
Unraveling Tumor Heterogeneity by Using DNA Barcoding Technologies to Develop Personalized Treatment Strategies in Advanced-Stage PDAC


Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumor Heterogeneity and Its Impact on Personalized Treatment Strategies in PDAC
2.1. Inter-Tumor Heterogeneity
2.1.1. Genomic Alterations and Subtypes
2.1.2. Transcriptomic Subtypes
2.1.3. Metabolic Subtypes
2.1.4. Immune-Landscape Heterogeneity
2.1.5. Inter-Tumor Heterogeneity between Tumor Sites
2.1.6. Inter-Tumor Heterogeneity in the Stroma
2.2. Intra-Tumor Heterogeneity
2.2.1. Intra-Tumor Heterogeneity among Tumor Cells
2.2.2. Intra-Tumor Heterogeneity in Stroma
3. Novel Personalized Treatment Advancements in PDAC
4. Studying Heterogeneity and Cell Fate
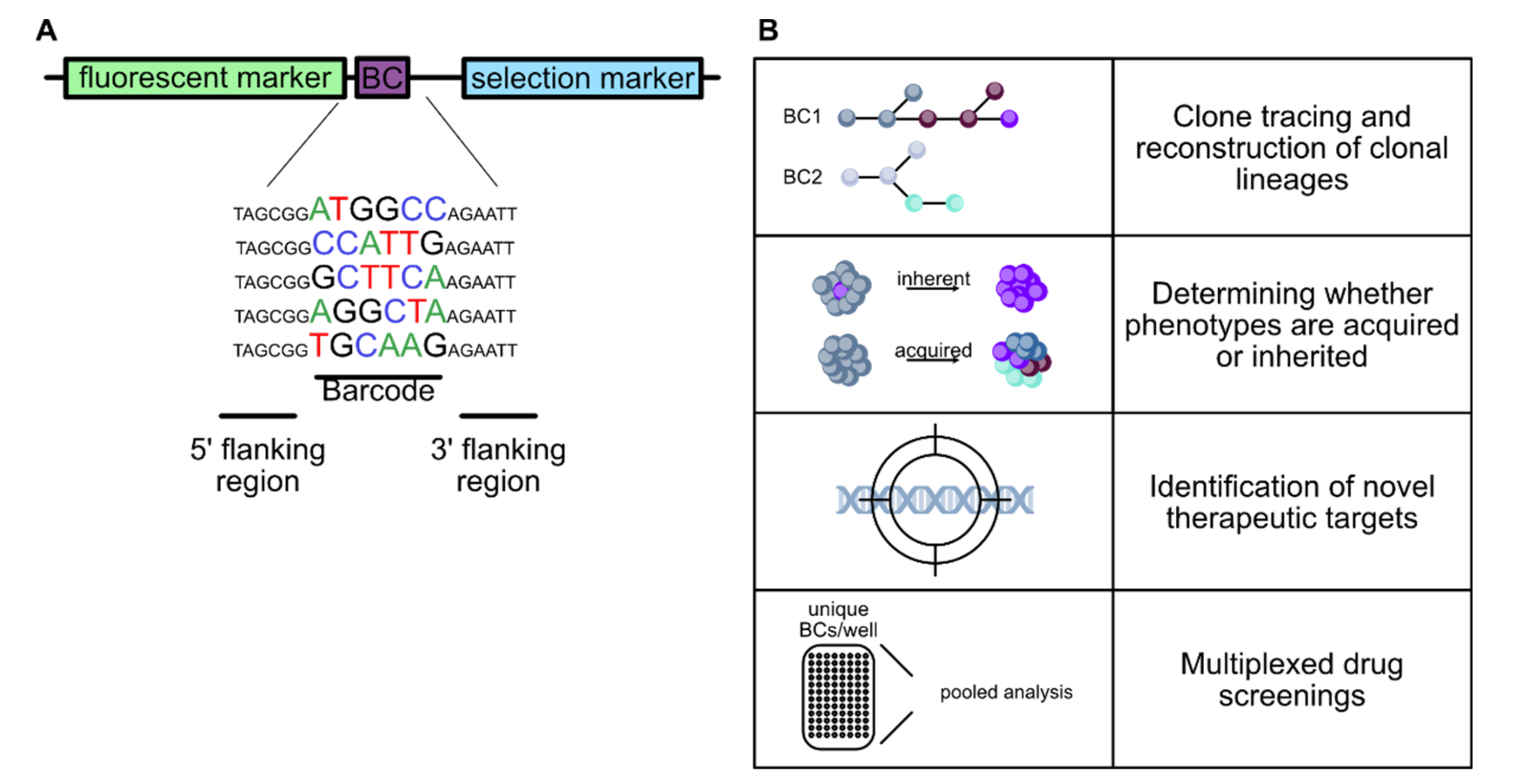
4.1. Molecular Barcoding
4.2. Applications of Molecular Barcoding
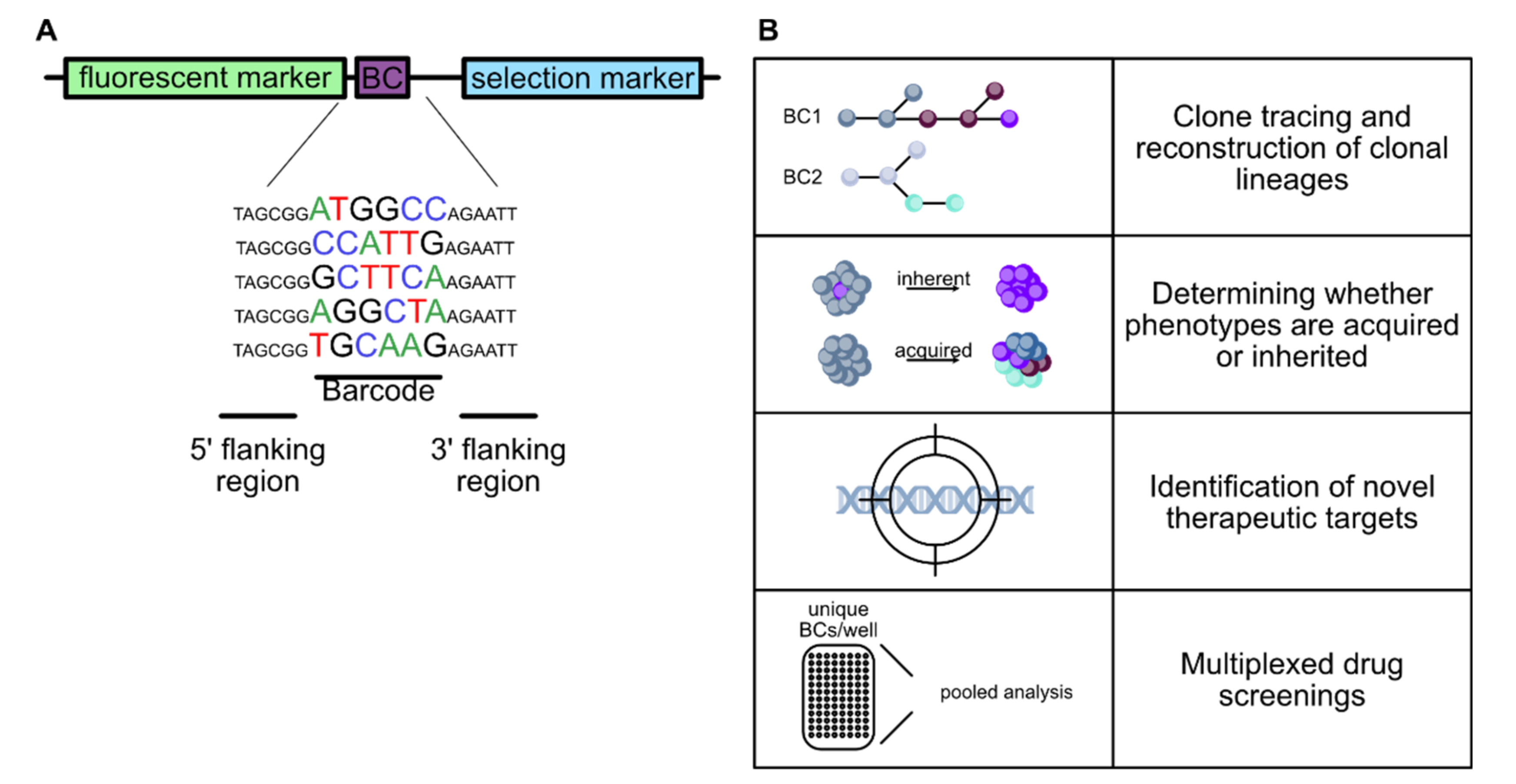
4.2.1. Lineage Tracing and Fate Mapping
4.2.2. Molecular Barcoding in Cancer Research
4.2.3. High Throughput Screens
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Facts & Figures. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf (accessed on 12 June 2020).
