
Molecular Mechanisms Associated with Brain Metastases in HER2-Positive and Triple Negative Breast Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Stages of Brain Metastasis Development: Key Factors Associated with HER2-Positive and Triple Negative BC Subtypes
2.1. Tissue Invasion, Intravasation, and Circulation
2.2. Extravasation—Overcoming the Blood-Brain Barrier
2.3. Intracerebral Metastatic Colonisation
2.4. DNA Repair Mechanisms in BCBM—Survival in the Brain Microenvironment
3. Potential Additional Molecular Alterations Associated with Brain Metastasis Development
3.1. TP53
3.2. PI3K/Akt/mTOR-Pathway
3.3. PTEN
3.4. Low Methylation Levels in TNBC
3.5. Metabolic Phenotype
4. Clinical Implications and Perspectives: How Is Our Current Understanding of BC Subtype and Its Influence on BCBM Formation Being Used to Combat BC Brain Metastasis?
4.1. XIST as a Therapy Target
4.2. Anti-HER2 Treatment in BCBM Patients
4.3. The PI3K/Akt Pathway Is Uniquely Active in BCBM
4.4. VEGF Antibodies in Combination with Anti-HER2 Therapy
4.5. Oestrogen Depletion as Potential BM Prevention in TNBC
5. Limitations and Unanswered Questions
5.1. Genetic Alterations during the Metastatic Process and/or Therapy
5.2. Difficulties in BCBM Therapy Development
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, M.B.; He, X.; Hoadley, K.A.; Hoyle, A.; Pearce, J.B.; Garrett, A.L.; Kumar, S.; Moylan, V.J.; Brady, C.M.; Van Swearingen, A.E.; et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J. Clin. Investig. 2018, 128, 1371–1383. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Eliyatkin, N.; Yalcin, E.; Zengel, B.; Aktas, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thurlimann, B.; Senn, H.J.; Panel, M. Strategies for subtypes—dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, J.S.; Kim, I.A. Molecular subtype predicts incidence and prognosis of brain metastasis from breast cancer in SEER database. J. Cancer Res. Clin. Oncol. 2018, 144, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Darlix, A.; Louvel, G.; Fraisse, J.; Jacot, W.; Brain, E.; Debled, M.; Mouret-Reynier, M.A.; Goncalves, A.; Dalenc, F.; Delaloge, S.; et al. Impact of breast cancer molecular subtypes on the incidence, kinetics and prognosis of central nervous system metastases in a large multicentre real-life cohort. Br. J. Cancer 2019, 121, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Arvold, N.D.; Oh, K.S.; Niemierko, A.; Taghian, A.G.; Lin, N.U.; Abi-Raad, R.F.; Sreedhara, M.; Harris, J.R.; Alexander, B.M. Brain metastases after breast-conserving therapy and systemic therapy: Incidence and characteristics by biologic subtype. Breast Cancer Res. Treat. 2012, 136, 153–160. [Google Scholar] [CrossRef]
- Rostami, R.; Mittal, S.; Rostami, P.; Tavassoli, F.; Jabbari, B. Brain metastasis in breast cancer: A comprehensive literature review. J. NeuroOncol. 2016, 127, 407–414. [Google Scholar] [CrossRef]
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S.; et al. Brain metastases. Nat. Rev. Dis. Primers 2019, 5, 5. [Google Scholar] [CrossRef]
