
The Renin–Angiotensin System in the Tumor Microenvironment of Glioblastoma

 , and
, and 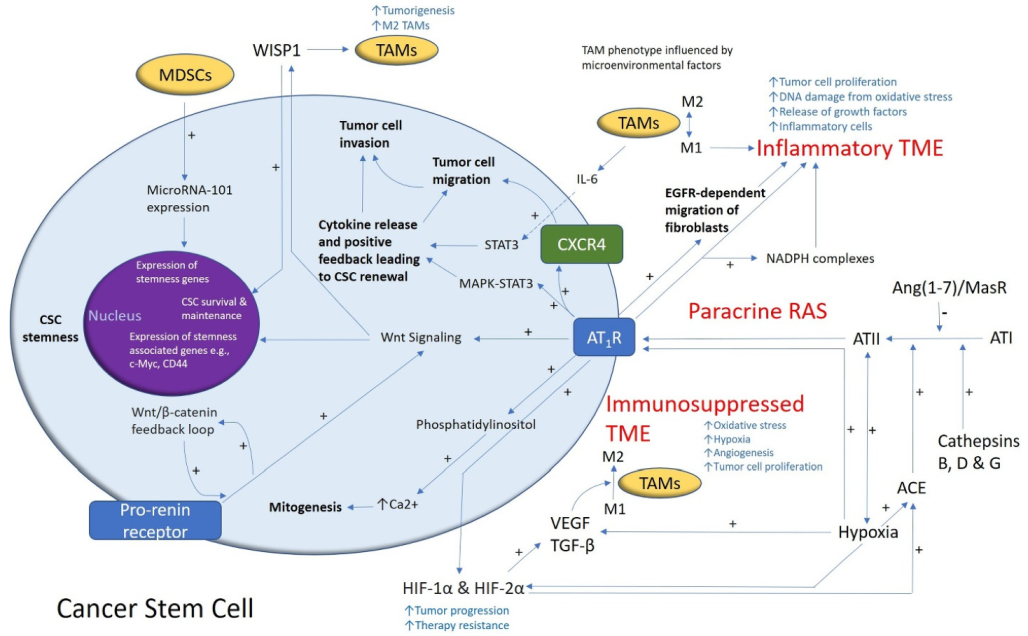{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. GB Tumor Microenvironment
3. Glioblastoma Cancer Stem Cells
4. The Renin–Angiotensin System and Convergent Signaling Pathways in Glioblastoma
5. Recent Developments
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino Mdel, C.; Otero, A.; Lopes, M.C.; De Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Zentgraf, H.; Balss, J.; Hartmann, C.; Von Deimling, A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009, 118, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.; Fujii, Y.; Osaka, N.; Ito, D.; Hirota, Y.; Sasaki, A.T. Emerging Roles of Wild-type and Mutant IDH1 in Growth, Metabolism and Therapeutics of Glioma. In Gliomas; Debinski, W., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- Gately, L.; McLachlan, S.A.; Dowling, A.; Philip, J. Life beyond a diagnosis of glioblastoma: A systematic review of the literature. J. Cancer Surviv. 2017, 11, 447–452. [Google Scholar] [CrossRef]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Awan, M.; Liu, S.; Sahgal, A.; Das, S.; Chao, S.T.; Chang, E.L.; Knisely, J.P.; Redmond, K.; Sohn, J.W.; Machtay, M.; et al. Extra-CNS metastasis from glioblastoma: A rare clinical entity. Expert Rev. Anticancer Ther. 2015, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Sullivan, R.; Maresh, G.; Zhang, X.; Salomon, C.; Hooper, J.; Margolin, D.; Li, L. The Emerging Roles of Extracellular Vesicles As Communication Vehicles within the Tumor Microenvironment and Beyond. Front. Endocrinol. 2017, 8, 194. [Google Scholar] [CrossRef]
- Haznedaroglu, I.C.; Malkan, U.Y. Local bone marrow renin-angiotensin system in the genesis of leukemia and other malignancies. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4089–4111. [Google Scholar]
- Kilmister, E.J.; Tan, S.T. The Role of the Renin-Angiotensin System in the Cancer Stem Cell Niche. J. Histochem. Cytochem. 2021, 00221554211026295. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, W.; Xue, B.Z.; Yi, D.Y.; Zhao, H.Y.; Fu, P. Growth factors contribute to the mediation of angiogenic capacity of glioma-associated mesenchymal stem cells. Oncol. Lett. 2021, 21, 215. [Google Scholar] [CrossRef] [PubMed]
- Monteforte, A.; Lam, B.; Sherman, M.B.; Henderson, K.; Sligar, A.D.; Spencer, A.; Tang, B.; Dunn, A.K.; Baker, A.B. (*) Glioblastoma Exosomes for Therapeutic Angiogenesis in Peripheral Ischemia. Tissue Eng. Part A 2017, 23, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Med. Oncol. 2019, 37, 2. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; Von Stechow, L.; Schanzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nister, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef]
- Guardia, G.D.A.; Correa, B.R.; Araujo, P.R.; Qiao, M.; Burns, S.; Penalva, L.O.F.; Galante, P.A.F. Proneural and mesenchymal glioma stem cells display major differences in splicing and lncRNA profiles. NPJ Genom. Med. 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef]
- Bar, E.E. Glioblastoma, cancer stem cells and hypoxia. Brain Pathol. 2011, 21, 119–129. [Google Scholar] [CrossRef]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; De Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro. Oncol. 2017, 19, 1435–1446. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; Van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12, aax6337. [Google Scholar] [CrossRef]
