
Exploring the Epigenome in Gastroenteropancreatic Neuroendocrine Neoplasias


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epigenetic Modifications in Cancer
2.1. DNA Methylation
2.2. Histone Modifications
2.3. MicroRNAs
3. Pancreatic Neuroendocrine Tumours
3.1. Promoter Hypermethylation in pNETs
3.2. Methylation of Other Loci in pNET
3.3. Histone and Chromatin Modification
3.4. MicroRNA (miRNA)
4. Gasterointestinal NETs
4.1. Promoter Methylation
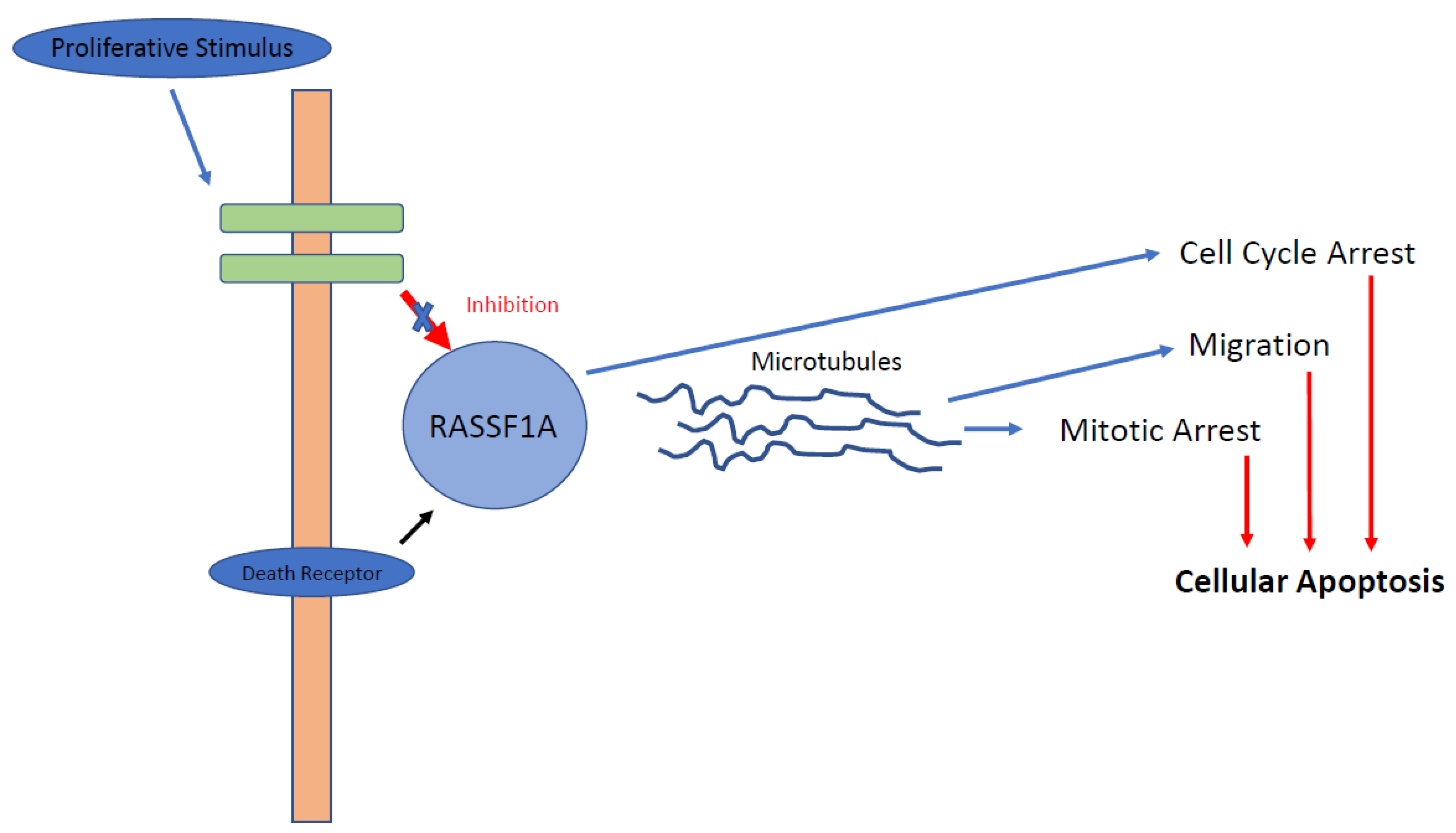
4.1.1. RASSF1A
4.1.2. Global Methylation Patterns
4.2. Histone and Chromatin Modification
4.3. MicroRNA
5. Targeting Epigenetic Changes for Therapeutic Benefit
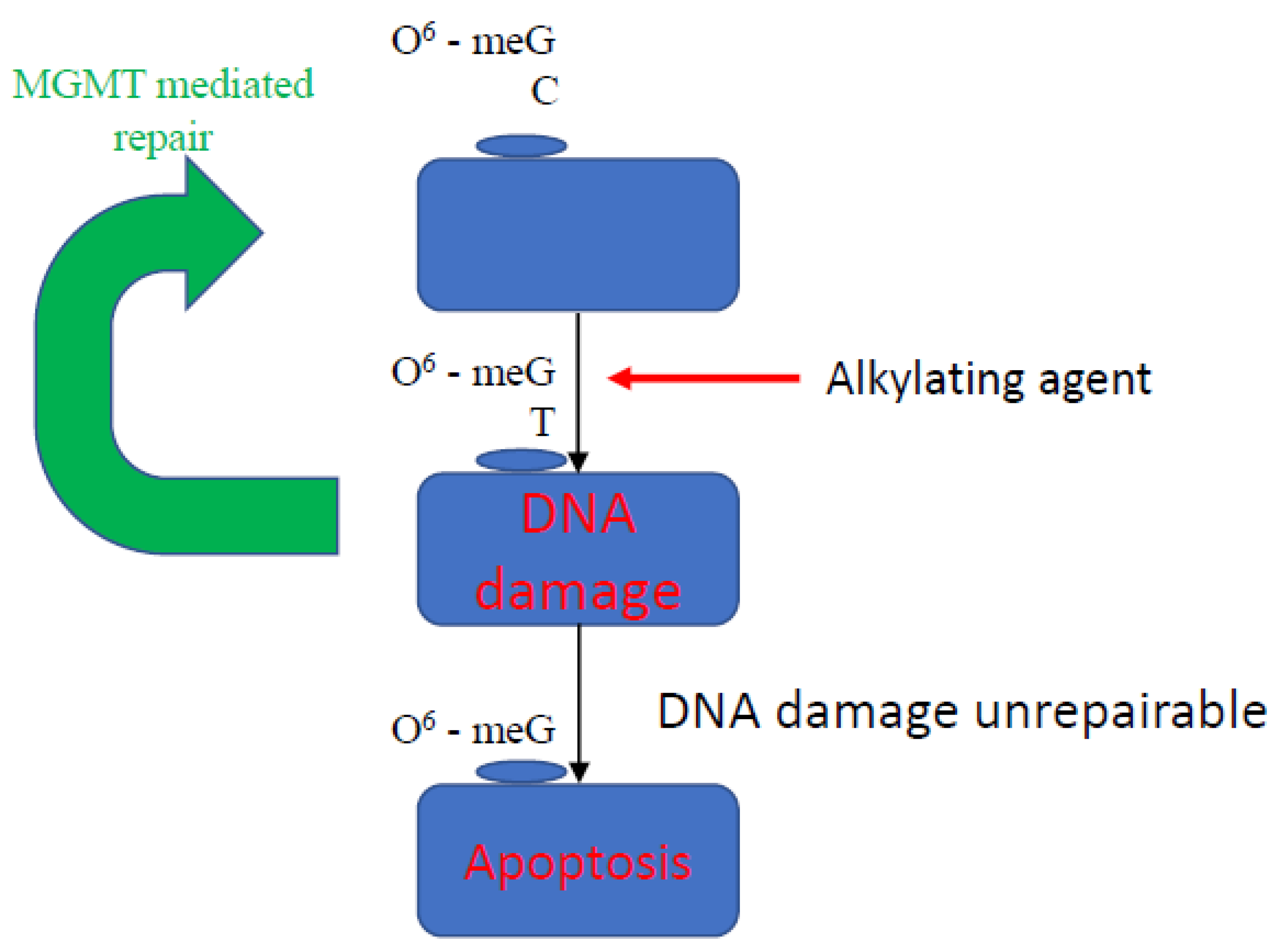
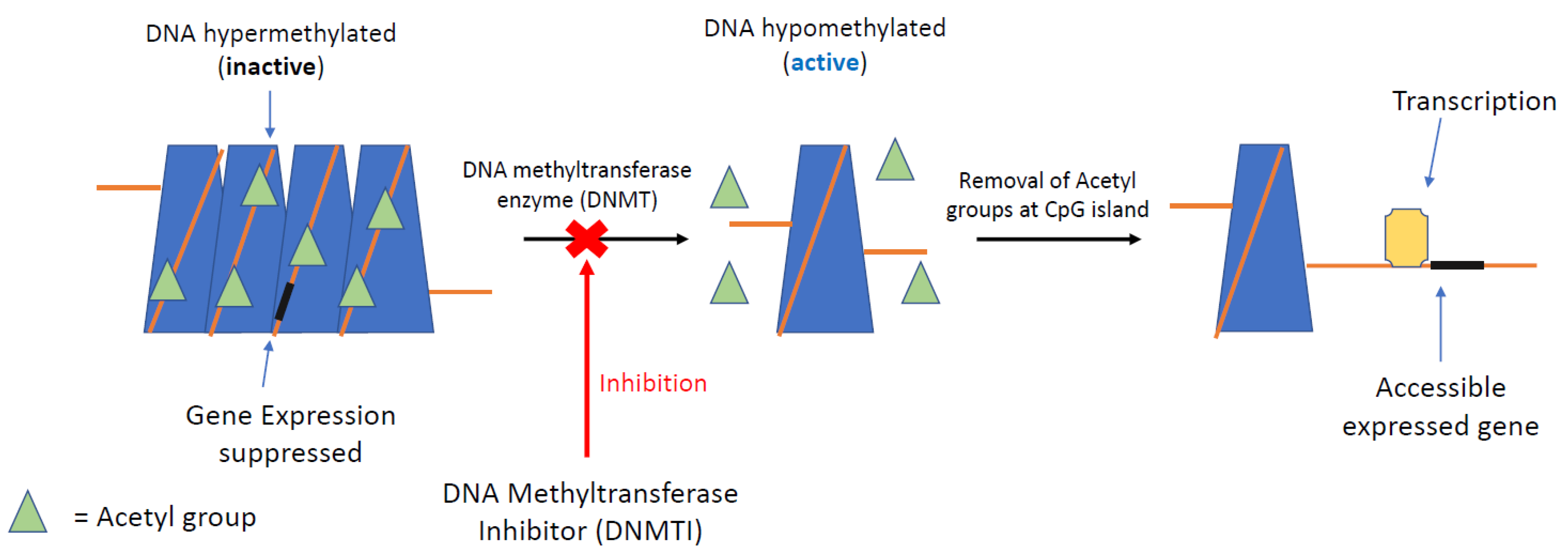
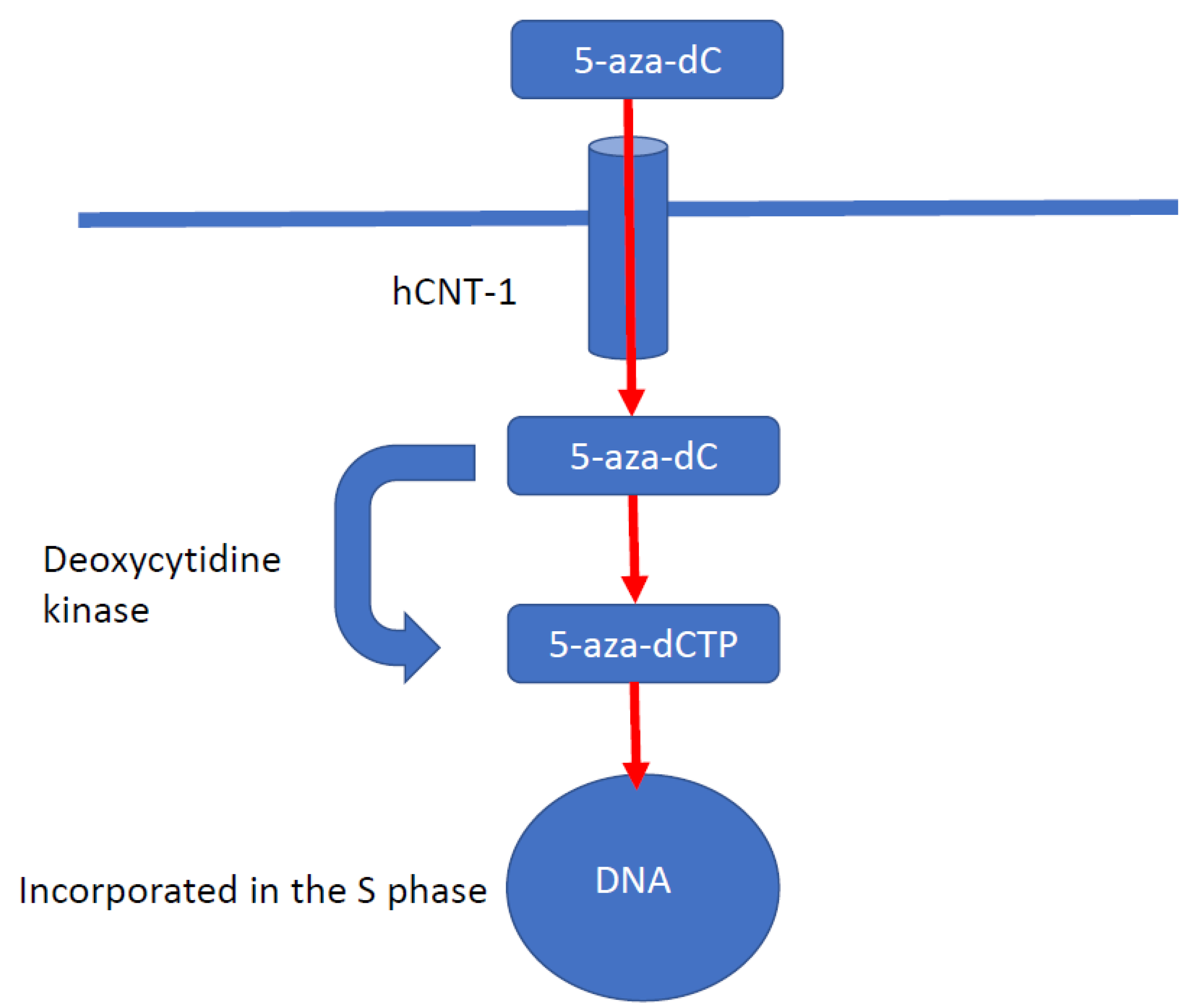
5.1. DNA Methyltransferase Inhibitors
5.2. Histone Deacetylation Inhibitors
6. Epigenetic Modification of the Somatostatin Receptor
7. Research Difficulties in GEP-NETs
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef] [Green Version]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Graham, J.S.; Kaye, S.B.; Brown, R. The promises and pitfalls of epigenetic therapies in solid tumours. Eur. J. Cancer 2009, 45, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widschwendter, M. 5-methylcytosine—The fifth base of DNA: The fifth wheel on a car or a highly promising diagnostic and therapeutic target in cancer? Dis. Markers 2007, 23, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chinnusamy, V.; Mohapatra, T. Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond. Front. Genet. 2018, 9, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundara, J.S.; Jamal, K.; Kurzawinski, T. Dictating genomic destiny: Epigenetic regulation of pancreatic neuroendocrine tumours. Mol. Cell. Endocrinol. 2018, 469, 85–91. [Google Scholar] [CrossRef]
- Chi, A.S.; Bernstein, B.E. Developmental biology. Pluripotent chromatin state. Science 2009, 323, 220–221. [Google Scholar] [CrossRef] [Green Version]
- Fishbein, L.; Nathanson, K.L. Inherited and Somatic Genetics of Pancreatic Neuroendocrine Tumors; Springer: New York, NY, USA, 2015. [Google Scholar]
- Di Domenico, A.; Wiedmer, T.; Marinoni, I.; Perren, A. Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocr. Relat. Cancer 2017, 24, R315–R334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M.; Calin, G.A. miRNAs, cancer, and stem cell division. Cell 2005, 122, 6–7. [Google Scholar] [CrossRef] [Green Version]
- Palanichamy, J.K.; Rao, D.S. miRNA dysregulation in cancer: Towards a mechanistic understanding. Front. Genet. 2014, 5, 54. [Google Scholar] [CrossRef]
- Fraenkel, M.; Kim, M.; Faggiano, A.; de Herder, W.W.; Valk, G.D.; NETwork, K. Incidence of gastroenteropancreatic neuroendocrine tumours: A systematic review of the literature. Endocr. Relat. Cancer 2014, 21, R153–R163. [Google Scholar] [CrossRef] [PubMed]
