonomous TH2 Program. Mucosal Immunol. 2019, 13, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, S.; Ikegame, K.; Ueno, K.; Narazaki, M.; Naka, T.; Yamamoto, H.; Matsumoto, T.; Saito, H.; Hosoe, S.; Kishimoto, T. Cloning and Functional Analysis of New Members of STAT Induced STAT Inhibitor (SSI) Family: SSI-2 and SSI-3. Biochem. Biophys. Res. Commun. 1997, 237, 79–83. [Google Scholar] [CrossRef]
- Masuhara, M.; Sakamoto, H.; Matsumoto, A.; Suzuki, R.; Yasukawa, H.; Mitsui, K.; Wakioka, T.; Tanimura, S.; Sasaki, A.; Misawa, H.; et al. Cloning and Characterization of Novel CIS Family Genes. Biochem. Biophys. Res. Commun. 1997, 239, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Rico-Bautista, E.; Flores-Morales, A.; Fernandez-Perez, L. Suppressor of Cytokine Signaling (SOCS) 2, A Protein with Multiple Functions. Cytokine Growth Factor Rev. 2006, 17, 431–439. [Google Scholar] [CrossRef]
- Dogusan, Z.; Hooghe-Peters, E.L.; Berus, D.; Velkeniers, B.; Hooghe, R. Expression of SOCS Genes in Normal and Leukemic Human Leukocytes Stimulated by Prolactin, Growth Hormone and Cytokines. J. Neuroimmunol. 2000, 109, 34–39. [Google Scholar] [CrossRef]
- Letellier, E.; Haan, S. SOCS2: Physiological and Pathological Functions. Front. Biosci. (Elite Ed.) 2016, 8. [Google Scholar]
- Nguyen, C.H.; Gluxam, T.; Schlerka, A.; Bauer, K.; Grandits, A.M.; Hackl, H. SOCS2 is Part of a Highly Prognostic 4-Gene Sig-nature in AML and Promotes Disease Aggressiveness. Sci. Rep. 2019, 9, 1–13. [Google Scholar]
- Kazi, J.U.; Rönnstrand, L. Suppressor of Cytokine Signaling 2 (SOCS2) Associates with FLT3 and Negatively Regulates Downstream Signaling. Mol. Oncol. 2013, 7, 693–703. [Google Scholar] [CrossRef]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene Expression Changes Associated with Progression and Response in Chronic Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Hansen, N.; Ågerstam, H.; Wahlestedt, M.; Landberg, N.; Askmyr, M.; Ehinger, M.; Rissler, M.; Lilljebjörn, H.; Johnels, P.; Ishiko, J.; et al. SOCS2 is Dispensable for BCR/ABL1-Induced Chronic Myeloid Leukemia-Like Disease and for Normal Hematopoietic Stem Cell Function. Leukemia 2012, 27, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Ortega, M.; Melo, S.; Meurens, F. Expression of SOCS1-7 and CIS mRNA in Porcine Tissues. Veter. Immunol. Immunopathol. 2011, 144, 493–498. [Google Scholar] [CrossRef]
- Kedzierski, L.; Linossi, E.M.; Kolesnik, T.B.; Day, E.B.; Bird, N.L.; Kile, B.T.; Belz, G.T.; Metcalf, D.; Nicola, N.A.; Kedzierska, K.; et al. Suppressor of Cytokine Signaling 4 (SOCS4) Protects against Severe Cytokine Storm and Enhances Viral Clearance during Influenza Infection. PLoS Pathog. 2014, 10, e1004134. [Google Scholar] [CrossRef]
- Kobayashi, D.; Nomoto, S.; Kodera, Y.; Fujiwara, M.; Koike, M.; Nakayama, G.; Ohashi, N.; Nakao, A. Suppressor of Cytokine Signaling 4 Detected as a Novel Gastric Cancer Suppressor Gene using Double Combination Array Analysis. World J. Surg. 2011, 36, 362–372. [Google Scholar] [CrossRef]
- Xiao, X.; Yang, D.; Gong, X.; Mo, D.; Pan, S.; Xu, J. miR-1290 Promotes Lung Adenocarcinoma Cell Proliferation and Invasion by Tar-geting SOCS4. Oncotarget 2018, 9, 11977–11988. [Google Scholar] [CrossRef] [Green Version]