- Palmieri, D.; Bronder, J.L.; Herring, J.M.; Yoneda, T.; Weil, R.J.; Stark, A.M.; Kurek, R.; Vega-Valle, E.; Feigenbaum, L.; Halverson, D.; et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007, 67, 4190–4198. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast cancer subtypes predispose the site of distant metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Sahin, A.A.; Hess, K.R.; Suki, D.; Aldape, K.D.; Sawaya, R.; Ibrahim, N.K. Breast cancer with brain metastases: Clinicopathologic features, survival, and paired biomarker analysis. Oncologist 2015, 20, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, K.; Lee, E.; Ahn, T.; Jung, H.H.; Lim, S.H.; Hong, M.; Do, I.G.; Cho, E.Y.; Kim, D.H.; et al. Gene Expression Profiling of Breast Cancer Brain Metastasis. Sci. Rep. 2016, 6, 28623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, J.C.; Prat, A.; Parker, J.S.; Fan, C.; He, X.; Carey, L.; Anders, C.; Ewend, M.; Perou, C.M. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res. Treat. 2012, 132, 523–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.; Lin, N.U. Updates on the management of breast cancer brain metastases. Oncology 2014, 28, 572–578. [Google Scholar]
- Martin, A.M.; Cagney, D.N.; Catalano, P.J.; Warren, L.E.; Bellon, J.R.; Punglia, R.S.; Claus, E.B.; Lee, E.Q.; Wen, P.Y.; Haas-Kogan, D.A.; et al. Brain Metastases in Newly Diagnosed Breast Cancer: A Population-Based Study. JAMA Oncol. 2017, 3, 1069–1077. [Google Scholar] [CrossRef]
- Sperduto, P.W.; Kased, N.; Roberge, D.; Chao, S.T.; Shanley, R.; Luo, X.; Sneed, P.K.; Suh, J.; Weil, R.J.; Jensen, A.W.; et al. The effect of tumor subtype on the time from primary diagnosis to development of brain metastases and survival in patients with breast cancer. J. NeuroOncol. 2013, 112, 467–472. [Google Scholar] [CrossRef]
- Qiu, J.; Xue, X.; Hu, C.; Xu, H.; Kou, D.; Li, R.; Li, M. Comparison of Clinicopathological Features and Prognosis in Triple-Negative and Non-Triple Negative Breast Cancer. J. Cancer 2016, 7, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Liu, Y.; Wu, S.Y.; Wu, K.; Sharma, S.; Mo, Y.Y.; Feng, J.; Sanders, S.; Jin, G.; Singh, R.; et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018, 78, 4316–4330. [Google Scholar] [CrossRef] [Green Version]
- Bos, P.D.; Zhang, X.H.F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Lo, H.W. Landscape of EGFR signaling network in human cancers: Biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012, 318, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Miller, L.; Metheny-Barlow, L.; Lo, H.W. EGFR and HER2 signaling in breast cancer brain metastasis. Front. BioSci. (Elite Ed.) 2016, 8, 245–263. [Google Scholar] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999. [Google Scholar] [CrossRef] [Green Version]
- Press, M.F.; Sauter, G.; Bernstein, L.; Villalobos, I.E.; Mirlacher, M.; Zhou, J.Y.; Wardeh, R.; Li, Y.T.; Guzman, R.; Ma, Y.; et al. Diagnostic evaluation of HER-2 as a molecular target: An assessment of accuracy and reproducibility of laboratory testing in large, prospective, randomized clinical trials. Clin. Cancer Res. 2005, 11, 6598–6607. [Google Scholar] [CrossRef] [Green Version]
- Hohensee, I.; Lamszus, K.; Riethdorf, S.; Meyer-Staeckling, S.; Glatzel, M.; Matschke, J.; Witzel, I.; Westphal, M.; Brandt, B.; Muller, V.; et al. Frequent Genetic Alterations in EGFR- and HER2-Driven Pathways in Breast Cancer Brain Metastases. Am. J. Pathol. 2013, 183, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Song, F.; Jin, T.; Qin, J.; Wu, J.; Wang, M.; Wang, Y.; Liu, J. Prognostic values of Notch receptors in breast cancer. Tumour Biol. 2016, 37, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.M.; Simedrea, C.; Ribot, E.J.; Foster, P.J.; Palmieri, D.; Steeg, P.S.; Allan, A.L.; Chambers, A.F. Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol. Cancer Res. 2011, 9, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Kobayashi, A.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Wilber, A.; Mo, Y.Y.; Moore, B.E.; et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol. Med. 2013, 5, 384–396. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouras, T.; Pal, B.; Vaillant, F.; Harburg, G.; Asselin-Labat, M.L.; Oakes, S.R.; Lindeman, G.J.; Visvader, J.E. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 2008, 3, 429–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolos, V.; Blanco, M.; Medina, V.; Aparicio, G.; Diaz-Prado, S.; Grande, E. Notch signalling in cancer stem cells. Clin. Transl. Oncol. 2009, 11, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Farnie, G.; Clarke, R.B. Mammary stem cells and breast cancer—Role of Notch signalling. Stem Cell Rev. 2007, 3, 169–175. [Google Scholar] [CrossRef]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W.; et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, M.; Wu, H.; Xu, H.; Han, N.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Expression of Notch1 Correlates with Breast Cancer Progression and Prognosis. PLoS ONE 2015, 10, e0131689. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Yagi, H.; Yotsumoto, F.; Kawarabayashi, T.; Mekada, E. Heparin-binding epidermal growth factor-like growth factor as a new target molecule for cancer therapy. Adv. Exp. Med. Biol. 2008, 622, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, C.A.; Hanna, C.T.; Gril, B.; Cruz, H.; Serkova, N.J.; Huber, K.M.; Kabos, P.; Schedin, T.B.; Borges, V.F.; Steeg, P.S.; et al. Estrogen promotes the brain metastatic colonization of triple negative breast cancer cells via an astrocyte-mediated paracrine mechanism. Oncogene 2016, 35, 2881–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollig-Fischer, A.; Michelhaugh, S.K.; Wijesinghe, P.; Dyson, G.; Kruger, A.; Palanisamy, N.; Choi, L.; Alosh, B.; Ali-Fehmi, R.; Mittal, S. Cytogenomic profiling of breast cancer brain metastases reveals potential for repurposing targeted therapeutics. Oncotarget 2015, 6, 14614–14624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Zheng, S.; Xie, X.; Li, X.; Zhang, L.; Yang, A.; Wang, J.; Tang, H.; Xie, X. SOX2 Promotes Brain Metastasis of Breast Cancer by Upregulating the Expression of FSCN1 and HBEGF. Mol. Ther. Oncolytics 2020, 17, 118–129. [Google Scholar] [CrossRef]
- Riebensahm, C.; Joosse, S.A.; Mohme, M.; Hanssen, A.; Matschke, J.; Goy, Y.; Witzel, I.; Lamszus, K.; Kropidlowski, J.; Petersen, C.; et al. Clonality of circulating tumor cells in breast cancer brain metastasis patients. Breast Cancer Res. 2019, 21, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaedcke, J.; Traub, F.; Milde, S.; Wilkens, L.; Stan, A.; Ostertag, H.; Christgen, M.; von Wasielewski, R.; Kreipe, H.H. Predominance of the basal type and HER-2/neu type in brain metastasis from breast cancer. Mod. Pathol. 2007, 20, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, R.; Mustafa, D.A.; Soffietti, R.; Kros, J.M. Breast cancer brain metastasis: Molecular mechanisms and directions for treatment. Neuro Oncol. 2018, 20, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Cai, B.; Zeng, M.; Hao, Y.; Giancotti, F.G.; Fu, B.M. Integrin beta4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann. Biomed. Eng. 2011, 39, 2223–2241. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G. Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol. Sci. 2007, 28, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-L.; Chu, P.-Y.; Lai, I.R.; Wang, M.-Y.; Tseng, H.-Y.; Guan, J.-L.; Liou, J.-Y.; Shen, T.-L. An EGFR/Src-dependent β4 integrin/FAK complex contributes to malignancy of breast cancer. Sci. Rep. 2015, 5, 16408. [Google Scholar] [CrossRef] [Green Version]
- Thind, A.; Wilson, C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J. Extracell Vesicles 2016, 5, 31292. [Google Scholar] [CrossRef]