- Wu, H.; Han, Y.; Rodriguez Sillke, Y.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Adams, J.M.; Strasser, A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008, 68, 4018–4021. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medecine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Najafi, M.; Mortezaee, K.; Ahadi, R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. 2019, 231, 116520. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Gurel, C.; Inetas, G.; Hortu, I.; Tunc, E.; Kuscu, G.C.; Dindaroglu, F.C.; Sahin, O.; Buhur, A.; Oktem, G. Cancer and Cancer Stem Cells: New Molecular Perspectives. Crit. Rev. Oncog. 2019, 24, 99–104. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Quail, D.F.; Taylor, M.J.; Postovit, L.M. Microenvironmental regulation of cancer stem cell phenotypes. Curr. Stem Cell Res. Ther. 2012, 7, 197–216. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. How does multistep tumorigenesis really proceed? Cancer Discov. 2015, 5, 22–24. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Baillie, R.; Itinteang, T.; Yu, H.H.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma. J. Clin. Pathol. 2016, 69, 742–744. [Google Scholar] [CrossRef]
- Yu, H.H.; Featherston, T.; Tan, S.T.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Itinteang, T. Characterization of cancer stem cells in moderately differentiated buccal mucosal squamous cell carcinoma. Front. Surg. 2016, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Brasch, H.D.; Dunne, J.C.; Davis, P.F.; Tan, S.T.; Itinteang, T. The identification of three cancer stem cell subpopulations within moderately differentiated lip squamous cell carcinoma. Front. Surg. 2017, 4, 12. [Google Scholar] [CrossRef]
- Koh, S.P.; Brasch, H.D.; De Jongh, J.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in moderately differentiated head and neck cutaneous squamous cell carcinoma. Heliyon 2019, 5, e02257. [Google Scholar] [CrossRef]
- Kilmister, E.J.; Patel, J.; Van Schaijik, B.; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T.; et al. Cancer Stem Cell Subpopulations Are Present Within Metastatic Head and Neck Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1091. [Google Scholar] [CrossRef]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Marsh, R.W.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in primary colon adenocarcinoma. PLoS ONE 2019, 14, e0221963. [Google Scholar] [CrossRef]
- Humphries, H.N.; Wickremesekera, S.K.; Marsh, R.W.; Brasch, H.D.; Mehrotra, S.; Tan, S.T.; Itinteang, T. Characterization of Cancer Stem Cells in Colon Adenocarcinoma Metastasis to the Liver. Front. Surg. 2018, 4, 76. [Google Scholar] [CrossRef]
- Wickremesekera, A.C.; Brasch, H.D.; Lee, V.M.; Davis, P.F.; Parker, A.; Koeck, H.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in metastatic melanoma to the brain express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 5, 62. [Google Scholar] [CrossRef]
- Yoganandarajah, V.; Patel, J.; Van Schaijik, B.; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T.; et al. Identification of Cancer Stem Cell Subpopulations in Head and Neck Metastatic Malignant Melanoma. Cells 2020, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.; Wickremesekera, A.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Glioblastoma Multiforme. Front. Surg. 2016, 3, 48. [Google Scholar] [CrossRef]
- Kalkan, R. Glioblastoma Stem Cells as a New Therapeutic Target for Glioblastoma. Clin. Med. Insights. Oncol. 2015, 9, 95–103. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Tejero, R.; Huang, Y.; Katsyv, I.; Kluge, M.; Lin, J.Y.; Tome-Garcia, J.; Daviaud, N.; Wang, Y.; Zhang, B.; Tsankova, N.M.; et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 2019, 42, 252–269. [Google Scholar] [CrossRef]