- Scheitz, C.J.F.; Lee, T.S.; McDermitt, D.J.; Tumbar, T. Defining a Tissue Stem Cell-Driven Runx1/Stat3 Signalling Axis in Epithelial Cancer. EMBO J. 2012, 31, 4124–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brender, C.; Columbus, R.; Metcalf, D.; Handman, E.; Starr, R.; Huntington, N.; Tarlinton, D.; Ødum, N.; Nicholson, S.E.; Nicola, N.A.; et al. SOCS5 Is Expressed in Primary B and T Lymphoid Cells but Is Dispensable for Lymphocyte Production and Function. Mol. Cell. Biol. 2004, 24, 6094–6103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D. Tumour-Secreted miR-9 Promotes Endothelial Cell Migration and Angiogenesis by Activating the JAK-STAT Pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing Activities of the R as and H Ippo Pathways Converge on Regulation of YAP Protein Turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Sharma, N.D.; Nickl, C.K.; Devidas, M.; Loh, M.L.; Hunger, S.P.; Dunsmore, K.P.; Winter, S.S.; Matlawska-Wasowska, K. Dysregulated Transcriptional Networks in KMT2A- and MLLT10-Rearranged T-ALL. Biomark. Res. 2018, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Wang, W.; Su, J.; Zhang, Y.; Luan, B.; Rao, H.; Cheng, T.; Zhang, W.; Xiao, S.; Zhang, M. SOCS6 Functions as a Tumor Suppressor by Inducing Apoptosis and Inhibiting Angiogenesis in Human Prostate Cancer. Curr. Cancer Drug Targets 2018, 18, 894–904. [Google Scholar] [CrossRef]
- Cheng, L.; Kong, B.H.; Zhao, Y.; Jiang, J. miR-494 Inhibits Cervical Cancer Cell Proliferation through Upregulation of SOCS6 Ex-Pression. Oncol. Lett. 2018, 15, 3075–3080. [Google Scholar]
- Tanaka, T.; Arai, M.; Jiang, X.; Sugaya, S.; Kanda, T.; Fujii, K.; Kita, K.; Sugita, K.; Imazeki, F.; Miyashita, T.; et al. Downregulation of microRNA-431 by Human Interferon-Beta Inhibits Viability of Medulloblastoma and Glioblastoma Cells via Upregulation of SOCS6. Int. J. Oncol. 2014, 44, 1685–1690. [Google Scholar] [CrossRef] [Green Version]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y. Identification of Mutations in the Coding Sequence of the Proto-Oncogene C-Kit in a Human Mast Cell Leukemia Cell Line Causing Ligand-Independent Activation of C-Kit Product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef]
- Ikeda, H.; Kanakura, Y.; Tamaki, T.; Kuriu, A.; Kitayama, H.; Ishikawa, J.; Kanayama, Y.; Yonezawa, T.; Tarui, S.; Griffin, J.D. Expression and Functional Role of the Proto-Oncogene C-Kit in Acute Myeloblastic Leukemia Cells. Blood 1991, 78, 2962–2968. [Google Scholar] [CrossRef] [Green Version]
- Zadjali, F.; Pike, A.C.; Vesterlund, M.; Sun, J.; Wu, C.; Li, S.S.; Rönnstrand, L.; Knapp, S.; Bullock, A.N.; Flores-Morales, A. Structural Basis for c-KIT Inhibition by the Suppressor of Cytokine Signaling 6 (SOCS6) Ubiquitin Ligase. J. Biol. Chem. 2011, 286, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayle, J.; Letard, S.; Frank, R.; Dubreuil, P.; De Sepulveda, P. Suppressor of Cytokine Signaling 6 Associates with KIT and Regulates KIT Receptor Signaling. J. Biol. Chem. 2004, 279, 12249–12259. [Google Scholar] [CrossRef] [Green Version]