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, 4410–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, E.; Safgren, S.L.; Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Pancreatic Cancer, A Mis-interpreter of the Epigenetic Language. Yale J. Biol. Med. 2016, 89, 575–590. [Google Scholar]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Heindl, A.; Nawaz, S.; Yuan, Y. Mapping spatial heterogeneity in the tumor microenvironment: A new era for digital pathology. Lab. Investig. 2015, 95, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Iacobuzio-Donahue, C.A. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [Green Version]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Favero, F.; De Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, K.; Miller, H.; Gordian, E.; Rocha-Lima, C.; Singal, R. Methylation-mediated silencing of TMS1 in pancreatic cancer and its potential contribution to chemosensitivity. Anticancer Res. 2010, 30, 3919–3925. [Google Scholar]
- Chakraborty, A.; Dorsett, K.A.; Trummell, H.Q.; Yang, E.S.; Oliver, P.G.; Bonner, J.A.; Buchsbaum, D.J.; Bellis, S.L. ST6Gal-I sialyltransferase promotes chemoresistance in pancreatic ductal adenocarcinoma by abrogating gemcitabine-mediated DNA damage. J. Biol. Chem. 2018, 293, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Basson, M.D.; Ito, H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301–51310. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, R.L.; Magnus, N.K.C.; Thapar, V.; Morris, R.; Szabolcs, A.; Neyaz, A.; Kulkarni, A.S.; Tai, E.; Chougule, A.; Hillis, A.; et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 26835–26845. [Google Scholar] [CrossRef] [Green Version]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [Green Version]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Roy, S.; Singh, A.P.; Gupta, D. Unsupervised subtyping and methylation landscape of pancreatic ductal adenocarcinoma. Heliyon 2021, 7, e06000. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Steuber, B.; Kopp, W.; Kari, V.; Urbach, L.; Wang, X.; Küffer, S.; Bohnenberger, H.; Spyropoulou, D.; Zhang, Z.; et al. EZH2 Regulates Pancreatic Cancer Subtype Identity and Tumor Progression via Transcriptional Repression of GATA6. Cancer Res. 2020, 80, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Paradise, B.D.; Barham, W.; Fernandez-Zapico, M.E. Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes? Cancers 2018, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, T.; Liao, Q.; Dai, M.; Guo, J.; Yang, X.; Tan, W.; Lin, D.; Wu, C.; Zhao, Y. Metformin inhibits pancreatic cancer metastasis caused by SMAD4 deficiency and consequent HNF4G upregulation. Protein Cell 2021, 12, 128–144. [Google Scholar] [CrossRef]
- Ren, D.; Qin, G.; Zhao, J.; Sun, Y.; Zhang, B.; Li, D.; Wang, B.; Jin, X.; Wu, H. Metformin activates the STING/IRF3/IFN-β pathway by inhibiting AKT phosphorylation in pancreatic cancer. Am. J. Cancer Res. 2020, 10, 2851–2864. [Google Scholar] [PubMed]
- Nie, K.; Li, J.; He, X.; Wang, Y.; Zhao, Q.; Du, M.; Sun, H.; Wang, J.; Lyu, J.; Fang, H.; et al. COX6B2 drives metabolic reprogramming toward oxidative phosphorylation to promote metastasis in pancreatic ductal cancer cells. Oncogenesis 2020, 9, 51. [Google Scholar] [CrossRef]