- Cui, C.; Chen, X.; Liu, Y.; Cao, B.; Xing, Y.; Liu, C.; Yang, F.; Li, Y.; Yang, T.; Hua, L.; et al. beta1,4-Galactosyltransferase V activates Notch1 signaling in glioma stem-like cells and promotes their transdifferentiation into endothelial cells. J. Biol. Chem. 2018, 293, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Guichet, P.O.; Guelfi, S.; Teigell, M.; Hoppe, L.; Bakalara, N.; Bauchet, L.; Duffau, H.; Lamszus, K.; Rothhut, B.; Hugnot, J.P. Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem Cells 2015, 33, 21–34. [Google Scholar] [CrossRef]
- Iwadate, Y. Plasticity in Glioma Stem Cell Phenotype and Its Therapeutic Implication. Neurol. Med. Chir. 2018, 58, 61–70. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, A.D.; Poisson, L.M.; Cherba, D.; Webb, C.P.; Transou, A.D.; Lemke, N.W.; Hong, X.; Hasselbach, L.A.; Irtenkauf, S.M.; Mikkelsen, T.; et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16, 193–206.e25. [Google Scholar] [CrossRef]
- Gangemi, R.M.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 222. [Google Scholar] [CrossRef] [PubMed]
- Senft, C.; Priester, M.; Polacin, M.; Schroder, K.; Seifert, V.; Kogel, D.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neurooncol. 2011, 101, 393–403. [Google Scholar] [CrossRef]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.; Van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J.; et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251–41264. [Google Scholar] [CrossRef]
- Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Robalo Cordeiro, C.; Medeiros, R. Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers 2020, 12, 1457. [Google Scholar] [CrossRef]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar] [CrossRef]
- Nakagawa, P.; Gomez, J.; Grobe, J.L.; Sigmund, C.D. The Renin-Angiotensin System in the Central Nervous System and Its Role in Blood Pressure Regulation. Curr. Hypertens. Rep. 2020, 22, 7. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef]
- Cassis, L.A.; Saye, J.; Peach, M.J. Location and regulation of rat angiotensinogen messenger RNA. Hypertension 1988, 11, 591–596. [Google Scholar] [CrossRef]
- Bodiga, V.L.; Bodiga, S. Renin Angiotensin System in Cognitive Function and Dementia. Asian J. Neurosci. 2013, 2013, 1–18. [Google Scholar] [CrossRef]
- Grobe, J.L.; Xu, D.; Sigmund, C.D. An intracellular renin-angiotensin system in neurons: Fact, hypothesis, or fantasy. Physiology 2008, 23, 187–193. [Google Scholar] [CrossRef]
- Kopp, U.C.; DiBona, G.F. Neural regulation of renin secretion. Semin. Nephrol. 1993, 13, 543–551. [Google Scholar] [PubMed]
- Riquier-Brison, A.D.M.; Sipos, A.; Prokai, A.; Vargas, S.L.; Toma, L.; Meer, E.J.; Villanueva, K.G.; Chen, J.C.M.; Gyarmati, G.; Yih, C.; et al. The macula densa prorenin receptor is essential in renin release and blood pressure control. Am. J. Physiol. Renal. Physiol. 2018, 315, F521–F534. [Google Scholar] [CrossRef]
- Jutras, I.; Reudelhuber, T.L. Prorenin processing by cathepsin B in vitro and in transfected cells. FEBS Lett. 1999, 443, 48–52. [Google Scholar] [CrossRef]
- Neves, F.A.; Duncan, K.G.; Baxter, J.D. Cathepsin B is a prorenin processing enzyme. Hypertension 1996, 27, 514–517. [Google Scholar] [CrossRef]
- Naseem, R.H.; Hedegard, W.; Henry, T.D.; Lessard, J.; Sutter, K.; Katz, S.A. Plasma cathepsin D isoforms and their active metabolites increase after myocardial infarction and contribute to plasma renin activity. Basic Res. Cardiol. 2005, 100, 139–146. [Google Scholar] [CrossRef]
