
The Function of the Mutant p53-R175H in Cancer

 and
and 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Gain-of-Function of Mutant p53-R175H
2.1. Inducing Genetic Instability
2.2. Promoting Tumor Cell Growth
2.3. Promoting Tumor Migration, Invasion, and Metastasis
2.4. Promoting Tumor Initiation and Conferring Stemness
2.5. Promoting Tumor Drug Resistance, Inflammation, Angiogenesis, and Metabolic Reprogramming
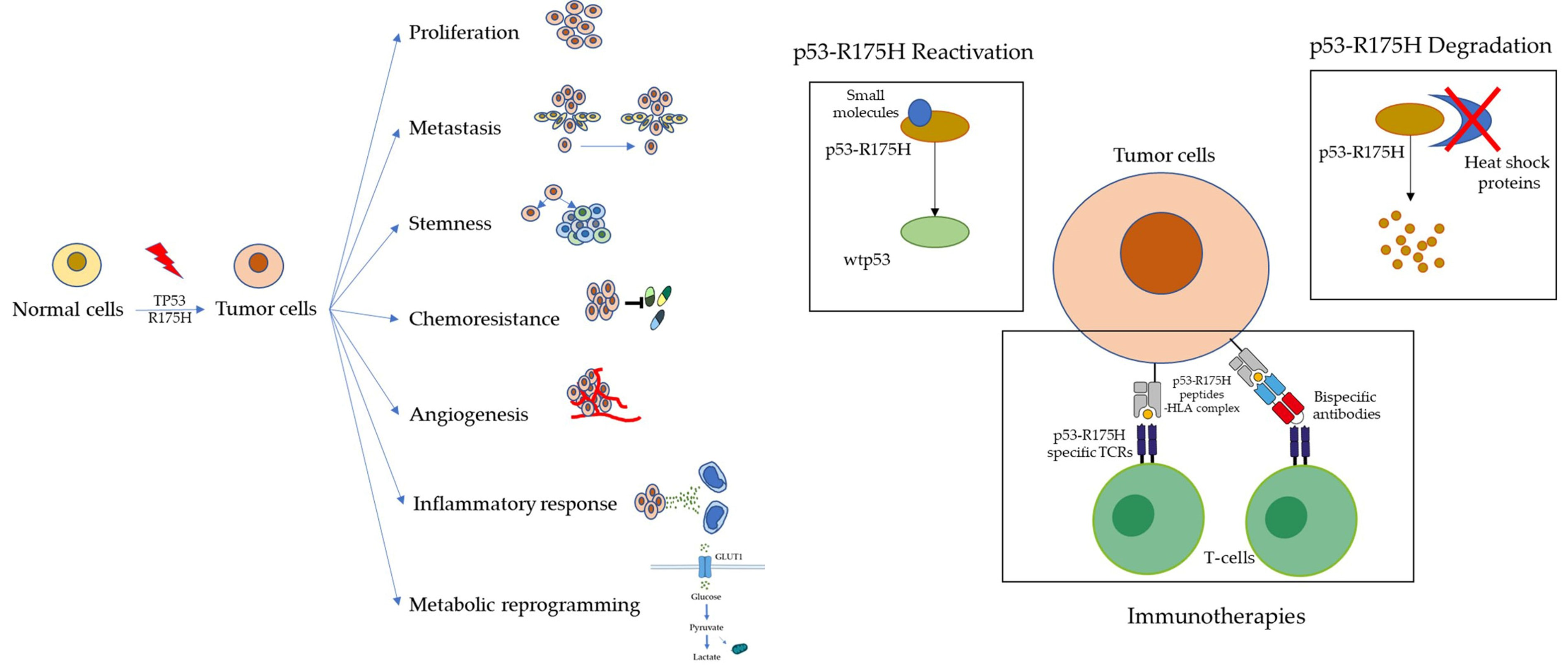
3. Targeting p53-R175H for Cancer Therapy
3.1. Reactivating Wild-Type p53 Function
3.2. Promoting Mutant p53 Degradation
3.3. Immunotherapy Based on Mutant p53 Neoantigen Recognition
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Guimaraes, D.P.; Hainaut, P. TP53: A key gene in human cancer. Biochimie 2002, 84, 83–93. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data (R20). Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Xu, A.; Gingold, J.; Strong, L.C.; Zhao, R.; Lee, D.F. Li-Fraumeni Syndrome Disease Model: A Platform to Develop Precision Cancer Therapy Targeting Oncogenic p53. Trends Pharmacol. Sci. 2017, 38, 908–927. [Google Scholar] [CrossRef]
- Figueiredo, B.C.; Sandrini, R.; Zambetti, G.P.; Pereira, R.M.; Cheng, C.; Liu, W.; Lacerda, L.; Pianovski, M.A.; Michalkiewicz, E.; Jenkins, J.; et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J. Med. Genet. 2006, 43, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T.; Kar, R.K.; Sa, G. Structural and sequential context of p53: A review of experimental and theoretical evidence. Prog. Biophys. Mol. Biol. 2015, 117, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Quante, T.; Otto, B.; Brazdova, M.; Kejnovska, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303. [Google Scholar] [CrossRef] [Green Version]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, X.; Tang, Y.; Lao, Z.; Lei, J.; Wei, G. Common cancer mutations R175H and R273H drive the p53 DNA-binding domain towards aggregation-prone conformations. Phys. Chem. Chem. Phys. 2020, 22, 9225–9232. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Scian, M.J.; Stagliano, K.E.; Ellis, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res. 2004, 64, 7447–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuarai, S.; Yamanaka, K.; Kotani, H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006, 66, 6319–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Chen, X. Identification of GRO1 as a critical determinant for mutant p53 gain of function. J. Biol. Chem. 2009, 284, 12178–12187. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef]
- Datta, A.; Ghatak, D.; Das, S.; Banerjee, T.; Paul, A.; Butti, R.; Gorain, M.; Ghuwalewala, S.; Roychowdhury, A.; Alam, S.K.; et al. p53 gain-of-function mutations increase Cdc7-dependent replication initiation. EMBO Rep. 2017, 18, 2030–2050. [Google Scholar] [CrossRef] [PubMed]
- Valentino, E.; Bellazzo, A.; Di Minin, G.; Sicari, D.; Apollonio, M.; Scognamiglio, G.; Di Bonito, M.; Botti, G.; Del Sal, G.; Collavin, L. Mutant p53 potentiates the oncogenic effects of insulin by inhibiting the tumor suppressor DAB2IP. Proc. Natl. Acad. Sci. USA 2017, 114, 7623–7628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 38, 198–211.e198. [Google Scholar] [CrossRef]