- Liu, S.H.; Yu, J.; Creeden, J.F.; Sutton, J.M.; Markowiak, S.; Sanchez, R.; Nemunaitis, J.; Kalinoski, A.; Zhang, J.T.; Damoiseaux, R.; et al. Repurposing metformin, simvastatin and digoxin as a combination for targeted therapy for pancreatic ductal adenocarcinoma. Cancer Lett. 2020, 491, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, H.; Zheng, C.; Gao, J.; Fu, Q.; Hu, N.; Shao, X.; Zhou, Y.; Xiong, J.; Nie, K.; et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.S.; Choi, J.Y. Synergistic anti-cancer effect of phenformin and oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Abdel Hadi, N.; Gicquel, T.; El Kaoutari, A.; Diémé, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, H.; Woo, S.M.; Jang, H.; Jeon, Y.; Kim, H.Y.; Song, J.; Lee, W.J.; Hong, E.K.; Park, S.J.; et al. Overall survival of pancreatic ductal adenocarcinoma is doubled by Aldh7a1 deletion in the KPC mouse. Theranostics 2021, 11, 3472–3488. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.R.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; De Oliveira, E.; Kamphorst, J.J.; Rabinowitz, J.D.; Tejero, H.; Al-Shahrour, F.; Hidalgo, M.; Maitra, A.; et al. Treatment of Pancreatic Cancer Patient-Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin. Cancer Res. 2017, 23, 5639–5647. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.H.; Dorsch, M.; Dujardin, P.; Silas, S.; Ueffing, K.; Hölken, J.M.; Yang, D.; Winslow, M.M.; Grüner, B.M. Altered Mitochondria Functionality Defines a Metastatic Cell State in Lung Cancer and Creates an Exploitable Vulnerability. Cancer Res. 2021, 81, 567–579. [Google Scholar] [CrossRef]
- Chakrabarti, G.; Moore, Z.R.; Luo, X.; Ilcheva, M.; Ali, A.; Padanad, M.; Zhou, Y.; Xie, Y.; Burma, S.; Scaglioni, P.P.; et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab. 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Quiles Del Rey, M.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef]
- Udupa, S.; Nguyen, S.; Hoang, G.; Nguyen, T.; Quinones, A.; Pham, K.; Asaka, R.; Nguyen, K.; Zhang, C.; Elgogary, A.; et al. Upregulation of the Glutaminase II Pathway Contributes to Glutamate Production upon Glutaminase 1 Inhibition in Pancreatic Cancer. Proteomics 2019, 19, e1800451. [Google Scholar] [CrossRef] [Green Version]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef]
- Lee, K.C.; Maturo, C.; Perera, C.N.; Luddy, J.; Rodriguez, R.; Shorr, R. Translational assessment of mitochondrial dysfunction of pancreatic cancer from in vitro gene microarray and animal efficacy studies, to early clinical studies, via the novel tumor-specific anti-mitochondrial agent, CPI-613. Ann. Transl. Med. 2014, 2, 91. [Google Scholar] [CrossRef]
- Gao, L.; Xu, Z.; Huang, Z.; Tang, Y.; Yang, D.; Huang, J.; He, L.; Liu, M.; Chen, Z.; Teng, Y. CPI-613 rewires lipid metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC signaling. J. Exp. Clin. Cancer Res. 2020, 39, 73. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Eriksson, E.; Moreno, R.; Milenova, I.; Liljenfeldt, L.; Dieterich, L.C.; Christiansson, L.; Karlsson, H.; Ullenhag, G.; Mangsbo, S.M.; Dimberg, A.; et al. Activation of myeloid and endothelial cells by CD40L gene therapy supports T-cell expansion and migration into the tumor microenvironment. Gene Ther. 2017, 24, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosewell Shaw, A.; Porter, C.E.; Yip, T.; Mah, W.C.; McKenna, M.K.; Dysthe, M.; Jung, Y.; Parihar, R.; Brenner, M.K.; Suzuki, M. Oncolytic adeno-immunotherapy modulates the immune system enabling CAR T-cells to cure pancreatic tumors. Commun. Biol. 2021, 4, 368. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Yang, M.-H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Raj, D.; Nikolaidi, M.; Garces, I.; Lorizio, D.; Castro, N.M.; Caiafa, S.G.; Moore, K.; Brown, N.F.; Kocher, H.M.; Duan, X.; et al. CEACAM7 Is an Effective Target for CAR T-cell Therapy of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1538–1552. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Fan, M.H.; Miao, C.H.; Liao, Y.J.; Yuan, R.H.; Liu, C.L. Engineering Chimeric Antigen Receptor T Cells against Immune Checkpoint Inhibitors PD-1/PD-L1 for Treating Pancreatic Cancer. Mol. Ther. Oncolytics 2020, 17, 571–585. [Google Scholar] [CrossRef]