- Rykl, J.; Thiemann, J.; Kurzawski, S.; Pohl, T.; Gobom, J.; Zidek, W.; Schluter, H. Renal cathepsin G and angiotensin II generation. J. Hypertens. 2006, 24, 1797–1807. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Metzger, R.; Sun, Z.L.; Berestetskaya, Y.V.; Albrecht, R.F.; Danilov, S.M. Development and characterization of rat monoclonal antibodies to denatured mouse angiotensin-converting enzyme. Tissue Antigens 2005, 65, 240–251. [Google Scholar] [CrossRef]
- Nakagawa, P.; Sigmund, C.D. How Is the Brain Renin-Angiotensin System Regulated? Hypertension 2017, 70, 10–18. [Google Scholar] [CrossRef]
- Hashemzehi, M.; Beheshti, F.; Hassanian, S.M.; Ferns, G.A.; Khazaei, M.; Avan, A. Therapeutic potential of renin angiotensin system inhibitors in cancer cells metastasis. Pathol. Res. Pract. 2020, 216, 153010. [Google Scholar] [CrossRef]
- Yu, C.; Tang, W.; Wang, Y.; Shen, Q.; Wang, B.; Cai, C.; Meng, X.; Zou, F. Downregulation of ACE2/Ang-(1-7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett. 2016, 376, 268–277. [Google Scholar] [CrossRef]
- Ziaja, M.; Urbanek, K.A.; Kowalska, K.; Piastowska-Ciesielska, A.W. Angiotensin II and Angiotensin Receptors 1 and 2-Multifunctional System in Cells Biology, What Do We Know? Cells 2021, 10, 381. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Lopes, D.H.; Shah, K.H.; Bernstein, E.A.; Fuchs, D.T.; Yu, J.J.; et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Investig. 2014, 124, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Elased, K.M.; Cunha, T.S.; Marcondes, F.K.; Morris, M. Brain angiotensin-converting enzymes: Role of angiotensin-converting enzyme 2 in processing angiotensin II in mice. Exp. Physiol. 2008, 93, 665–675. [Google Scholar] [CrossRef]
- Tetzner, A.; Gebolys, K.; Meinert, C.; Klein, S.; Uhlich, A.; Trebicka, J.; Villacañas, Ó.; Walther, T. G-Protein–Coupled Receptor MrgD Is a Receptor for Angiotensin-(1–7) Involving Adenylyl Cyclase, cAMP, and Phosphokinase A. Hypertension 2016, 68, 185–194. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Lanciego, J.L.; Guerra, M.J. Brain Renin-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Front. Aging Neurosci. 2017, 9, 129. [Google Scholar] [CrossRef]
- Xu, Q.; Jensen, D.D.; Peng, H.; Feng, Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol. Ther. 2016, 164, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Yatabe, M.S. The (pro)renin receptor in health and disease. Nat. Rev. Nephrol. 2019, 15, 693–712. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Xu, P.; Li, G.; Qiao, Y.; Han, W.; Geng, C.; Liao, D.; Yang, M.; Chen, D.; Jiang, P. Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: Role of renin-angiotensin system. Redox Biol. 2019, 26, 101295. [Google Scholar] [CrossRef] [PubMed]
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millan, S.; Sanchez-Corona, J. The renin-angiotensin system meets the hallmarks of cancer. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 227–233. [Google Scholar] [CrossRef]
- Beitia, M.; Solano-Iturri, J.D.; Errarte, P.; Sanz, B.; Perez, I.; Etxezarraga, M.C.; Loizate, A.; Asumendi, A.; Larrinaga, G. Altered expression of renin-angiotensin system receptors throughout colorectal adenoma-adenocarcinoma sequence. Int. J. Med. Sci. 2019, 16, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Renziehausen, A.; Wang, H.; Rao, B.; Weir, L.; Nigro, C.L.; Lattanzio, L.; Merlano, M.; Vega-Rioja, A.; Del Carmen Fernandez-Carranco, M.; Hajji, N.; et al. The renin angiotensin system (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene 2019, 38, 2320–2336. [Google Scholar] [CrossRef]