- Oberg, K. Neuroendocrine tumors (NETs): Historical overview and epidemiology. Tumori 2010, 96, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, L.; Minna, J.; Sakamaki, T.; Pestell, R.; White, M.A. The RASSF1A Tumor Suppressor Blocks Cell Cycle Progression and Inhibits Cyclin D1 Accumulation. Mol. Cell. Biol. 2002, 22, 4309–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dammann, R.; Schagdarsurengin, U.; Strunnikova, M.; Rastetter, M.; Seidel, C.; Liu, L.; Tommasi, S.; Pfeifer, G.P. Epigenetic inactivation of the Ras-association domain family 1 (RASSF1A) gene and its function in human carcinogenesis. Histol. Histopathol. 2003, 18, 2. [Google Scholar]
- Pizzi, S.; Azzoni, C.; Bottarelli, L.; Campanini, N.; D’Adda, T.; Pasquali, C.; Rossi, G.; Rindi, G.; Bordi, C. RASSF1A promoter methylation and 3p21.3 loss of heterozygosity are features of foregut, but not midgut and hindgut, malignant endocrine tumours. J. Pathol. 2005, 206, 409–416. [Google Scholar] [CrossRef] [PubMed]
- House, M.G.; Herman, J.G.; Guo, M.Z.; Hooker, C.M.; Schulick, R.D.; Lillemoe, K.D.; Cameron, J.L.; Hruban, R.H.; Maitra, A.; Yeo, C.J. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann. Surg. 2003, 238, 423–431. [Google Scholar] [CrossRef]
- Arnold, C.N.; Sosnowski, A.; Schmitt-Graff, A.; Arnold, R.; Blum, H.E. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int. J. Cancer 2007, 120, 2157–2164. [Google Scholar] [CrossRef]
- Liu, L.; Broaddus, R.R.; Yao, J.C.; Xie, S.; White, J.A.; Wu, T.T.; Hamilton, S.R.; Rashid, A. Epigenetic alterations in neuroendocrine tumors: Methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod. Pathol. 2005, 18, 1632–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpeli, G.; Amato, E.; Dandrea, M.; Fumagalli, C.; Debattisti, V.; Boninsegna, L.; Pelosi, G.; Falconi, M.; Scarpa, A. Methylation-associated down-regulation of RASSF1A and up-regulation of RASSF1C in pancreatic endocrine tumors. BMC Cancer 2011, 11, 351. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, D.; Kajiho, H.; Negishi, T.; Ura, S.; Watanabe, T.; Wada, T.; Ichijo, H.; Katada, T.; Nishina, H. Release of RASSF1C from the nucleus by Daxx degradation links DNA damage and SAPK/JNK activation. EMBO J. 2006, 25, 3286–3297. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.J.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the Cdkn2/P16/Mts1 Gene Is Frequently Associated with Aberrant DNA Methylation in All Common Human Cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar]
- Lubomierski, N.; Kersting, M.; Bert, T.; Muench, K.; Wulbrand, U.; Schuermann, M.; Bartsch, D.; Simon, B. Tumor suppressor genes in the 9p21 gene cluster are selective targets of inactivation in neuroendocrine gastroenteropancreatic tumors. Cancer Res. 2001, 61, 5905–5910. [Google Scholar] [PubMed]
- Arnold, C.N.; Nagasaka, T.; Goel, A.; Scharf, I.; Grabowski, P.; Sosnowski, A.; Schmitt-Graff, A.; Boland, C.R.; Arnold, R.; Blum, H.E. Molecular characteristics and predictors of survival in patients with malignant neuroendocrine tumors. Int. J. Cancer 2008, 123, 1556–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, J.; Goebel, S.U.; Peghini, P.L.; Lubensky, I.A.; Gibril, F.; Jensen, R.T. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J. Clin. Endocrinol. Metab. 2000, 85, 4146–4156. [Google Scholar] [CrossRef]