- Kazi, J.U.; Sun, J.; Phung, B.; Zadjali, F.; Flores-Morales, A.; Rönnstrand, L. Suppressor of Cytokine Signaling 6 (SOCS6) Negatively Regulates Flt3 Signal Transduction through Direct Binding to Phosphorylated Tyrosines 591 and 919 of Flt3. J. Biol. Chem. 2012, 287, 36509–36517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matuoka, K.; Miki, H.; Takahashi, K.; Takenawa, T. A Novel Ligand for an SH3 Domain of the Adaptor Protein Nck Bears an SH2 Domain and Nuclear Signaling Motifs. Biochem. Biophys. Res. Commun. 1997, 239, 488–492. [Google Scholar] [CrossRef]
- Krebs, D.L.; Uren, R.; Metcalf, D.; Rakar, S.; Zhang, J.-G.; Starr, R.; DE Souza, D.; Hanzinikolas, K.; Eyles, J.; Connolly, L.M.; et al. SOCS-6 Binds to Insulin Receptor Substrate 4, and Mice Lacking the SOCS-6 Gene Exhibit Mild Growth Retardation. Mol. Cell. Biol. 2002, 22, 4567–4578. [Google Scholar] [CrossRef] [Green Version]
- Martens, N.; Uzan, G.; Wery, M.; Hooghe, R.; Hooghe-Peters, E.L.; Gertler, A. Suppressor of Cytokine Signaling 7 Inhibits Prolactin, Growth Hormone, and Leptin Signaling by Interacting with STAT5 or STAT3 and Attenuating Their Nuclear Translocation. J. Biol. Chem. 2005, 280, 13817–13823. [Google Scholar] [CrossRef] [Green Version]
- Ge, D.; Gao, A.C.; Zhang, Q.; Liu, S.; Xue, Y.; You, Z. LNCaP Prostate Cancer Cells with Autocrine Interleukin-6 Expression are Resistant to IL-6-Induced Neuroendocrine Differentiation due to Increased Expression of Suppressors of Cytokine Signaling. Prostate 2011, 72, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.H.; Xu, S.B.; Yuan, J.; Li, B.H.; Zhang, Y.; Yuan, Q. Defective Interleukin-4/Stat6 Activity Correlates with Increased consti-tutive Expression of Negative Regulators SOCS-3, SOCS-7, and CISH in Colon Cancer Cells. J. Interferon Cytokine Res. 2009, 29, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Lindeman, G.J.; Choong, D.Y.H.; Wittlin, S.; Brentzell, L.; Phillips, W.; Campbell, I.G.; E Visvader, J. Differential Hypermethylation of SOCS Genes in Ovarian and Breast Carcinomas. Oncogene 2004, 23, 7726–7733. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, H.; Matsubara, K.; Qian, G.-S.; E Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a Negative Regulator of the JAK/STAT Pathway, is Silenced by Methylation in Human Hepatocellular Carcinoma and Shows Growth-Suppression Activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Fukushima, N.; Sato, N.; Sahin, F.; Su, G.; Hruban, R.H.; Goggins, M. Aberrant Methylation of Suppressor of Cytokine Signalling-1 (SOCS-1) Gene in Pancreatic Ductal Neoplasms. Br. J. Cancer 2003, 89, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.-C.; Chen, M.-L.; Yang, F.; Gao, B.-Q.; Yang, Q.-H.; Zheng, W.-W.; Hao, S. Promoter Methylation and Expression of SOCS-1 Affect Clinical Outcome and Epithelial-Mesenchymal Transition in Colorectal Cancer. Biomed. Pharmacother. 