- He, J.; Zhang, Z.; Lv, S.; Liu, X.; Cui, L.; Jiang, D.; Zhang, Q.; Li, L.; Qin, W.; Jin, H.; et al. Engineered CAR T cells targeting mesothelin by piggyBac transposon system for the treatment of pancreatic cancer. Cell Immunol. 2018, 329, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Song, B.; Wang, P.; Shi, B.; Li, Q.; Fan, M.; Di, S.; Yang, J.; Li, Z. Efficient growth suppression in pancreatic cancer PDX model by fully human anti-mesothelin CAR-T cells. Protein Cell 2017, 8, 926–931. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Muiños, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupinacci, R.M.; Goloudina, A.; Buhard, O.; Bachet, J.B.; Maréchal, R.; Demetter, P.; Cros, J.; Bardier-Dupas, A.; Collura, A.; Cervera, P.; et al. Prevalence of Microsatellite Instability in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2018, 154, 1061–1065. [Google Scholar] [CrossRef]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, e122092. [Google Scholar] [CrossRef] [Green Version]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF-Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J.; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3578–3588. [Google Scholar] [CrossRef] [Green Version]
- Nair, N.; Chen, S.Y.; Lemmens, E.; Chang, S.; Le, D.T.; Jaffee, E.M.; Murphy, A.; Whiting, C.; Müller, T.; Brockstedt, D.G. Single-Cell Immune Competency Signatures Associate with Survival in Phase II GVAX and CRS-207 Randomized Studies in Patients with Metastatic Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Luheshi, N.M.; Coates-Ulrichsen, J.; Harper, J.; Mullins, S.; Sulikowski, M.G.; Martin, P.; Brown, L.; Lewis, A.; Davies, G.; Morrow, M.; et al. Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget 2016, 7, 18508–18520. [Google Scholar] [CrossRef] [Green Version]
- Le, K.; Wang, J.; Zhang, T.; Guo, Y.; Chang, H.; Wang, S.; Zhu, B. Overexpression of Mesothelin in Pancreatic Ductal Adenocarcinoma (PDAC). Int. J. Med. Sci. 2020, 17, 422–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hahn, O.; Rappl, G.; Nowak, M.; Schmidt-Wolf, I.H.; Hombach, A.A.; Abken, H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 2012, 143, 1095–1107. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algül, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Zhao, J.; Wen, X.; Tian, L.; Li, T.; Xu, C.; Wen, X.; Melancon, M.P.; Gupta, S.; Shen, B.; Peng, W.; et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat. Commun. 2019, 10, 899. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Campbell, P.J.; Pleasance, E.D.; Stephens, P.J.; Dicks, E.; Rance, R.; Goodhead, I.; Follows, G.A.; Green, A.R.; Futreal, P.A.; Stratton, M.R. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc. Natl. Acad. Sci. USA 2008, 105, 13081–13086. [Google Scholar] [CrossRef] [Green Version]