- Featherston, T.; Yu, H.H.; Dunne, J.C.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Moderately Differentiated Buccal Mucosal Squamous Cell Carcinoma Express Components of the Renin-Angiotensin System. Front. Surg. 2016, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Itinteang, T.; Dunne, J.C.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma express components of the renin-angiotensin system. J. Clin. Pathol. 2016, 69, 942–945. [Google Scholar] [CrossRef]
- Siljee, S.; Milne, B.; Brasch, H.D.; Bockett, N.; Patel, J.; Davis, P.F.; Kennedy-Smith, A.; Itinteang, T.; Tan, S.T. Expression of Components of the Renin-Angiotensin System by Cancer Stem Cells in Renal Clear Cell Carcinoma. Biomolecules 2021, 11, 537. [Google Scholar] [CrossRef]
- Nallaiah, S.; Lee, V.M.Y.; Brasch, H.D.; De Jongh, J.; Schaijik, B.V.; Marsh, R.; Tan, S.T.; Itinteang, T. Cancer stem cells within moderately differentiated head and neck cutaneous squamous cell carcinoma express components of the renin-angiotensin system. J. Plast. Reconstr. Aesthet. Surg. 2019, 72, 1484–1493. [Google Scholar] [CrossRef]
- Siljee, S.; Buchanan, O.; Brasch, H.D.; Bockett, N.; Patel, J.; Paterson, E.; Purdie, G.L.; Davis, P.F.; Itinteang, T.; Tan, S.T. Cancer Stem Cells in Metastatic Head and Neck Cutaneous Squamous Cell Carcinoma Express Components of the Renin-Angiotensin System. Cells 2021, 10, 243. [Google Scholar] [CrossRef]
- Narayanan, A.; Wickremesekera, S.K.; Van Schaijik, B.; Marsh, R.W.; Brasch, H.D.; Tan, S.T.; Itinteang, T. Cancer stem cells in liver metastasis from colon adenocarcinoma express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 5, 36. [Google Scholar] [CrossRef]
- Bradshaw, A.R.; Wickremesekera, A.C.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Glioblastoma Multiforme Cancer Stem Cells Express Components of the Renin-Angiotensin System. Front. Surg. 2016, 3, 51. [Google Scholar] [CrossRef]
- Kouchi, M.; Shibayama, Y.; Ogawa, D.; Miyake, K.; Nishiyama, A.; Tamiya, T. (Pro)renin receptor is crucial for glioma development via the Wnt/beta-catenin signaling pathway. J. Neurosurg. 2017, 127, 819–828. [Google Scholar] [CrossRef]
- Wang, J.; Nishiyama, A.; Matsuyama, M.; Wang, Z.; Yuan, Y. The (pro)renin receptor: A novel biomarker and potential therapeutic target for various cancers. Cell Commun. Signal. 2020, 18, 39. [Google Scholar] [CrossRef]
- Nguyen, G. Renin, (pro)renin and receptor: An update. Clin. Sci. 2011, 120, 169–178. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Ohkawara, B.; Acebron, S.P.; Karaulanov, E.; Reinhard, C.; Ingelfinger, D.; Boutros, M.; Niehrs, C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 2010, 327, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Wiedemeyer, R.; Yan, H.; Quayle, S.N.; Ivanova, E.V.; Paik, J.-H.; Zhang, H.; Xiao, Y.; Perry, S.R.; et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell 2010, 17, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Pu, P.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009, 16, 351–361. [Google Scholar] [CrossRef]
- Pulvirenti, T.; Van Der Heijden, M.; Droms, L.A.; Huse, J.T.; Tabar, V.; Hall, A. Dishevelled 2 Signaling Promotes Self-Renewal and Tumorigenicity in Human Gliomas. Cancer Res. 2011, 71, 7280. [Google Scholar] [CrossRef] [PubMed]
- Ariza, A.; Fernandez, L.A.; Inagami, T.; Kim, J.H.; Manuelidis, E.E. Renin in glioblastoma multiforme and its role in neovascularization. Am. J. Clin. Pathol. 1988, 90, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Y.; Jiang, Y. Podocalyxin promotes glioblastoma multiforme cell invasion and proliferation by inhibiting angiotensin-(1-7)/Mas signaling. Oncol. Rep. 2015, 33, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.; Malla, R.; Alapati, K.; Gorantla, B.; Gujrati, M.; Dinh, D.H.; Rao, J.S. Cathepsin B and uPAR regulate self-renewal of glioma-initiating cells through GLI-regulated Sox2 and Bmi1 expression. Carcinogenesis 2013, 34, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Ding, Y.M.; Hueng, D.Y.; Chen, J.Y.; Chen, Y. Caffeine suppresses the progression of human glioblastoma via cathepsin B and MAPK signaling pathway. J. Nutr. Biochem. 2016, 33, 63–72. [Google Scholar] [CrossRef]