2016, 80, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Watari, J.; Zhang, X.X.; Ran, Y.; Tomita, T.; Oshima, T.; Hirota, S.; Miwa, H. Phosphorylated STAT3 Expression linked to SOCS3 Methylation is Associated with Proliferative Ability of Gastric Mucosa in Patients with Early Gastric Cancer. Oncol. Lett. 2020, 19, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- Letellier, E.; Schmitz, M.; Baig, K.; Beaume, N.; Schwartz, C.; Frasquilho, S.; Antunes, L.; Marcon, N.; Nazarov, P.; Vallar, L.; et al. Identification of SOCS2 and SOCS6 as Biomarkers in Human Colorectal Cancer. Br. J. Cancer 2014, 111, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Ren, S.; Howell, P.; Fodstad, O.; Riker, A.I. Identification of Novel Epigenetically Modified Genes in Human Melanoma via Promoter Methylation Gene Profiling. Pigment. Cell Melanoma Res. 2008, 21, 545–558. [Google Scholar] [CrossRef]
- Fandy, T.E.; Herman, J.G.; Kerns, P.; Jiemjit, A.; Sugar, E.A.; Choi, S.-H.; Yang, A.S.; Aucott, T.; Dauses, T.; Odchimar-Reissig, R.; et al. Early Epigenetic Changes and DNA Damage do not Predict Clinical Response in an Overlapping Schedule of 5-Azacytidine and Entinostat in Patients with Myeloid Malignancies. Blood 2009, 114, 2764–2773. [Google Scholar] [CrossRef]
- Wu, Q.-Y.; Zhu, Y.-Y.; Liu, Y.; Wei, F.; Tong, Y.-X.; Cao, J.; Zhou, P.; Niu, M.-S.; Li, Z.-Y.; Zeng, L.-Y.; et al. CUEDC2, A Novel Interacting Partner of the SOCS1 Protein, Plays Important Roles in the Leukaemogenesis of Acute Myeloid Leukaemia. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Saudemont, A.; Hamrouni, A.; Marchetti, P.; Liu, J.; Jouy, N.; Hetuin, D.; Colucci, F.; Quesnel, B. Dormant Tumor Cells Develop Cross-Resistance to Apoptosis Induced by CTLs or Imatinib Mesylate via Methylation of Suppressor of Cytokine Signaling 1. Cancer Res. 2007, 67, 4491–4498. [Google Scholar] [CrossRef] [Green Version]
- Pena, M.C.R.; Pardini, M.I.M.C.; Colturato, V.A.R.; Pinheiro, N.A. Methylation Status of the SOCS 1 and JUNB Genes in Chronic Myeloid Leukemia Patients. Rev. Bras. De Hematol. E Hemoter. 2009, 31, 147–152. [Google Scholar] [CrossRef]
- Elias, M.H.; Azlan, H.; Baba, A.A.; Ankathil, R. Aberrant DNA Methylation of SOCS1 Gene is Not Associated with Resistance to Imatinib Mesylate among Chronic Myeloid Leukemia Patients. Cardiovasc. Hematol. Disord. Targets 2018, 18, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Naser, S.A. MiR-146a rs2910164 G> C Polymorphism Modulates Notch-1/IL-6 Signaling During Infection: A Possible Risk Factor for CROHN’S Disease. Gut Pathogens 2020, 12, 1–10. [Google Scholar]
- Mendell, J.T.; Olson, E.N. MicroRNAs in Stress Signaling and Human Disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardani, R.; Jafari Najaf Abadi, M.H.; Motieian, M.; Taghizadeh-Boroujeni, S.; Bayat, A.; Farsinezhad, A.; Hayat, S.M.G.; Motieian, M.; Pourghadamyari, H. MicroRNA in Leukemia: Tumor Suppressors and Oncogenes with Prognostic Potential. J. Cell. Physiol. 2019, 234, 8465–8486. [Google Scholar] [CrossRef]
- Eis, P.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in Human B Cell Lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar] [CrossRef] [Green Version]