- Rodriguez-Pinilla, S.M.; Sarrio, D.; Moreno-Bueno, G.; Rodriguez-Gil, Y.; Martinez, M.A.; Hernandez, L.; Hardisson, D.; Reis-Filho, J.S.; Palacios, J. Sox2: A possible driver of the basal-like phenotype in sporadic breast cancer. Mod. Pathol. 2007, 20, 474. [Google Scholar] [CrossRef]
- Ren, D.; Zhu, X.; Kong, R.; Zhao, Z.; Sheng, J.; Wang, J.; Xu, X.; Liu, J.; Cui, K.; Zhang, X.H.; et al. Targeting Brain-Adaptive Cancer Stem Cells Prohibits Brain Metastatic Colonization of Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 2052–2064. [Google Scholar] [CrossRef] [Green Version]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Doheny, D.; Zhu, D.; Aguayo, N.R.; Xing, F.; Chan, M.; Ruiz, J.; Metheny-Barlow, L.J.; et al. TGLI1 transcription factor mediates breast cancer brain metastasis via activating metastasis-initiating cancer stem cells and astrocytes in the tumor microenvironment. Oncogene 2020, 39, 64–78. [Google Scholar] [CrossRef]
- Cao, X.; Geradts, J.; Dewhirst, M.W.; Lo, H.W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 2012, 31, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Paw, I.; Zhu, H.; Sirkisoon, S.; Xing, F.; Watabe, K.; Debinski, W.; Lo, H.W. The gain-of-function GLI1 transcription factor TGLI1 enhances expression of VEGF-C and TEM7 to promote glioblastoma angiogenesis. Oncotarget 2015, 6, 22653–22665. [Google Scholar] [CrossRef] [Green Version]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sullivan, P.; Suyama, J.; Marchetti, D. Epidermal growth factor-induced heparanase nucleolar localization augments DNA topoisomerase I activity in brain metastatic breast cancer. Mol. Cancer Res. 2010, 8, 278–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Sullivan, P.S.; Goodman, J.C.; Gunaratne, P.H.; Marchetti, D. MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res. 2011, 71, 645–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Zhang, Q.; Zhao, S.; Wang, J.; Lu, K.; Song, Y.; Zhao, L.; Kang, X.; Wang, J.; Xu, S.; et al. The expression and clinical significance of microRNA-1258 and heparanase in human breast cancer. Clin. Biochem. 2013, 46, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Maxhimer, J.B.; Quiros, R.M.; Stewart, R.; Dowlatshahi, K.; Gattuso, P.; Fan, M.; Prinz, R.A.; Xu, X. Heparanase-1 expression is associated with the metastatic potential of breast cancer. Surgery 2002, 132, 326–333. [Google Scholar] [CrossRef]
- Woditschka, S.; Evans, L.; Duchnowska, R.; Reed, L.T.; Palmieri, D.; Qian, Y.; Badve, S.; Sledge, G., Jr.; Gril, B.; Aladjem, M.I.; et al. DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Hao, L.; Jiang, D.; Wu, B.; He, T.; Tang, Y. The diagnostic value of DNA repair gene in breast cancer metastasis. Sci. Rep. 2020, 10, 19626. [Google Scholar] [CrossRef]
- Ferguson, S.D.; Zheng, S.; Xiu, J.; Zhou, S.; Khasraw, M.; Brastianos, P.K.; Kesari, S.; Hu, J.; Rudnick, J.; Salacz, M.E.; et al. Profiles of brain metastases: Prioritization of therapeutic targets. Int. J. Cancer 2018, 143, 3019–3026. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.J.; Giannoudis, A.; Palmieri, C. The genomic landscape of breast cancer brain metastases: A systematic review. Lancet Oncol. 2021, 22, e7–e17. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Ng, C.K.Y.; Piscuoglio, S.; Gonzalez-Cao, M.; Lim, R.S.; De Filippo, M.R.; Fusco, N.; Schultheis, A.M.; Ortiz, C.; Viteri, S.; et al. Genetic heterogeneity and actionable mutations in HER2-positive primary breast cancers and their brain metastases. Oncotarget 2018, 9, 20617–20630. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, M.; Strappazzon, F.; Arisi, I.; Brandi, R.; D’Onofrio, M.; Sambucci, M.; Manic, G.; Vitale, I.; Barila, D.; Stagni, V. ATM kinase sustains breast cancer stem-like cells by promoting ATG4C expression and autophagy. Oncotarget 2017, 8, 21692–21709. [Google Scholar] [CrossRef] [PubMed]