- Koh, S.P.; Wickremesekera, A.C.; Brasch, H.D.; Marsh, R.; Tan, S.T.; Itinteang, T. Expression of Cathepsins B, D, and G in Isocitrate Dehydrogenase-Wildtype Glioblastoma. Front. Surg. 2017, 4, 28. [Google Scholar] [CrossRef]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef]
- Krop, M.; Lu, X.; Danser, A.H.; Meima, M.E. The (pro)renin receptor. A decade of research: What have we learned? Pflugers Arch. 2013, 465, 87–97. [Google Scholar] [CrossRef]
- Arundhathi, A.; Chuang, W.H.; Chen, J.K.; Wang, S.E.; Shyr, Y.M.; Chen, J.Y.; Liao, W.N.; Chen, H.W.; Teng, Y.M.; Pai, C.C.; et al. Prorenin receptor acts as a potential molecular target for pancreatic ductal adenocarcinoma diagnosis. Oncotarget 2016. [Google Scholar] [CrossRef]
- Pinter, M.; Kwanten, W.J.; Jain, R.K. Renin-Angiotensin System Inhibitors to Mitigate Cancer Treatment-Related Adverse Events. Clin. Cancer Res. 2018, 24, 3803–3812. [Google Scholar] [CrossRef]
- Shen, J.; Huang, Y.M.; Wang, M.; Hong, X.Z.; Song, X.N.; Zou, X.; Pan, Y.H.; Ling, W.; Zhu, M.H.; Zhang, X.X.; et al. Renin-angiotensin system blockade for the risk of cancer and death. J. Renin Angiotensin Aldosterone Syst. 2016, 17. [Google Scholar] [CrossRef]
- Van der Knaap, R.; Siemes, C.; Coebergh, J.W.; Van Duijn, C.M.; Hofman, A.; Stricker, B.H. Renin-angiotensin system inhibitors, angiotensin I-converting enzyme gene insertion/deletion polymorphism, and cancer: The Rotterdam Study. Cancer 2008, 112, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Makar, G.A.; Holmes, J.H.; Yang, Y.X. Angiotensin-converting enzyme inhibitor therapy and colorectal cancer risk. J. Natl. Cancer Inst. 2014, 106, djt374. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.N.; Wang, J.H.; Zhu, J.Z.; Lin, J.Q.; Yu, C.H.; Li, Y.M. Angiotensin-converting enzyme inhibitors/angiotensin receptor blockers therapy and colorectal cancer: A systematic review and meta-analysis. Cancer Causes Control 2015, 26, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.Y.; Chen, K.B.; Tsai, T.H.; Tsai, W.C. Lowered cancer risk with ACE inhibitors/ARBs: A population-based cohort study. J. Clin. Hypertens. 2014, 16, 27–33. [Google Scholar] [CrossRef]
- Liu, H.; Naxerova, K.; Pinter, M.; Incio, J.; Lee, H.; Shigeta, K.; Ho, W.W.; Crain, J.A.; Jacobson, A.; Michelakos, T.; et al. Use of Angiotensin System Inhibitors Is Associated with Immune Activation and Longer Survival in Nonmetastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 5959–5969. [Google Scholar] [CrossRef]
- Rosenthal, T.; Gavras, I. Renin-Angiotensin Inhibition in Combating Malignancy: A Review. Anticancer Res. 2019, 39, 4597–4602. [Google Scholar] [CrossRef]
- Happold, C.; Gorlia, T.; Nabors, L.B.; Erridge, S.C.; Reardon, D.A.; Hicking, C.; Picard, M.; Stupp, R.; Weller, M.; Group, E.B.T.; et al. Do statins, ACE inhibitors or sartans improve outcome in primary glioblastoma? J. Neurooncol. 2018, 138, 163–171. [Google Scholar] [CrossRef]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef]
- Levin, V.A.; Chan, J.; Datta, M.; Yee, J.L.; Jain, R.K. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J. Neurooncol. 2017, 134, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; Pavlakis, N.; Wheeler, H.; Grant, R.; Simes, J.; Khasraw, M. Anti-angiogenic therapy for high-grade glioma. Cochrane Database Syst. Rev. 2018, 11, CD008218. [Google Scholar] [CrossRef] [PubMed]