- Cammarata, G.; Augugliaro, L.; Salemi, D.; Agueli, C.; La Rosa, M.; Dagnino, L.; Civiletto, G.; Messana, F.; Marfia, A.; Bica, M.G.; et al. Differential Expression of Specific microRNA and their Targets in Acute Myeloid Leukemia. Am. J. Hematol. 2010, 85, 331–339. [Google Scholar] [CrossRef]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.; Callen, D. The Oncogenic Role of miR-155 in Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1236–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Shen, H.; Qiu, C.; Ni, Y.; Wang, L.; Dong, W. High Expression of miR-21 and miR-155 Predicts Recurrence and Un-Favourable Survival in Non-Small Cell Lung Cancer. Eur. J. Cancer 2013, 49, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Xie, M.; Wang, X.; Jiang, X.; Li, J.; Huang, H. miR-155 Modulates TNF-α-Inhibited Osteogenic Differentiation by Targeting SOCS1 Expression. Bone 2012, 51, 498–505. [Google Scholar] [CrossRef]
- Yao, R.; Ma, Y.-L.; Liang, W.; Li, H.-H.; Ma, Z.-J.; Yu, X.; Liao, Y.-H. MicroRNA-155 Modulates Treg and Th17 Cells Differentiation and Th17 Cell Function by Targeting SOCS1. PLoS ONE 2012, 7, e46082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, R.M.; Rao, D.; Chaudhuri, A.A.; Boldin, M.; Taganov, K.D.; Nicoll, J.; Paquette, R.L.; Baltimore, D. Sustained Expression of microRNA-155 in Hematopoietic Stem Cells Causes a Myeloproliferative Disorder. J. Exp. Med. 2008, 205, 585–594. [Google Scholar] [CrossRef]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M. Nanoparticle-based Therapy in an In Vivo mi-croRNA-155 (miR-155)-Dependent Mouse Model of Lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [Green Version]
- Di Iasio, M.G.; Norcio, A.; Melloni, E.; Zauli, G. SOCS1 is Significantly Up-Regulated in Nutlin-3-Treated p53 Wild-Type B Chronic Lymphocytic Leukemia (B-CLL) Samples and Shows an Inverse Correlation with miR-155. Investig. New Drugs 2012, 30, 2403–2406. [Google Scholar] [CrossRef] [PubMed]
- Mignacca, L.; Saint-Germain, E.; Benoit, A.; Bourdeau, V.; Moro, A.; Ferbeyre, G. Sponges Against miR-19 and miR-155 Reactivate the p53-Socs1 Axis in Hematopoietic Cancers. Cytokine 2016, 82, 80–86. [Google Scholar] [CrossRef]
- Zanette, D.L.; Rivadavia, F.; Molfetta, G.A.; Barbuzano, F.G.; Proto-Siqueira, R.; Silva, W.A., Jr. miRNA Expression Profiles in Chronic Lymphocytic and Acute Lymphocytic Leukemia. Braz J. Med. Biol Res. 2007, 40, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ai, F.; Ji, W.-F.; Rao, W.; Zhang, H.-C.; Yao, W.-J. miR-19a Promotes Cell Growth and Tumorigenesis through Targeting SOCS1 in Gastric Cancer. Asian Pac. J. Cancer Prev. 2013, 14, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.G.; Chen, Z. MicroRNA-19a Functions as an Oncogenic microRNA in Non-Small Cell Lung Cancer by Targeting the Sup-pressor of Cytokine Signaling 1 and Mediating STAT3 Activation. Int. J. Mol. Med. 2015, 35, 839–846. [Google Scholar] [CrossRef] [Green Version]
- Pichiorri, F.; Suh, S.-S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.; et al. MicroRNAs Regulate Critical Genes Associated with Multiple Myeloma Pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.S.; McCoy, C.; Lloyd, A.T.; O’Farrelly, C.; Stevenson, N.J. miR-19a: An Effective Regulator of SOCS3 and Enhancer of JAK-STAT Signalling. PLoS ONE 2013, 8, e69090. [Google Scholar] [CrossRef]