- Muscarella, P.; Melvin, W.S.; Fisher, W.E.; Foor, J.; Ellison, E.C.; Herman, J.G.; Schirmer, W.J.; Hitchcock, C.L.; DeYoung, B.R.; Weghorst, C.M. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: An analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998, 58, 237–240. [Google Scholar] [PubMed]
- Bartsch, D.K.; Kersting, M.; Wild, A.; Ramaswamy, A.; Gerdes, B.; Schuermann, M.; Simon, B.; Rothmund, M. Low frequency of p(16INK4a) alterations in insulinomas. Digestion 2000, 62, 171–177. [Google Scholar] [CrossRef]
- Wild, A.; Ramaswamy, A.; Langer, P.; Celik, I.; Fendrich, V.; Chaloupka, B.; Simon, B.; Bartsch, D.K. Frequent methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene in pancreatic endocrine tumors. J. Clin. Endocrinol. Metab. 2003, 88, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Stefanoli, M.; La Rosa, S.; Sahnane, N.; Romualdi, C.; Pastorino, R.; Marando, A.; Capella, C.; Sessa, F.; Furlan, D. Prognostic relevance of aberrant DNA methylation in g1 and g2 pancreatic neuroendocrine tumors. Neuroendocrinology 2014, 100, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Frauenhoffer, C.S.; Hooshmand, S.M.; Ryan, D.P.; Enzinger, P.C.; Meyerhardt, J.A.; Clark, J.W.; Hornick, J.; Fuchs, C.S.; Redston, M.S. Prediction of response to temozolomide (TMZ)-based therapy by loss of MGMT expression in patients with advanced neuroendocrine tumors (NET). J. Clin. Oncol. 2007, 25, 4505. [Google Scholar] [CrossRef]
- Liu, L.; Gerson, S.L. Targeted modulation of MGMT: Clinical implications. Clin. Cancer Res. 2006, 12, 328–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, K.S.; Dogliotti, E.; Connors, T.D.; Basu, A.K.; Essigmann, J.M. Site-specific mutagenesis by O6-alkylguanines located in the chromosomes of mammalian cells: Influence of the mammalian O6-alkylguanine-DNA alkyltransferase. Proc. Natl. Acad. Sci. USA 1989, 86, 8620–8624. [Google Scholar] [CrossRef] [Green Version]
- Bonfanti, M.; Broggni, M.; Prontera, C.; D’incalci, M. O6-methylguanine inibits the binding of transcription factors to DNA. Nucleic Acids Res. 1991, 19, 5739–5742. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.F.; Yaya-Tur, R.; Rojas-Marcos, I.; Reynes, G.; Pollan, M.; Aguirre-Cruz, L.; García-Lopez, J.L.; Piquer, J.; Safont, M.J.; Balaña, C.; et al. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin. Cancer Res. 2004, 10, 4933–4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, T.; Van Brakel, B.; Vercherat, C.; Hervieu, V.; Forestier, J.; Chayvialle, J.A.; Molin, Y.; Lombard-Bohas, C.; Joly, M.O.; Scoazec, J.Y. O6-Methylguanine-DNA methyltransferase status in neuroendocrine tumours: Prognostic relevance and association with response to alkylating agents. Br. J. Cancer 2015, 112, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Kulke, M.H.; Hornick, J.L.; Frauenhoffer, C.; Hooshmand, S.; Ryan, D.P.; Enzinger, P.C.; Meyerhardt, J.A.; Clark, J.W.; Stuart, K.; Fuchs, C.S.; et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin. Cancer Res. 2009, 15, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Raj, N.; Klimstra, D.S.; Horvat, N.; Zhang, L.Y.; Chou, J.F.; Capanu, M.; Basturk, O.; Do, R.K.G.; Allen, P.J.; Reidy-Lagunes, D. O-6-Methylguanine DNA Methyltransferase Status Does Not Predict Response or Resistance to Alkylating Agents in Well-Differentiated Pancreatic Neuroendocrine Tumors. Pancreas 2017, 46, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Kunz, P.L.; Morse, B.; Coppola, D.; Schell, M.J.; Campos, T.; Nguyen, P.T.; Nandoskar, P.; Khandelwal, V.; Strosberg, J.R. Phase II clinical trial of pasireotide long-acting repeatable in patients with metastatic neuroendocrine tumors. Endocr. Relat. Cancer 2015, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lemelin, A.; Barritault, M.; Hervieu, V.; Payen, L.; Peron, J.; Couvelard, A.; Cros, J.; Scoazec, J.Y.; Bin, S.; Villeneuve, L.; et al. O6-methylguanine-DNA methyltransferase (MGMT) status in neuroendocrine tumors: A randomized phase II study (MGMT-NET). Dig. Liver Dis. 2019, 51, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C. Epigenetic aspects on therapy development for gastroenteropancreatic neuroendocrine tumors. Neuroendocrinology 2013, 97, 19–25. [Google Scholar] [CrossRef]
- Dejeux, E.; Olaso, R.; Dousset, B.; Audebourg, A.; Gut, I.G.; Terris, B.; Tost, J. Hypermethylation of the IGF2 differentially methylated region 2 is a specific event in insulinomas leading to loss-of-imprinting and overexpression. Endocr. Relat. Cancer 2009. [Google Scholar] [CrossRef] [Green Version]
- House, M.G.; Herman, J.G.; Guo, M.Z.; Hooker, C.M.; Schulick, R.D.; Cameron, J.L.; Hruban, R.H.; Maitra, A.; Yeo, C.J. Prognostic value of hMLH1 methylation and microsatellite instability in pancreatic endocrine neoplasms. Surgery 2003, 134, 902–908, discussion 909. [Google Scholar] [CrossRef]
- Mei, M.; Deng, D.; Liu, T.H.; Sang, X.T.; Lu, X.; Xiang, H.D.; Zhou, J.; Wu, H.; Yang, Y.; Chen, J.; et al. Clinical implications of microsatellite instability and MLH1 gene inactivation in sporadic insulinomas. J. Clin. Endocrinol. Metab. 2009, 94, 3448–3457. [Google Scholar] [CrossRef]
- Karpathakis, A.; Dibra, H.; Thirlwell, C. Neuroendocrine tumours: Cracking the epigenetic code. Endocr. Relat. Cancer 2013, 20, R65–R82. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.S.; Estecio, M.R.; Nagano, Y.; Kim, D.H.; White, J.A.; Yao, J.C.; Issa, J.P.; Rashid, A. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod. Pathol. 2007, 20, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.K.; Jothi, R. Genome-wide characterization of menin-dependent H3K4me3 reveals a specific role for menin in the regulation of genes implicated in MEN1-like tumors. PLoS ONE 2012, 7, e37952. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; Livolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.J.; Song, T.Y.; Park, J.; Lee, J.; Lim, J.; Jang, H.; Kim, Y.N.; Yang, J.H.; Song, Y.; Choi, A.; et al. Menin mediates epigenetic regulation via histone H3 lysine 9 methylation. Cell Death Dis. 2013, 4, e583. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, A.B.; Xing, B.; Liu, C.; Naji, A.; Ma, X.; Simmons, R.A.; Hua, X. Menin and PRMT5 suppress GLP1 receptor transcript and PKA-mediated phosphorylation of FOXO1 and CREB. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E148–E166. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; He, J.; Li, J.; Tian, D.; Gu, L.; Zhou, M. Methylation of RASSF1A gene promoter is regulated by p53 and DAXX. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.J.; Perren, A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014, 146, 453–460.e5. [Google Scholar] [CrossRef]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 2006, 24, 4677–4684. [Google Scholar] [CrossRef] [PubMed]
- Thorns, C.; Schurmann, C.; Gebauer, N.; Wallaschofski, H.; Kumpers, C.; Bernard, V.; Feller, A.C.; Keck, T.; Habermann, J.K.; Begum, N.; et al. Global microRNA profiling of pancreatic neuroendocrine neoplasias. Anticancer Res. 2014, 34, 2249–2254. [Google Scholar]
- Grolmusz, V.K.; Kovesdi, A.; Borks, K.; Igaz, P.; Patocs, A. Prognostic relevance of proliferation-related miRNAs in pancreatic neuroendocrine neoplasms. Eur. J. Endocrinol. 2018, 179, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadanandam, A.; Wullschleger, S.; Lyssiotis, C.A.; Grotzinger, C.; Barbi, S.; Bersani, S.; Korner, J.; Wafy, I.; Mafficini, A.; Lawlor, R.T.; et al. A Cross-Species Analysis in Pancreatic Neuroendocrine Tumors Reveals Molecular Subtypes with Distinctive Clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov. 2015, 5, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Rumilla, K.M.; Jin, L.; Nakamura, N.; Stilling, G.A.; Ruebel, K.H.; Hobday, T.J.; Erlichman, C.; Erickson, L.A.; Lloyd, R.V. Association of DNA methylation and epigenetic inactivation of RASSF1A and beta-catenin with metastasis in small bowel carcinoid tumors. Endocrine 2006, 30, 299–306. [Google Scholar] [CrossRef]
- Fotouhi, O.; Adel Fahmideh, M.; Kjellman, M.; Sulaiman, L.; Hoog, A.; Zedenius, J.; Hashemi, J.; Larsson, C. Global hypomethylation and promoter methylation in small intestinal neuroendocrine tumors: An in vivo and in vitro study. Epigenetics 2014, 9, 987–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stricker, I.; Tzivras, D.; Nambiar, S.; Wulf, J.; Liffers, S.T.; Vogt, M.; Verdoodt, B.; Tannapfel, A.; Mirmohammadsadegh, A. Site- and grade-specific diversity of LINE1 methylation pattern in gastroenteropancreatic neuroendocrine tumours. Anticancer Res. 2012, 32, 3699–3706. [Google Scholar] [PubMed]