- Khoshchehreh, R.; Totonchi, M.; Carlos Ramirez, J.; Torres, R.; Baharvand, H.; Aicher, A.; Ebrahimi, M.; Heeschen, C. Epigenetic reprogramming of primary pancreatic cancer cells counteracts their in vivo tumourigenicity. Oncogene 2019, 38, 6226–6239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Zhong, Y.; Yachida, S.; Rajeshkumar, N.V.; Abel, M.L.; Marimuthu, A.; Mudgal, K.; Hruban, R.H.; Poling, J.S.; Tyner, J.W.; et al. Heterogeneity of pancreatic cancer metastases in a single patient revealed by quantitative proteomics. Mol. Cell Proteom. 2014, 13, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-Targeting Therapy in Pancreatic Cancer: One Coin With Two Sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. Faseb. J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef]
- Nakamura, T.; Kuwai, T.; Kitadai, Y.; Sasaki, T.; Fan, D.; Coombes, K.R.; Kim, S.J.; Fidler, I.J. Zonal heterogeneity for gene expression in human pancreatic carcinoma. Cancer Res. 2007, 67, 7597–7604. [Google Scholar] [CrossRef] [Green Version]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Seth, S.; Li, C.Y.; Ho, I.L.; Corti, D.; Loponte, S.; Sapio, L.; Del Poggetto, E.; Yen, E.Y.; Robinson, F.S.; Peoples, M.; et al. Pre-existing Functional Heterogeneity of Tumorigenic Compartment as the Origin of Chemoresistance in Pancreatic Tumors. Cell Rep. 2019, 26, 1518–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2015, 28, 831–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Blair, A.B.; Groot, V.P.; Gemenetzis, G.; Wei, J.; Cameron, J.L.; Weiss, M.J.; Goggins, M.; Wolfgang, C.L.; Yu, J.; He, J. BRCA1/BRCA2 Germline Mutation Carriers and Sporadic Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2018, 226, 630–637.e1. [Google Scholar] [CrossRef]
- Reiss, K.A.; Yu, S.; Judy, R.; Symecko, H.; Nathanson, K.L.; Domchek, S.M. Retrospective Survival Analysis of Patients with Advanced Pancreatic Ductal Adenocarcinoma and Germline BRCA or PALB2 Mutations. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer 2020, 122, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-H.; Wang, W.-Q.; Han, X.; Gao, H.-L.; Li, T.-J.; Xu, S.-S.; Li, S.; Xu, H.-X.; Li, H.; Ye, L.-Y.; et al. Advances on diagnostic biomarkers of pancreatic ductal adenocarcinoma: A systems biology perspective. Comput. Struct. Biotechnol. J. 2020, 18, 3606–3614. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202. [Google Scholar] [CrossRef] [Green Version]
- Capello, M.; Bantis, L.E.; Scelo, G.; Zhao, Y.; Li, P.; Dhillon, D.S.; Patel, N.J.; Kundnani, D.L.; Wang, H.; Abbruzzese, J.L.; et al. Sequential Validation of Blood-Based Protein Biomarker Candidates for Early-Stage Pancreatic Cancer. JNCI 2017, 109, 4. [Google Scholar] [CrossRef]
- Kaur, S.; Smith, L.M.; Patel, A.; Menning, M.; Watley, D.C.; Malik, S.S.; Krishn, S.R.; Mallya, K.; Aithal, A.; Sasson, A.R.; et al. A Combination of MUC5AC and CA19-9 Improves the Diagnosis of Pancreatic Cancer: A Multicenter Study. Off. J. Am. Coll. Gastroenterol. ACG 2017, 112, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Bamlet, W.R.; Oberg, A.L.; Chaffee, K.G.; Donahue, G.; Cao, X.-J.; Chari, S.; Garcia, B.A.; Petersen, G.M.; Zaret, K.S. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci. Transl. Med. 2017, 9, eaah5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Aquila, E.; Fulgenzi, C.A.M.; Minelli, A.; Citarella, F.; Stellato, M.; Pantano, F.; Russano, M.; Cursano, M.C.; Napolitano, A.; Zeppola, T.; et al. Prognostic and predictive factors in pancreatic cancer. Oncotarget 2020, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Diao, Z.; Yue, X.; Chen, Y.; Zhao, H.; Cheng, L.; Sun, J. Construction and analysis of dysregulated lncRNA-associated ceRNA network identified novel lncRNA biomarkers for early diagnosis of human pancreatic cancer. Oncotarget 