- Stagni, V.; Manni, I.; Oropallo, V.; Mottolese, M.; Di Benedetto, A.; Piaggio, G.; Falcioni, R.; Giaccari, D.; Di Carlo, S.; Sperati, F.; et al. ATM kinase sustains HER2 tumorigenicity in breast cancer. Nat. Commun. 2015, 6, 6886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullin, R.P.; Wittner, B.S.; Yang, C.; Denton-Schneider, B.R.; Hicks, D.; Singavarapu, R.; Moulis, S.; Lee, J.; Akbari, M.R.; Narod, S.A.; et al. A BRCA1 deficient-like signature is enriched in breast cancer brain metastases and predicts DNA damage-induced poly (ADP-ribose) polymerase inhibitor sensitivity. Breast Cancer Res. 2014, 16, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyran, M.; Carbuccia, N.; Garnier, S.; Guille, A.; Adelaide, J.; Finetti, P.; Toulzian, J.; Viens, P.; Tallet, A.; Goncalves, A.; et al. A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases. Cancers 2019, 11, 665. [Google Scholar] [CrossRef] [Green Version]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Toth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, K.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.N.; Hong, M.; Do, I.G.; Ahn, T.; et al. Mutational profiling of brain metastasis from breast cancer: Matched pair analysis of targeted sequencing between brain metastasis and primary breast cancer. Oncotarget 2015, 6, 43731–43742. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network; Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar] [CrossRef] [Green Version]
- Langerod, A.; Zhao, H.; Borgan, O.; Nesland, J.M.; Bukholm, I.R.; Ikdahl, T.; Karesen, R.; Borresen-Dale, A.L.; Jeffrey, S.S. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9, R30. [Google Scholar] [CrossRef]
- Adamo, B.; Deal, A.M.; Burrows, E.; Geradts, J.; Hamilton, E.; Blackwell, K.L.; Livasy, C.; Fritchie, K.; Prat, A.; Harrell, J.C.; et al. Phosphatidylinositol 3-kinase pathway activation in breast cancer brain metastases. Breast Cancer Res. 2011, 13, R125. [Google Scholar] [CrossRef] [Green Version]
- Dillon, R.L.; White, D.E.; Muller, W.J. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene 2007, 26, 1338–1345. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Engelman, J.A.; Irie, H.Y.; Luo, J.; Brachmann, S.M.; Pearline, R.V.; Cantley, L.C.; Brugge, J.S. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005, 65, 10992–11000. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Stemke-Hale, K.; Sahin, A.; Liu, S.; Barrera, J.A.; Burgues, O.; Lluch, A.M.; Chen, H.; Hortobagyi, G.N.; et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol. Cancer Ther. 2011, 10, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohensee, I.; Chuang, H.N.; Grottke, A.; Werner, S.; Schulte, A.; Horn, S.; Lamszus, K.; Bartkowiak, K.; Witzel, I.; Westphal, M.; et al. PTEN mediates the cross talk between breast and glial cells in brain metastases leading to rapid disease progression. Oncotarget 2017, 8, 6155–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Li, T.; Ding, Y.; Deng, Y.; Zhang, W.; Yang, H.; Zhou, J.; Liu, C.; Lin, J.; Ding, Y. Methylation status of T-lymphoma invasion and metastasis 1 promoter and its overexpression in colorectal cancer. Hum. Pathol. 