- Edfeldt, K.; Ahmad, T.; Akerstrom, G.; Janson, E.T.; Hellman, P.; Stalberg, P.; Bjorklund, P.; Westin, G. TCEB3C a putative tumor suppressor gene of small intestinal neuroendocrine tumors. Endocr. Relat. Cancer 2014, 21, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Bollard, J.; Massoma, P.; Vercherat, C.; Blanc, M.; Lepinasse, F.; Gadot, N.; Couderc, C.; Poncet, G.; Walter, T.; Joly, M.O.; et al. The axon guidance molecule semaphorin 3F is a negative regulator of tumor progression and proliferation in ileal neuroendocrine tumors. Oncotarget 2015, 6, 36731–36745. [Google Scholar] [CrossRef] [Green Version]
- Verdugo, A.D.; Crona, J.; Starker, L.; Stalberg, P.; Akerstrom, G.; Westin, G.; Hellman, P.; Bjorklund, P. Global DNA methylation patterns through an array-based approach in small intestinal neuroendocrine tumors. Endocr. Relat. Cancer 2014, 21, L5–L7. [Google Scholar] [CrossRef]
- Karpathakis, A.; Dibra, H.; Pipinikas, C.; Feber, A.; Morris, T.; Francis, J.; Oukrif, D.; Mandair, D.; Pericleous, M.; Mohmaduvesh, M.; et al. Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor. Clin. Cancer Res. 2016, 22, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Karpathakis, A.; Dibra, H.; Pipinikas, C.; Feber, A.; Morris, T.; Francis, J.; Oukrif, D.; Mandair, D.; Pericleous, M.; Mohmaduvesh, M.; et al. Progressive epigenetic dysregulation in neuroendocrine tumour liver metastases. Endocr. Relat. Cancer 2017, 24, L21–L25. [Google Scholar] [CrossRef]
- Rahman, M.M.; Qian, Z.R.; Wang, E.L.; Yoshimoto, K.; Nakasono, M.; Sultana, R.; Yoshida, T.; Hayashi, T.; Haba, R.; Ishida, M.; et al. DNA methyltransferases 1, 3a, and 3b overexpression and clinical significance in gastroenteropancreatic neuroendocrine tumors. Hum. Pathol. 2010, 41, 1069–1078. [Google Scholar] [CrossRef]
- Warneboldt, J.; Haller, F.; Horstmann, O.; Danner, B.C.; Fuzesi, L.; Doenecke, D.; Happel, N. Histone H1x is highly expressed in human neuroendocrine cells and tumours. BMC Cancer 2008, 8, 388. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Magerl, C.; Ellinger, J.; Braunschweig, T.; Kremmer, E.; Koch, L.K.; Holler, T.; Buttner, R.; Luscher, B.; Gutgemann, I. H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum. Pathol. 2010, 41, 181–189. [Google Scholar] [CrossRef]
- Ruebel, K.; Leontovich, A.A.; Stilling, G.A.; Zhang, S.; Righi, A.; Jin, L.; Lloyd, R.V. MicroRNA expression in ileal carcinoid tumors: Downregulation of microRNA-133a with tumor progression. Mod. Pathol. 2010, 23, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Ji, X. The Impact of MicroRNA-133a on Prognosis and Clinicopathological Parameters for Digestive System Cancers: A Comprehensive Study Based on Meta-Analysis and TCGA Database. Pathol. Oncol. Res. 2020, 26, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.C.; Frampton, A.E.; Malczewska, A.; Ottaviani, S.; Stronach, E.A.; Flora, R.; Kaemmerer, D.; Schwach, G.; Pfragner, R.; Faiz, O.; et al. MicroRNAs associated with small bowel neuroendocrine tumours and their metastases. Endocr. Relat. Cancer 2016, 23, 711–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.C.; Essaghir, A.; Martijn, C.; Lloyd, R.V.; Demoulin, J.B.; Oberg, K.; Giandomenico, V. Global microRNA profiling of well-differentiated small intestinal neuroendocrine tumors. Mod. Pathol. 2013, 26, 685–696. [Google Scholar] [CrossRef]