- Liu, G.; McDonnell, T.J.; Luna, R.M.D.; Kapoor, M.; Mims, B.; El-Naggar, A.K.; Lozano, G. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4174–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.t.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: Regulators and effectors. Hum. Mutat. 2014, 35, 702–714. [Google Scholar] [CrossRef]
- Ferraiuolo, M.; Di Agostino, S.; Blandino, G.; Strano, S. Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front. Oncol. 2016, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Zawacka-Pankau, J.E. The Undervalued Avenue to Reinstate Tumor Suppressor Functionality of the p53 Protein Family for Improved Cancer Therapy-Drug Repurposing. Cancers 2020, 12, 2717. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef]
- Dong, P.X.; Xu, Z.J.; Jia, N.; Li, D.J.; Feng, Y.J. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol. Cancer 2009, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, C.; Ma, B.; Xu, M.; Xu, S.; Liu, J.; Tian, Y.; Fu, Y.; Luo, Y. Mutant p53 Drives Cancer Metastasis via RCP-Mediated Hsp90alpha Secretion. Cell Rep. 2020, 32, 107879. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, J.D.; Morton, J.P.; Wilczynska, A.; Sansom, O.J.; Bushell, M.D. MiR-142-3p is downregulated in aggressive p53 mutant mouse models of pancreatic ductal adenocarcinoma by hypermethylation of its locus. Cell Death Dis. 2018, 9, 14. [Google Scholar] [CrossRef]
- Capaci, V.; Bascetta, L.; Fantuz, M.; Beznoussenko, G.V.; Sommaggio, R.; Cancila, V.; Bisso, A.; Campaner, E.; Mironov, A.A.; Wisniewski, J.R.; et al. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat. Commun. 2020, 11, 3945. [Google Scholar] [CrossRef]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.X.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012, 33, 442–451. [Google Scholar] [CrossRef]
- Yue, X.; Zhang, C.; Zhao, Y.; Liu, J.; Lin, A.W.; Tan, V.M.; Drake, J.M.; Liu, L.; Boateng, M.N.; Li, J.; et al. Gain-of-function mutant p53 activates small GTPase Rac1 through SUMOylation to promote tumor progression. Genes Dev. 2017, 31, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e318. [Google Scholar] [CrossRef] [Green Version]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef]
- Grespi, F.; Landre, V.; Molchadsky, A.; Di Daniele, N.; Marsella, L.T.; Melino, G.; Rotter, V. Differential regulated microRNA by wild type and mutant p53 in induced pluripotent stem cells. Cell Death Dis. 2016, 7, e2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Liu, D.P.; Xu, Y. The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene 2013, 32, 2900–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef] [PubMed]
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018, 37, 1669–1684. [Google Scholar] [CrossRef] [Green Version]
- Polireddy, K.; Singh, K.; Pruski, M.; Jones, N.C.; Manisundaram, N.V.; Ponnela, P.; Ouellette, M.; Van Buren, G.; Younes, M.; Bynon, J.S.; et al. Mutant p53(R175H) promotes cancer initiation in the pancreas by stabilizing HSP70. Cancer Lett. 2019, 453, 122–130. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.Z.; He, Y.Y.; Wang, H.H.; Zhang, H.L.; Zhang, J.; Yan, X.F.; Wang, X.J.; Che, Q.; Ke, J.Q.; Chen, Z.; et al. Mutant p53 induces EZH2 expression and promotes epithelial-mesenchymal transition by disrupting p68-Drosha complex assembly and attenuating miR-26a processing. Oncotarget 2015, 6, 44660–44674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Donnenberg, V.S.; Donnenberg, A.D. Multiple drug resistance in cancer revisited: The cancer stem cell hypothesis. J. Clin. Pharmacol. 