2011, 42, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends NeuroSci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, S.; Wang, X. The Metabolic Mechanisms of Breast Cancer Metastasis. Front. Oncol. 2020, 10, 602416. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Kim, D.H.; Jung, W.H.; Koo, J.S. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res. 2013, 15, R78. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Chiu, L.L.; Sethi, S.K.; Koay, E.S. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol. Cell Proteom. 2005, 4, 1686–1696. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.J.; Cook, R.S.; Manning, H.C.; Hicks, D.J.; Lafontant, A.; Arteaga, C.L.; Skala, M.C. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013, 73, 6164–6174. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl Acad Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, A.S.; Almeida, C.R.; Helguero, L.A.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; McKinney, A.M.; Brace, J.R.; Santacruz, K. Clinical and imaging features of fludarabine neurotoxicity. J. Neuroophthalmol. 2010, 30, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Broglio, K.; Buzdar, A.U.; Hortobagyi, G.N.; Giordano, S.H. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: An institutional-based review. J. Clin. Oncol. 2010, 28, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchnowska, R.; Loibl, S.; Jassem, J. Tyrosine kinase inhibitors for brain metastases in HER2-positive breast cancer. Cancer Treat. Rev. 2018, 67, 71–77. [Google Scholar] [CrossRef]
- Bonneau, C.; Paintaud, G.; Tredan, O.; Dubot, C.; Desvignes, C.; Dieras, V.; Taillibert, S.; Tresca, P.; Turbiez, I.; Li, J.; et al. Phase I feasibility study for intrathecal administration of trastuzumab in patients with HER2 positive breast carcinomatous meningitis. Eur. J. Cancer 2018, 95, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, W.T.; Bell, P.; Richman, L.K.; Limberis, M.P.; Tretiakova, A.P.; Li, M.; Wilson, J.M. Intrathecal Viral Vector Delivery of Trastuzumab Prevents or Inhibits Tumor Growth of Human HER2-Positive Xenografts in Mice. Cancer Res. 2018, 78, 6171–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, C.C.; Davarpanah, N.N.; Abraham, J.; Bates, S.E. Current challenges in the management of breast cancer brain metastases. Semin. Oncol. 2017, 44, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Miles, D.; Kim, S.B.; Im, Y.H.; Im, S.A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Figura, N.B.; Rizk, V.T.; Mohammadi, H.; Evernden, B.; Mokhtari, S.; Yu, H.M.; Robinson, T.J.; Etame, A.B.; Tran, N.D.; Liu, J.; et al. Clinical outcomes of breast leptomeningeal disease treated with intrathecal trastuzumab, intrathecal chemotherapy, or whole brain radiation therapy. Breast Cancer Res. Treat. 2019, 175, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Borges, V.; Anders, C.; Murthy, R.K.; Paplomata, E.; Hamilton, E.; Hurvitz, S.; Loi, S.; Okines, A.; Abramson, V.; et al. Intracranial Efficacy and Survival With Tucatinib Plus Trastuzumab and Capecitabine for Previously Treated HER2-Positive Breast Cancer With Brain Metastases in the HER2CLIMB Trial. J. Clin. Oncol. 2020, 38, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.; Singh, R.; Adams, B.; Taguchi, J.A.; Chan, D.; Dichmann, R.A.; Castrellon, A.; Hu, E.; Berkowitz, J.; Mani, A.; et al. Phase Ib/II single-arm trial evaluating the combination of everolimus, lapatinib and capecitabine for the treatment of HER2-positive breast cancer with brain metastases (TRIO-US B-09). Ther. Adv. Med. Oncol. 2018, 10, 1758835918807339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodack, D.P.; Askoxylakis, V.; Ferraro, G.B.; Sheng, Q.; Badeaux, M.; Goel, S.; Qi, X.; Shankaraiah, R.; Cao, Z.A.; Ramjiawan, R.R.; et al. The brain microenvironment mediates resistance in luminal breast cancer to PI3K inhibition through HER3 activation. Sci. Transl. Med. 2017, 9, eaal4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimandi, M.; Romano, A.; Curia, M.C.; Muraro, R.; Fedi, P.; Aaronson, S.A.; Di Fiore, P.P.; Kraus, M.H. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 1995, 10, 1813–1821. [Google Scholar]