- Hamfjord, J.; Stangeland, A.M.; Hughes, T.; Skrede, M.L.; Tveit, K.M.; Ikdahl, T.; Kure, E.H. Differential expression of miRNAs in colorectal cancer: Comparison of paired tumor tissue and adjacent normal mucosa using high-throughput sequencing. PLoS ONE 2012, 7, e34150. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, Y.; Rehammar, A.; Bergstrom, A.; Andersson, E.; Altiparmak, G.; Sward, C.; Wangberg, B.; Kristiansson, E.; Nilsson, O. miRNA profiling of small intestinal neuroendocrine tumors defines novel molecular subtypes and identifies miR-375 as a biomarker of patient survival. Mod. Pathol. 2018, 31, 1302–1317. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Modlin, I.M.; Drozdov, I. Gene network-based analysis identifies two potential subtypes of small intestinal neuroendocrine tumors. BMC Genom. 2014, 15, 595. [Google Scholar] [CrossRef] [Green Version]
- Bowden, M.; Zhou, C.W.; Zhang, S.; Brais, L.; Rossi, A.; Naudin, L.; Thiagalingam, A.; Sicinska, E.; Kulke, M.H. Profiling of metastatic small intestine neuroendocrine tumors reveals characteristic miRNAs detectable in plasma. Oncotarget 2017, 8, 54331–54344. [Google Scholar] [CrossRef]
- Issa, J.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Alexander, V.M.; Roy, M.; Steffens, K.A.; Kunnimalaiyaan, M.; Chen, H. Azacytidine induces cell cycle arrest and suppression of neuroendocrine markers in carcinoids. Int. J. Clin. Exp. Med. 2010, 3, 95–102. [Google Scholar]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Savona, M.R.; Odenike, O.; Amrein, P.C.; Steensma, D.P.; DeZern, A.E.; Michaelis, L.C.; Faderl, S.; Harb, W.; Kantarjian, H.; Lowder, J.; et al. An oral fixed-dose combination of decitabine and cedazuridine in myelodysplastic syndromes: A multicentre, open-label, dose-escalation, phase 1 study. Lancet Haematol. 2019, 6, e194–e203. [Google Scholar] [CrossRef]
- Wagner, J.M.; Hackanson, B.; Lubbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenetics 2010, 1, 117–136. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Adachi, M.; Kawamura, R.; Imai, K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006, 13, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Baradari, V.; Huether, A.; Hopfner, M.; Schuppan, D.; Scherubl, H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr. Relat. Cancer 2006, 13, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, T.C.; Lin, A.J.; Ververis, K.; Chang, L.; Tang, M.M.; Okabe, J.; El-Osta, A. Trichostatin A accentuates doxorubicin-induced hypertrophy in cardiac myocytes. Aging 2010, 2, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Kunnimalaiyaan, M.; Chen, H. Tumor suppressor role of Notch-1 signaling in neuroendocrine tumors. Oncologist 2007, 12, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, T.A.; Holen, K.D.; Jaskula-Sztul, R.; Mulkerin, D.; Lubner, S.J.; Schelman, W.R.; Eickhoff, J.; Chen, H.; Loconte, N.K. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist 2011, 16, 835–843. [Google Scholar] [CrossRef]