2005, 45, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.W.; Bruns, C. Cancer stem cells and angiogenesis. Int. J. Dev. Biol. 2011, 55, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Ju, D.T.; Chang, C.F.; Muralidhar Reddy, P.; Velmurugan, B.K. A review on the effects of current chemotherapy drugs and natural agents in treating non-small cell lung cancer. Biomedicine 2017, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Shigdar, S.; Li, Y.; Bhattacharya, S.; O’Connor, M.; Pu, C.; Lin, J.; Wang, T.; Xiang, D.; Kong, L.; Wei, M.Q.; et al. Inflammation and cancer stem cells. Cancer Lett. 2014, 345, 271–278. [Google Scholar] [CrossRef]
- Scian, M.J.; Stagliano, K.E.R.; Anderson, M.A.E.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-kappa B2 gene expression. Mol. Cell. Biol. 2005, 25, 10097–10110. [Google Scholar] [CrossRef] [Green Version]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Wang, Z.; Fu, J.; Ji, L.; Liu, J.; Li, L.; Wang, H.; Chen, J.; Caulin, C.; Myers, J.N.; et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat. Commun. 2013, 4, 2667. [Google Scholar] [CrossRef] [Green Version]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.K.; Yadav, V.K.; Bajaj, S.; Datta, A.; Dutta, S.K.; Bhattacharyya, M.; Bhattacharya, S.; Debnath, S.; Roy, S.; Boardman, L.A.; et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016, 23, 707–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, 1086–1093. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nistico, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zache, N.; Lambert, J.M.; Rokaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Wassman, C.D.; Baronio, R.; Demir, O.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.L.; Liang, Y.; Tang, Y.G.; Song, H.X.; Wu, L.L.; Xing, Y.F.; Yan, N.; Li, Y.T.; Wang, Z.Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e228. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, A.T.Q.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell. Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiue, E.H.C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef] [PubMed]
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, M.Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arner, E.S.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, T.; Sebert, M.; Rahme, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myelodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [Green Version]
- King, F.W.; Wawrzynow, A.; Hohfeld, J.; Zylicz, M. Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 2001, 20, 6297–6305. [Google Scholar] [CrossRef] [Green Version]
- Hiraki, M.; Hwang, S.Y.; Cao, S.G.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Hwang, L.A.; Phang, B.H.; Liew, O.W.; Iqbal, J.; Koh, X.H.; Koh, X.Y.; Othman, R.; Xue, Y.Z.; Richards, A.M.; Lane, D.P.; et al. Monoclonal Antibodies against Specific p53 Hotspot Mutants as Potential Tools for Precision Medicine. Cell Rep. 2018, 22, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torikai, H.; Akatsuka, Y.; Miyauchi, H.; Terakura, S.; Onizuka, M.; Tsujimura, K.; Miyamura, K.; Morishima, Y.; Kodera, Y.; Kuzushima, K.; et al. The HLA-A*0201-restricted minor histocompatibility antigen HA-1H peptide can also be presented by another HLA-A2 subtype, A*0206. Bone Marrow Transpl. 