- Kodack, D.P.; Chung, E.; Yamashita, H.; Incio, J.; Duyverman, A.M.; Song, Y.; Farrar, C.T.; Huang, Y.; Ager, E.; Kamoun, W.; et al. Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc. Natl Acad Sci. USA 2012, 109, E3119–E3127. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Moulder, S.; Naing, A.; Wheler, J.J.; Hong, D.S.; Piha-Paul, S.A.; Tsimberidou, A.M.; Fu, S.; Zinner, R.; Janku, F.; et al. A phase I trial of combination trastuzumab, lapatinib, and bevacizumab in patients with advanced cancer. Investig. New Drugs 2015, 33, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsbergen, A.F.C.; Claes, A.; Kavouridis, V.K.; Ansaripour, A.; Nogarede, C.; Hughes, M.E.; Smith, T.R.; Brastianos, P.K.; Verhoeff, J.J.C.; Lin, N.U.; et al. Subtype switching in breast cancer brain metastases: A multicenter analysis. Neuro Oncol. 2020, 22, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Cejalvo, J.M.; Martinez de Duenas, E.; Galvan, P.; Garcia-Recio, S.; Burgues Gasion, O.; Pare, L.; Antolin, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic Subtypes and Gene Expression Profiles in Primary and Metastatic Breast Cancer. Cancer Res. 2017, 77, 2213–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperduto, P.W.; Mesko, S.; Li, J.; Cagney, D.; Aizer, A.; Lin, N.U.; Nesbit, E.; Kruser, T.J.; Chan, J.; Braunstein, S.; et al. Estrogen/progesterone receptor and HER2 discordance between primary tumor and brain metastases in breast cancer and its effect on treatment and survival. Neuro Oncol. 2020, 22, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Schwarzenbach, H.; Pantel, K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Weigelt, B.; Hu, Z.; He, X.; Livasy, C.; Carey, L.A.; Ewend, M.G.; Glas, A.M.; Perou, C.M.; Van’t Veer, L.J. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005, 65, 9155–9158. [Google Scholar] [CrossRef] [Green Version]
- Timmer, M.; Werner, J.M.; Rohn, G.; Ortmann, M.; Blau, T.; Cramer, C.; Stavrinou, P.; Krischek, B.; Mallman, P.; Goldbrunner, R. Discordance and Conversion Rates of Progesterone-, Estrogen-, and HER2/neu-Receptor Status in Primary Breast Cancer and Brain Metastasis Mainly Triggered by Hormone Therapy. Anticancer Res. 2017, 37, 4859–4865. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D. The blood-brain barrier: Its influence in the treatment of brain tumors metastases. Curr. Cancer Drug Targets 2012, 12, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Kodack, D.P.; Askoxylakis, V.; Ferraro, G.B.; Fukumura, D.; Jain, R.K. Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 2015, 27, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
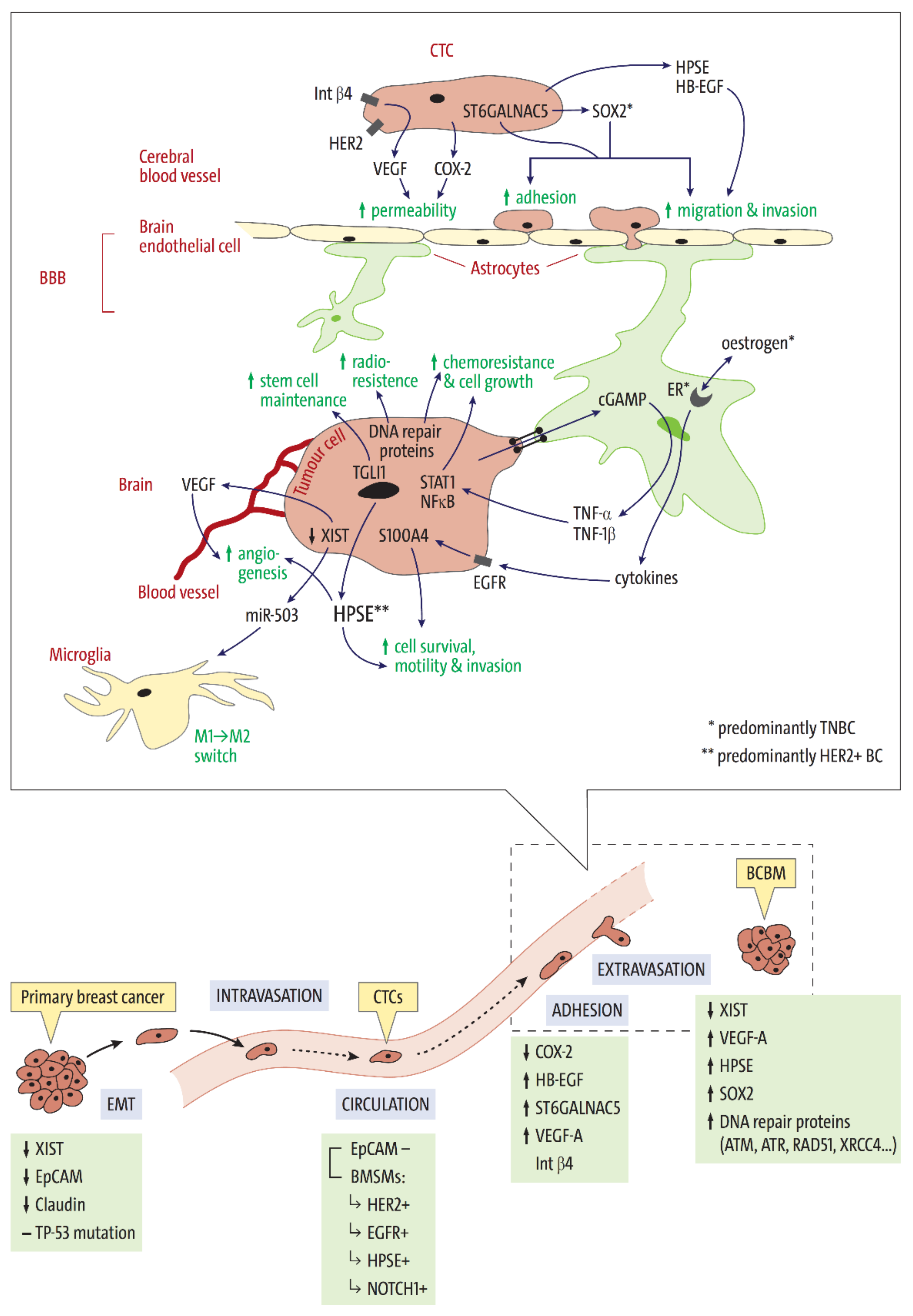

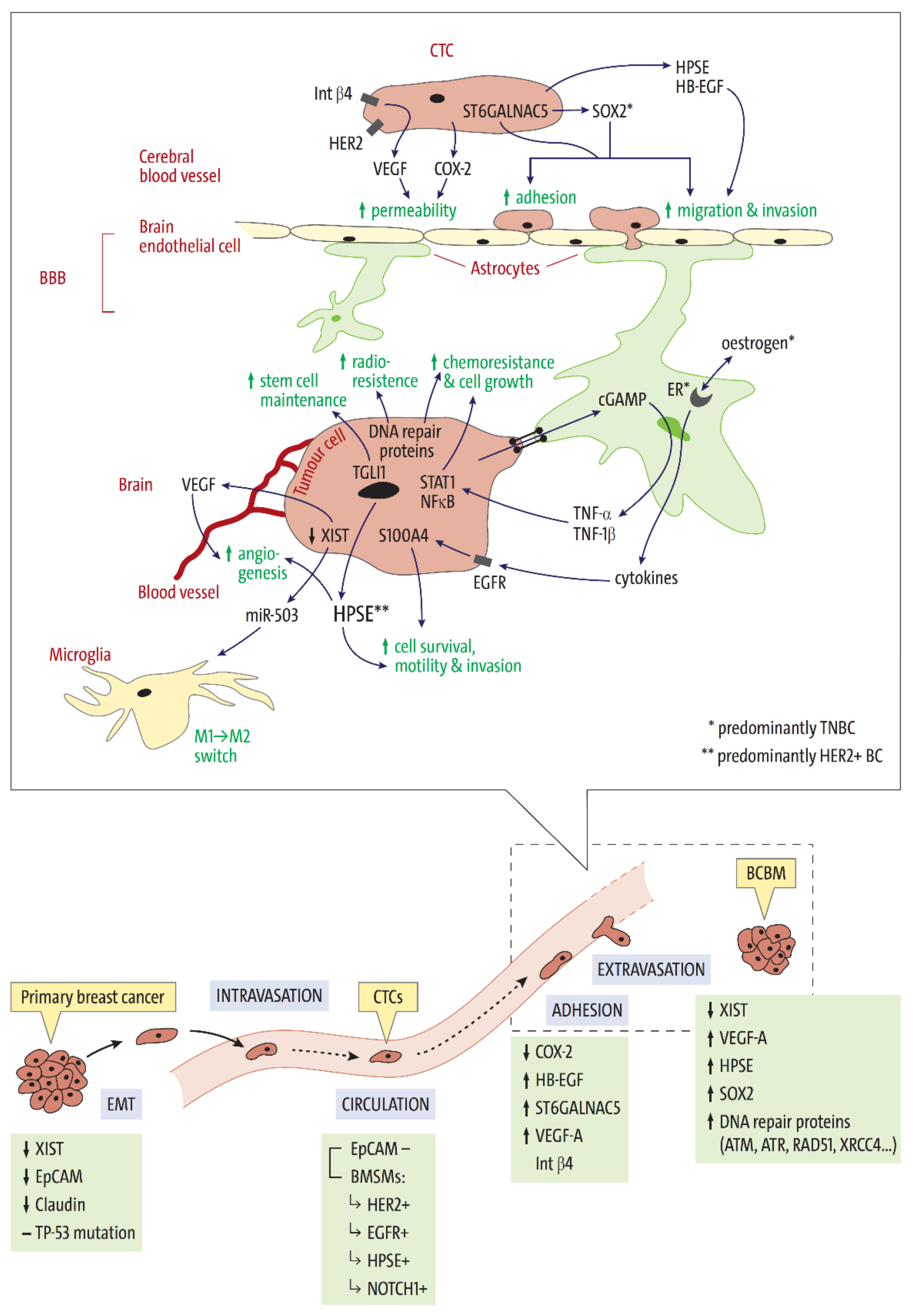{kind=link}
| Subtype | Hormone Receptor | Growth Factor Receptor | Proliferation Marker | % of Invasive BC | 5-Year-Risk (%) of Brain Metastases after BC Diagnosis | Literature | |
|---|---|---|---|---|---|---|---|
| ER | PR | HER2 | Ki-67 | ||||
| Luminal A | (+) | (+) | − | low | 50% | 0.1% | [9,10,11] |
| Luminal B | (+) | (+) | − | high | 20% | ~3.3% | |