- Srirajaskanthan, R.; Watkins, J.; Marelli, L.; Khan, K.; Caplin, M.E. Expression of somatostatin and dopamine 2 receptors in neuroendocrine tumours and the potential role for new biotherapies. Neuroendocrinology 2009, 89, 308–314. [Google Scholar] [CrossRef]
- Rinke, A.; Muller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Blaker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Ruszniewski, P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 1556–1557. [Google Scholar] [CrossRef]
- Strosberg, J.; Krenning, E. 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 1391–1392. [Google Scholar] [CrossRef]
- Torrisani, J.; Hanoun, N.; Laurell, H.; Lopez, F.; Maoret, J.J.; Souque, A.; Susini, C.; Cordelier, P.; Buscail, L. Identification of an upstream promoter of the human somatostatin receptor, hSSTR2, which is controlled by epigenetic modifications. Endocrinology 2008, 149, 3137–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taelman, V.F.; Radojewski, P.; Marincek, N.; Ben-Shlomo, A.; Grotzky, A.; Olariu, C.I.; Perren, A.; Stettler, C.; Krause, T.; Meier, L.P.; et al. Upregulation of Key Molecules for Targeted Imaging and Therapy. J. Nucl. Med. 2016, 57, 1805–1810. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, S.; Carpenter, J.E.; Sippel, R.S.; Kunnimalaiyaan, M.; Chen, H. A mouse model of carcinoid syndrome and heart disease. J. Surg. Res. 2005, 126, 102–105. [Google Scholar] [CrossRef]
- Capdevila, J.; Casanovas, O.; Salazar, R.; Castellano, D.; Segura, A.; Fuster, P.; Aller, J.; Garcia-Carbonero, R.; Jimenez-Fonseca, P.; Grande, E.; et al. Translational research in neuroendocrine tumors: Pitfalls and opportunities. Oncogene 2017, 36, 1899–1907. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [Green Version]

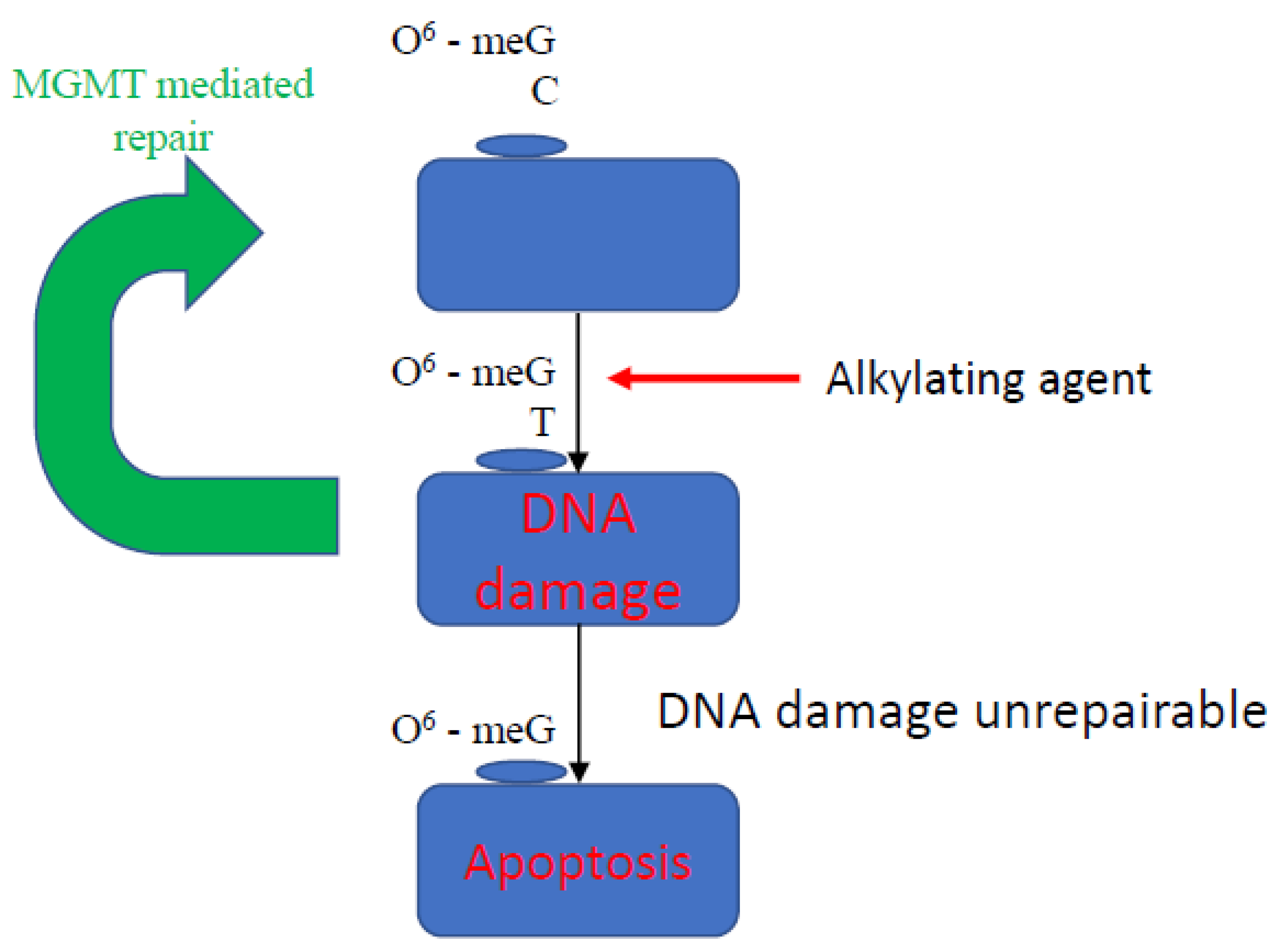
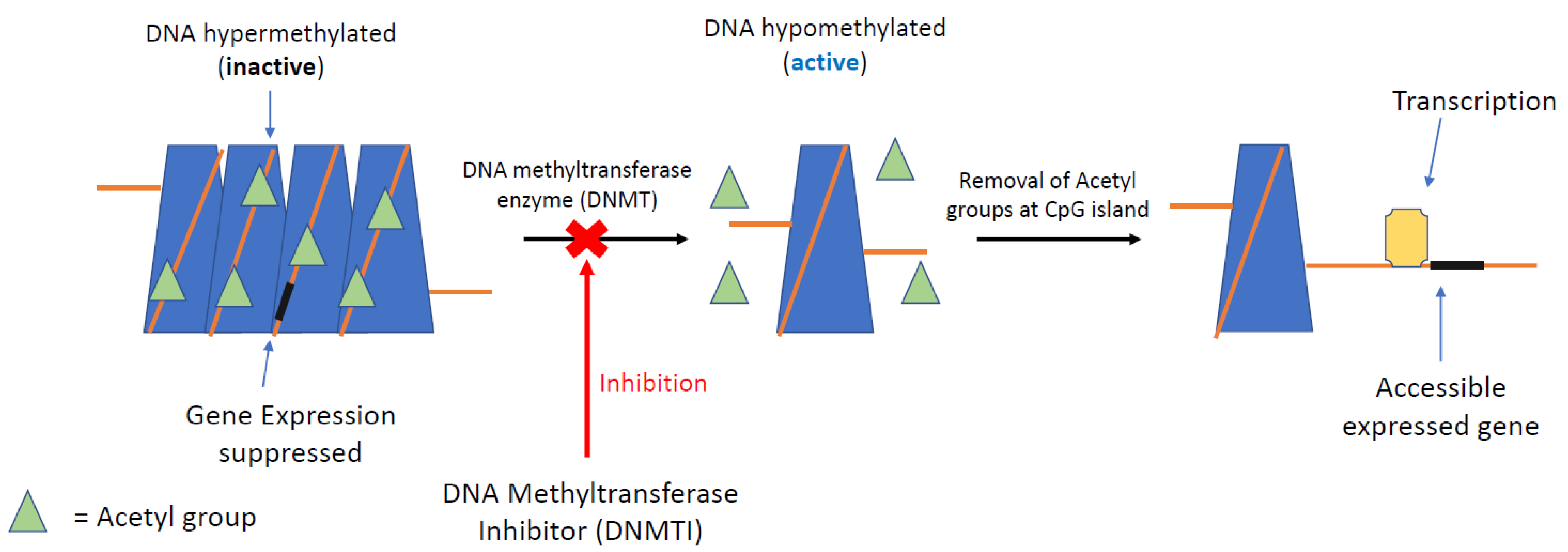
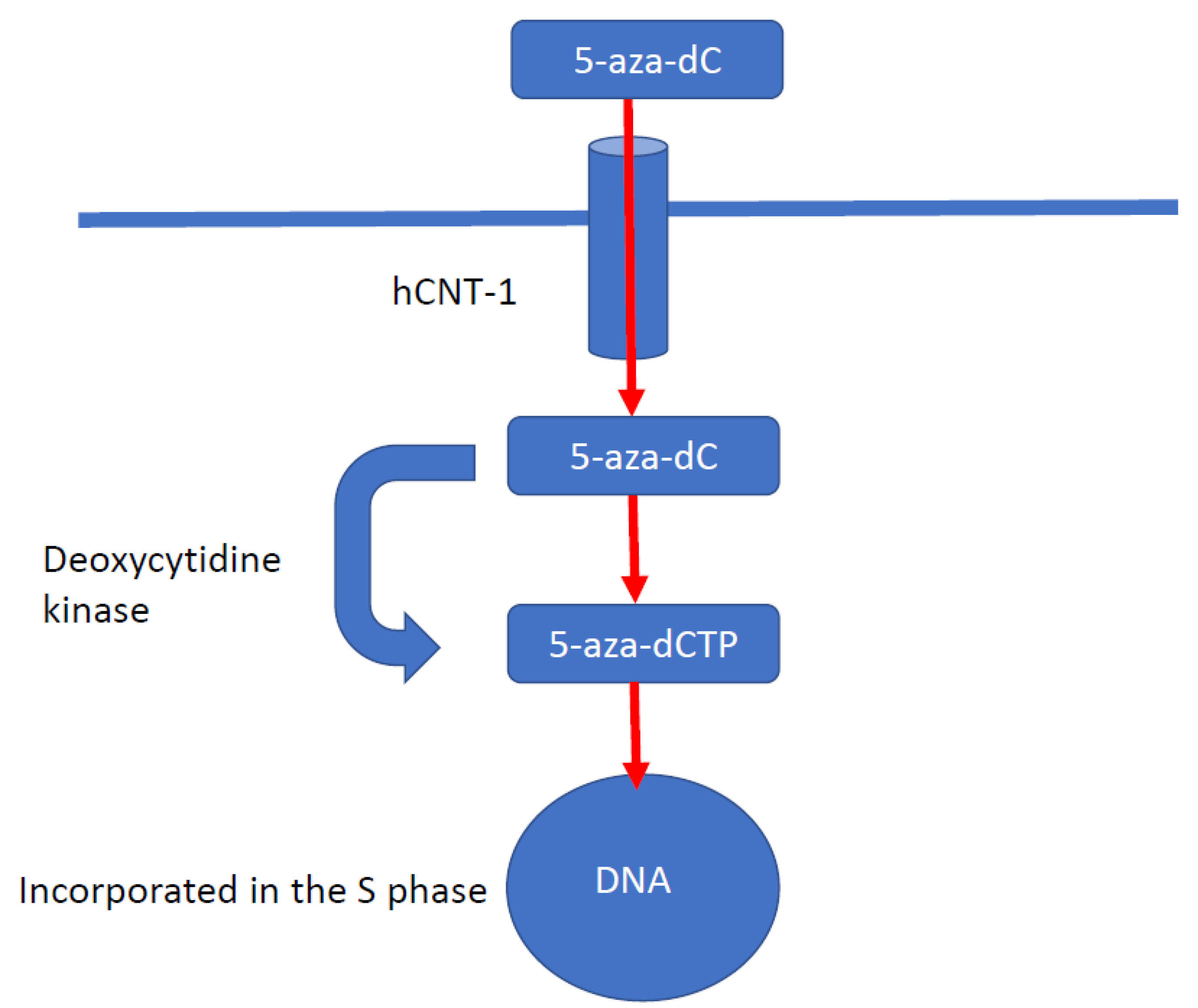
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R.; Lythgoe, M.P.; Slaich, B.; Patel, N. Exploring the Epigenome in Gastroenteropancreatic Neuroendocrine Neoplasias. Cancers 2021, 13, 4181. https://doi.org/10.3390/cancers13164181
Sharma R, Lythgoe MP, Slaich B, Patel N. Exploring the Epigenome in Gastroenteropancreatic Neuroendocrine Neoplasias. Cancers. 2021; 13(16):4181. https://doi.org/10.3390/cancers13164181
Chicago/Turabian StyleSharma, Rohini, Mark P. Lythgoe, Bhavandeep Slaich, and Nishil Patel. 2021. "Exploring the Epigenome in Gastroenteropancreatic Neuroendocrine Neoplasias" Cancers 13, no. 16: 4181. https://doi.org/10.3390/cancers13164181
APA StyleSharma, R., Lythgoe, M. P., Slaich, B., & Patel, N. (2021). Exploring the Epigenome in Gastroenteropancreatic Neuroendocrine Neoplasias. Cancers, 13(16), 4181. https://doi.org/10.3390/cancers13164181