2007, 40, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Yossef, R.; Cafri, G.; Paria, B.C.; Lowery, F.J.; Jafferji, M.; Good, M.L.; Sachs, A.; Copeland, A.; Kim, S.P.; et al. Antigen Experienced T Cells from Peripheral Blood Recognize p53 Neoantigens. Clin. Cancer Res. 2020, 26, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.C.; Gallagher, D.T.; Gowthaman, R.; Pierce, B.G.; Mariuzza, R.A. Structural basis for oligoclonal T cell recognition of a shared p53 cancer neoantigen. Nat. Commun. 2020, 11, 2908. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| TCGA Database | IARC TP53 Somatic Mutations Database | ||
|---|---|---|---|
| Mutants | Percentage in TP53 Mutant Cases | Mutants | Percentage in TP53 Mutant Cases |
| Missense TP53 R175H | 3.69% | Missense R175H | 4.21% |
| Missense TP53 R248Q | 2.88% | Missense R248Q | 3.28% |
| Missense TP53 R273C | 2.80% | Missense R273H | 2.97% |
| Missense TP53 R273H | 2.25% | Missense R248W | 2.65% |
| Missense TP53 R248W | 2.19% | Missense R273C | 2.45% |
| Missense TP53 R282W | 2.05% | Missense R282W | 2.10% |
| Stop Gained TP53 R213 * | 1.78% | Missense G245S | 1.58% |
| Missense TP53 Y220C | 1.60% | Missense R249S | 1.52% |
| Stop Gained TP53 R196 * | 1.30% | Missense Y220C | 1.39% |
| Missense TP53 G245S | 1.05% | Stop Gained TP53 R213 1 * | 1.14% |
| Tumor Type | Same Function Mutations | R175h Interacting Protein | Affected Downstream Molecule or Pathway | Ref. |
|---|---|---|---|---|
| Pancreatic cancer | - | - | - | [16] |
| None (mouse embryonic fibroblast) | R248W, R273H | Mre11 | MRN/ATM | [17] |
| Tumor Type | Same Function Mutations | R175H Interacting Protein | Affected Downstream Molecule or Pathway | Ref. |
|---|---|---|---|---|
| Breast cancer | R248Q, R248W, R249S, R273H | ETS2 | MLL1, MLL2, MOZ/Global chromatin remodeling | [21] |
| Breast cancer | R248Q, R273H, R280K | NF-Y, YAP | Cyclin A, cyclin B, and CDK1 genes | [22] |
| Pancreatic cancer | - | - | hnRNPK/GAP17 isoforms/KRAS signaling pathway | [25] |
| Bladder, bone, and ovarian cancer | V157F, 248Q | - | GEF-H1 | [19] |
| Colorectal and pancreatic cancer | - | - | GRO1 | [20] |
| Breast, colorectal, and lung cancer | R273H | - | CDC7, Dbf4 | [23] |
| Breast and prostate cancer | R280K | DAB2IP | PI3K/AKT1 | [24] |
| Tumor Type | Same Function Mutations | R175H Interacting Protein | Affected Downstream Molecule or Pathway | Ref. |
|---|---|---|---|---|
| Breast cancer | - | p63, Smad2 | p63 target genes | [32] |
| Breast cancer | R273H | - | RCP/EGFR and integrin/PI3K/AKT | [34] |
| Breast cancer | R273H | - | RCP/Hsp90a secretion | [35] |
| Breast cancer | R273H, R280K | HIF-1α | miR-30d/accelerated vesicular trafficking | [38] |
| Colorectal cancer | R248W, R273H | SENP1 | Activation of Rac1 by SUMOylation | [41] |
| Endometrial cancer | R273H, C135Y | - | miR-130b/ZEB1/Snail/E-cad | [36] |
| Pancreatic cancer | - | p73 | NF-YA, NF-YB, and NF-YC complex/PDGFRb | [28] |
| Pancreatic cancer | - | - | Dnmt1/MiR-142-3p | [37] |
| Prostate cancer | - | - | BMI-1/Twist1 | [39] |
| Breast, lung, and skin cancer | R273H, D281G | - | CXCL5, CXCL8, and CXCL12 | [40] |
| Tumor Type | Same Function Mutations | R175H Interacting Protein | Affected Downstream Molecule or Pathway | Ref. |
|---|---|---|---|---|
| Colorectal cancer | R248W, R273H | - | Stem cell markers CD44, LGR5, and ALDH1 | [47] |
| Colorectal cancer | R273H | TCF4 | β-catenin/WNT signaling pathway | [51] |
| Leukemia | R248W, R273H | EZH2 | Global increase of H3K27me3 | [49] |