- Lerut, E.; Gessler, M.; Schubert, M.; Kalogirou, C.; Kneitz, S.; Kneitz, B.; Van Poppel, H.; Riedmiller, H.; Scholz, C.J.; Spahn, M.; et al. Survival in Patients with High-Risk Prostate Cancer is Predicted by miR-221, Which Regulates Proliferation, Apoptosis, and Invasion of Prostate Cancer Cells by Inhibiting IRF2 and SOCS3. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Zhang, J.W.; Wang, X.; Li, G.C.; Wang, D.; Han, S.; Zhang, Y.D.; Luo, C.H.; Wang, H.W.; Jiang, W.J.; Li, C.X.; et al. MiR-30a-5p Promotes Cholangiocarcinoma Cell Proliferation through Targeting SOCS3. J. Cancer 2020, 11, 3604–3614. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-X.; Wang, L.; Liu, W.-J.; Zhang, H.-T.; Xue, J.-H.; Zhang, Z.-W.; Gao, C.-J. MiR-124-3p/B4GALT1 Axis Plays an Important Role in SOCS3-Regulated Growth and Chemo-Sensitivity of CML. J. Hematol. Oncol. 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Miao, F.; Zhu, J.; Chen, Y.; Tang, N.; Wang, X.; Li, X. MicroRNA-183-5p Promotes the Proliferation, Invasion and Metastasis of Human Pancreatic Adenocarcinoma Cells. Oncol. Lett. 2015, 11, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sha, C.; Jia, G.; Jingjing, Z.; Yapeng, H.; Zhi, L.; Guanghui, X. miR-486 is Involved in the Pathogenesis of Acute Myeloid Leukemia by Regulating JAK-STAT Signaling. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 394, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-M.; Guo, C.-L.; Zhang, Y.-F.; Chen, J.-F.; Liang, Z.-P.; Yang, L.-H. Leonurine-Repressed miR-18a-5p/SOCS5/JAK2/STAT3 Axis Activity Disrupts CML Malignancy. Front. Pharmacol. 2021, 12. [Google Scholar]
- Zhou, X.; Xia, Y.; Li, L.; Zhang, G. MiR-101 Inhibits Cell Growth and Tumorigenesis of Helicobacter Pylori Related Gastric Cancer by Repression of SOCS2. Cancer Biol. Ther. 2014, 16, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Gene | Cancer Type | Expression | Function | Ref. |
|---|---|---|---|---|
| SOCS1 | Breast cancer | Up-regulation | Associated with better clinical outcomes | [19] |
| Down-regulation | Enhances cell proliferation and colony formation | [20] | ||
| Colorectal tumor | Up-regulation | Reduces tumor cell invasion | [21] | |
| Multiple myeloma | Down-regulation | Supports the survival and expansion of multiple myeloma cells | [22] | |
| Prostate cancer | Down-regulation | Associated with regional lymph node invasion | [23] | |
| SOCS2 | Hepatocellular carcinoma | Down-regulation | Associated with aggressive tumor progression and poor prognosis | [15] |
| Prostate cancer | Down-regulation | Promotes cancer metastasis | [24] | |
| SOCS3 | Prostate cancer | Down-regulation | Associated with unfavorable clinical outcome | [25] |
| Colorectal cancer | Up-regulation | Inhibits proliferation, migration, and invasion, while increasing cell apoptosis | [26] | |
| SOCS4 | Thyroid cancer | Down-regulation | Induces cell migration and invasion | [27] |
| Renal cancer | Down-regulation | Promotes cells proliferation and migration | [28] | |
| SOCS5 | Pancreatic cancer | Down-regulation | Promotes tumor growth, invasion, and metastasis | [29] |
| Hepatocellular carcinoma | Down-regulation | Induces autophagy, reduces cell invasion and metastasis | [30] | |
| SOCS6 | Breast cancer | Down-regulation | Promotes cell proliferation, tumor growth and induces tamoxifen resistance | [31] |
| Gastric cancer | Down-regulation | Inhibits cell proliferation and colony formation | [32] | |