| HER2-positive | ± | ± | + | high | 15% | 3.2%/3.7% * | |
| TNBC | − | − | − | high | ~15% | 7.4% | |
| Genetic Marker | Primary BC Subtype | Tissue Type Other than Primary BC | Mechanism | Associated with | Literature | |
|---|---|---|---|---|---|---|
| Increased | Reduced | |||||
| EMT and Circulation (CTCs) | ||||||
| XISTlow | TNBC (48%), HER2+ (28%), Lum. A (19%) | BCBM | ↓ XIST → ↑ c-Met → STAT3 and PI3K pathway activation | tumour cell proliferation, motility, migration, and invasion | [27] | |
| EGFR | 25–30% of BC, most often TNBC | EpCAM negative CTCs, BCBM | STAT3, Ras-MAPK and PI3K pathway activation | tumour cell proliferation, invasiveness, brain metastases | apoptosis, patient survival | [14,17,28,29,30,31,32] |
| HER2 | 15–35% of BC | |||||
| NOTCH1 | TNBC | Gene mutation → ↑ Notch1 receptor → ↑ Notch signaling | primary BC tumorigenesis, stem cell and CTC maintenance | overall survival | [33,34,35,36,37,38,39,40] | |
| Extravasation | ||||||
| COX2 | ER-negative tumour cells | BCBM-cell lines | BBB permeability | [41] | ||
| HBEGF | ↑ EGFR-ligand HB-EGF | tumour cell motility and invasiveness | [41,42,43] | |||
| ST6GALNAC5 | BC-cell adhesion to brain endothelial cells | [41,43] | ||||
| ITGB4 | HER2+ | β4 integrin interaction with HER2, VEGF production | adhesion and extravasation of BC cells through BBB, vascular growth in BCBM microenvironment | endothelial tight and adherence junctions, VEGF-dependent endothelial integrity | [44] | |
| Cerebral Colonization | ||||||
| XISTlow | BLBC (48%), HER2+ (28%), Lum. A (19%) | BCBM | ↓ XIST → ↑ miR-503 → ↑ STAT3 and NF-κB signaling → microglia phenotype switch M1 to M2 | microglial defence against invading tumour cells | [27] | |
| S100A4 | TNBC cells | ER-expressing astrocytes following oestrogen stimulation (TNBC model) | Oestrogen stimulation → cytokines → ↑ EGFR-signaling → ↑ S100A4 | cell survival, motility and invasion | [45] | |
| TGLI1 | HER2+ and TNBC (low exp.) | BCBM (high exp.), CSCs, radioresistant BCBM cell lines | transcription factor → activation of several genes incl. SOX2 | CSC renewal, astrocyte activation in BCBM microenvironment | BM-free survival | [46] |
| HPSE | HER2+ | CTCs (all subtypes), BCBM, brain endothelial and glial cells | EGFR/HER2 signaling → HPSE | tumorigenesis, angiogenesis, and metastasis | [28,42,47,48,49,50] | |
| SOX2 | TNBC BLBC | BCBM | transcription factor | stem-cell maintenance in the CNS, tumour cell plasticity and endothelial cell adhesion, trans-endothelial migration, and migration across the BBB | BM-free survival | [18,48,49,50,51,52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryan, S.; Witzel, I.; Borgmann, K.; Oliveira-Ferrer, L. Molecular Mechanisms Associated with Brain Metastases in HER2-Positive and Triple Negative Breast Cancers. Cancers 2021, 13, 4137. https://doi.org/10.3390/cancers13164137
Bryan S, Witzel I, Borgmann K, Oliveira-Ferrer L. Molecular Mechanisms Associated with Brain Metastases in HER2-Positive and Triple Negative Breast Cancers. Cancers. 2021; 13(16):4137. https://doi.org/10.3390/cancers13164137
Chicago/Turabian StyleBryan, Sarah, Isabell Witzel, Kerstin Borgmann, and Leticia Oliveira-Ferrer. 2021. "Molecular Mechanisms Associated with Brain Metastases in HER2-Positive and Triple Negative Breast Cancers" Cancers 13, no. 16: 4137. https://doi.org/10.3390/cancers13164137
APA StyleBryan, S., Witzel, I., Borgmann, K., & Oliveira-Ferrer, L. (2021). Molecular Mechanisms Associated with Brain Metastases in HER2-Positive and Triple Negative Breast Cancers. Cancers, 13(16), 4137. https://doi.org/10.3390/cancers13164137