| Brain and breast cancer | R273H | - | RCP/EGFR and integrin/PI3K/AKT/WIP/YAP, TAZ | [46] |
| Gain-of-Function | Tumor Type | Same Function Mutations | R175H Interacting Protein | Affected Downstream Molecule or Pathway | Ref. |
|---|---|---|---|---|---|
| Chemoresistance | Colorectal cancer | R273H, R248W | NF-Y, p300 | Ephrin-B2/SRC/FAK/ MEKK/MEK/JNK/c-JUN/ABCG | [63] |
| Lung cancer | R273H, D281G | - | NF-κB2 | [59] | |
| Lung cancer | - | - | miR128-2/E2F5/p21/pro-caspase-3 | [60] | |
| Breast, colorectal, head and neck, and lung cancer | R248W, R273H, R280K, R282W | SMDA3 | REGγ/proteasome activation/p53, p21, and p16 degradation | [61] | |
| Breast and lung cancer | G245A, D281G, R273H, R280K | - | SLC25A1 | [62] | |
| Angiogenesis | Breast cancer | R273H, R280K | E2F1 | ID4/IL8, GRO-α | [64] |
| Inflammatory response | Colorectal cancer | R273H | - | NF-κB expression in response to TNFα | [66] |
| Breast, colorectal, and liver cancer | R273H, R280K | MAFF | sIL-1Ra/IL-1β | [67] | |
| Metabolic reprogramming | Breast and lung cancer | R248Q, R273H | - | RhoA, ROCK/GLUT1 translocation/Warburg effect | [68] |
| Head and neck cancer | P151S, G245C, R282W | AMPK | AMPK inactivation/Warburg effect | [69] |
| Concept | Strategy | Drug | Other Targeted p53 Mutants | Clinical Trial | Phase | Status | Ref. |
|---|---|---|---|---|---|---|---|
| Reactivating wild-type p53 function | targeting the cysteine residues of p53 | PRIMA-1 | R273H | - | - | - | [70] |
| APR-246 | R273H | NCT03931291 | 2 | Active, not recruiting | [71] | ||
| NCT04214860 | 1 | Recruiting | |||||
| NCT04383938 | 1, 2 | Recruiting | |||||
| NCT03745716 | 3 | Active, not recruiting | |||||
| NCT04419389 | 1, 2 | Recruiting | |||||
| NCT03588078 | 1, 2 | Active, not recruiting | |||||
| NCT03072043 | 1, 2 | Active, not recruiting | |||||
| MIRA-1 | R273H | - | - | - | [72] | ||
| STIMA-1 | R273H | - | - | - | [73] | ||
| Stictic acid | - | - | - | - | [74] | ||
| KSS-9 | - | - | - | - | [75] | ||
| Arsenic trioxide | Structural p53 mutants | NCT04695223 | 2 | Recruiting | [76] | ||
| NCT03855371 | 1 | Recruiting | |||||
| NCT03377725 | 3 | unknown | |||||
| NCT04869475 | 2 | Recruiting | |||||
| Other strategies | ZMC1 | - | - | - | - | [77] | |
| ReACp53 | R248Q | - | - | - | [78] | ||
| Promoting mutant p53 degradation | Targeting the mevalonate pathway | Atorvastatin | Structural p53 mutants | NCT03560882 | 1 | Recruiting | [79,80] |
| NCT04767984 | 2 | Not yet recruiting | |||||
| NCT03358017 | 2 | Recruiting | |||||
| HDAC inhibitor | SAHA | Broad effect | NCT02042989 | 1 | Active, not recruiting | [81] | |
| Hsp90 inhibitor | Ganetespib | Broad effect | - | - | - | [82] | |
| Immuno- therapy | Bispecific antibody | H2-scDb | - | - | - | - | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, Y.-T.; Chien, Y.-C.; Lin, Y.-H.; Wu, H.-H.; Lee, D.-F.; Yu, Y.-L. The Function of the Mutant p53-R175H in Cancer. Cancers 2021, 13, 4088. https://doi.org/10.3390/cancers13164088
Chiang Y-T, Chien Y-C, Lin Y-H, Wu H-H, Lee D-F, Yu Y-L. The Function of the Mutant p53-R175H in Cancer. Cancers. 2021; 13(16):4088. https://doi.org/10.3390/cancers13164088
Chicago/Turabian StyleChiang, Yen-Ting, Yi-Chung Chien, Yu-Heng Lin, Hui-Hsuan Wu, Dung-Fang Lee, and Yung-Luen Yu. 2021. "The Function of the Mutant p53-R175H in Cancer" Cancers 13, no. 16: 4088. https://doi.org/10.3390/cancers13164088