| Hepatocellular carcinoma | Down-regulation | Induces aggressive tumor progression and poor prognosis | [15] | |
| SOCS7 | Bladder cancer | Up-regulation | Induces tumor growth | [33] |
| Breast cancer | Down-regulation | Increases tumor growth and migration | [34] | |
| CIS | Breast cancer | Up-regulation | Increases cell proliferation | [17] |
| Leukemia type | Gene | Expression | Function | Ref |
|---|---|---|---|---|
| Lymphocytic leukemia | ||||
| ALL | SOCS2 | Up-regulation | Correlated with the enrichment in hematopoietic and leukemic stemness genes. | [69,70] |
| SOCS3 | Down-regulation | Associated with constitutive activation of JAK/STAT3 signaling and negatively regulated anti-tumor immunity. | [71] | |
| SOCS5 | Down-regulation | Associated with T-ALL and B-ALL harboring KMT2A rearrangements. | [72] | |
| SOCS6 | Up-regulation | Negatively correlated with chemotherapy-induced remission in ALL patients. | [73] | |
| CIS | Identified as one of the synergistic key regulators in Ph-like B-ALL. | [74] | ||
| CLL | SOCS3 | Down-regulation | Forced expression of SOCS3 reduced cell migration and increased leukemic cell death. | [75] |
| SOCS5 | Up-regulation | Associated with immune suppression in CLL. | [76] | |
| Myelogenous leukemia | ||||
| AML | SOCS1 | Down-regulation | Associated with relapsed/refractory AML compared to remission and normal control samples. | [77] |
| SOCS2 | Up-regulation | Associated with poor overall survival in pediatric AML. | [78] | |
| SOCS3 | Inhibited the CXCL12/CXCR4 signaling axis and reduced the migratory capacity of AML blasts. | [79] | ||
| SOCS5 | Down-regulation | Associated with AML samples harboring KMT2A rearrangements. | [72] | |
| CIS | Deletion of CIS in human pluripotent stem cell-derived natural killer cells enhanced anti-tumor immunity | [80] | ||
| CML | SOCS1 | Down-regulation | Associated with constitutive activation of JAK/STAT signaling, increased leukemic stem cell proliferation, and poor prognosis. | [81,82] |
| UP-regulation | Subverted cytogenetic response to IFN-α and linked to poor prognosis. | [83] | ||
| SOCS2 | Up-regulation | Associated with blast crisis compared with chronic phase patients and healthy individuals | [18] | |
| SOCS3 | Down-regulation | Linked to imatinib resistance in BCR-ABL positive CML | [81] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keewan, E.; Matlawska-Wasowska, K. The Emerging Role of Suppressors of Cytokine Signaling (SOCS) in the Development and Progression of Leukemia. Cancers 2021, 13, 4000. https://doi.org/10.3390/cancers13164000
Keewan E, Matlawska-Wasowska K. The Emerging Role of Suppressors of Cytokine Signaling (SOCS) in the Development and Progression of Leukemia. Cancers. 2021; 13(16):4000. https://doi.org/10.3390/cancers13164000
Chicago/Turabian StyleKeewan, Esra’a, and Ksenia Matlawska-Wasowska. 2021. "The Emerging Role of Suppressors of Cytokine Signaling (SOCS) in the Development and Progression of Leukemia" Cancers 13, no. 16: 4000. https://doi.org/10.3390/cancers13164000
APA StyleKeewan, E., & Matlawska-Wasowska, K. (2021). The Emerging Role of Suppressors of Cytokine Signaling (SOCS) in the Development and Progression of Leukemia. Cancers, 13(16), 4000. https://doi.org/10.3390/cancers13164000