- Burcklé, C.; Bader, M. Prorenin and its ancient receptor. Hypertension 2006, 48, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Juillerat-Jeanneret, L.; Celerier, J.; Chapuis Bernasconi, C.; Nguyen, G.; Wostl, W.; Maerki, H.P.; Janzer, R.C.; Corvol, P.; Gasc, J.M. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br. J. Cancer 2004, 90, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Roth, I.M.; Wickremesekera, A.C.; Wickremesekera, S.K.; Davis, P.F.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells via Modulation of the Renin-Angiotensin System. Front. Oncol. 2019, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.C.; Roth, I.M.; Wickremesekera, A.C.; Davis, P.F.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells 2019, 8, 1364. [Google Scholar] [CrossRef] [PubMed]
- Abiodun, O.A.; Ola, M.S. Role of brain renin angiotensin system in neurodegeneration: An update. Saudi J. Biol. Sci. 2020, 27, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef]
- Pan, Y.; Lu, P.; Yin, L.; Chen, K.; He, Y. Effect of fluoride on the proteomic profile of the hippocampus in rats. Z. Naturforsch. C. J. Biosci. 2015, 70, 151–157. [Google Scholar] [CrossRef]
- Adilijiang, A.; Hirano, M.; Okuno, Y.; Aoki, K.; Ohka, F.; Maeda, S.; Tanahashi, K.; Motomura, K.; Shimizu, H.; Yamaguchi, J.; et al. Next Generation Sequencing-Based Transcriptome Predicts Bevacizumab Efficacy in Combination with Temozolomide in Glioblastoma. Molecules 2019, 24, 3046. [Google Scholar] [CrossRef]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Blue, R.; Miranda, S.P.; Gu, B.J.; Chen, H.I. A Primer on Human Brain Organoids for the Neurosurgeon. Neurosurgery 2020, 87, 620–629. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef]
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 2020, 369, aaz5626. [Google Scholar] [CrossRef]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro. Oncol. 2020, 22, 1138–1149. [Google Scholar] [CrossRef]
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov. 2020, 10, 964–979. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F.P.; Garofano, L.; D’Angelo, F.; Yu, K.; Tang, F.; Yuan, J.; Zhang, J.; Cerulo, L.; Pagnotta, S.M.; Bedognetti, D.; et al. A map of tumor-host interactions in glioma at single-cell resolution. Gigascience 2020, 9, giaa109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jin, M.; Zhao, J.; Chen, J.; Jin, W. Organoid models of glioblastoma: Advances, applications and challenges. Am. J. Cancer Res. 2020, 10, 2242–2257. [Google Scholar] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Rawe, M.; Kilmister, E.J.; Mantamadiotis, T.; Kaye, A.H.; Tan, S.T.; Wickremesekera, A.C. The Renin–Angiotensin System in the Tumor Microenvironment of Glioblastoma. Cancers 2021, 13, 4004. https://doi.org/10.3390/cancers13164004
O’Rawe M, Kilmister EJ, Mantamadiotis T, Kaye AH, Tan ST, Wickremesekera AC. The Renin–Angiotensin System in the Tumor Microenvironment of Glioblastoma. Cancers. 2021; 13(16):4004. https://doi.org/10.3390/cancers13164004
Chicago/Turabian StyleO’Rawe, Michael, Ethan J. Kilmister, Theo Mantamadiotis, Andrew H. Kaye, Swee T. Tan, and Agadha C. Wickremesekera. 2021. "The Renin–Angiotensin System in the Tumor Microenvironment of Glioblastoma" Cancers 13, no. 16: 4004. https://doi.org/10.3390/cancers13164004
APA StyleO’Rawe, M., Kilmister, E. J., Mantamadiotis, T., Kaye, A. H., Tan, S. T., & Wickremesekera, A. C. (2021). The Renin–Angiotensin System in the Tumor Microenvironment of Glioblastoma. Cancers, 13(16), 4004. https://doi.org/10.3390/cancers13164004