2016, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Raimondo, M.; Guha, S.; Chen, J.; Diao, L.; Dong, X.; Wallace, M.B.; Killary, A.M.; Frazier, M.L.; Woodward, T.A.; et al. Circulating microRNAs in Pancreatic Juice as Candidate Biomarkers of Pancreatic Cancer. J. Cancer 2014, 5, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reizel, Y.; Itzkovitz, S.; Adar, R.; Elbaz, J.; Jinich, A.; Chapal-Ilani, N.; Maruvka, Y.E.; Nevo, N.; Marx, Z.; Horovitz, I.; et al. Cell lineage analysis of the mammalian female germline. PLoS Genet. 2012, 8, e1002477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmel, C.B.; Warga, R.M.; Schilling, T.F. Origin and organization of the zebrafish fate map. Development 1990, 108, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Chapple, R.H.; Tseng, Y.J.; Hu, T.; Kitano, A.; Takeichi, M.; Hoegenauer, K.A.; Nakada, D. Lineage tracing of murine adult hematopoietic stem cells reveals active contribution to steady-state hematopoiesis. Blood Adv. 2018, 2, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Neff, N.F.; Quake, S.R.; Weissman, I.L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 2011, 29, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Gerrits, A.; Dykstra, B.; Kalmykowa, O.J.; Klauke, K.; Verovskaya, E.; Broekhuis, M.J.C.; De Haan, G.; Bystrykh, L.V. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood 2010, 115, 2610–2618. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 1979, 26, 557–573. [Google Scholar] [CrossRef] [Green Version]
- Vogt, W. Gestaltungsanalyse am Amphibienkeim mit Örtlicher Vitalfärbung. Wilhelm Roux’ Archiv Für Entwicklungsmechanik der Organismen 1929, 120, 384–706. [Google Scholar] [CrossRef]
- Serbedzija, G.N.; Bronner-Fraser, M.; Fraser, S.E. A vital dye analysis of the timing and pathways of avian trunk neural crest cell migration. Development 1989, 106, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef]
- Skardelly, M.; Hempel, E.; Hirrlinger, J.; Wegner, F.; Meixensberger, J.; Milosevic, J. Fluorescent Protein-Expressing Neural Progenitor Cells as a Tool for Transplantation Studies. PLoS ONE 2014, 9, e99819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef]
- Cai, D.; Cohen, K.B.; Luo, T.; Lichtman, J.W.; Sanes, J.R. Improved tools for the Brainbow toolbox. Nat. Methods 2013, 10, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Weissman, T.A.; Pan, Y.A. Brainbow: New resources and emerging biological applications for multicolor genetic labeling and analysis. Genetics 2015, 199, 293–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.; Cepko, C.L. Widespread dispersion of neuronal clones across functional regions of the cerebral cortex. Science 1992, 255, 434–440. [Google Scholar] [CrossRef]
- Schepers, K.; Swart, E.; Van Heijst, J.W.; Gerlach, C.; Castrucci, M.; Sie, D.; Heimerikx, M.; Velds, A.; Kerkhoven, R.M.; Arens, R.; et al. Dissecting T cell lineage relationships by cellular barcoding. J. Exp. Med. 2008, 205, 2309–2318. [Google Scholar] [CrossRef] [Green Version]
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef]
- Grüner, B.M.; Schulze, C.J.; Yang, D.; Ogasawara, D.; Dix, M.M.; Rogers, Z.N.; Chuang, C.-H.; McFarland, C.D.; Chiou, S.-H.; Brown, J.M.; et al. An in vivo multiplexed small-molecule screening platform. Nat. Methods 2016, 13, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Bhang, H.-e.C.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Pei, W.; Feyerabend, T.B.; Rössler, J.; Wang, X.; Postrach, D.; Busch, K.; Rode, I.; Klapproth, K.; Dietlein, N.; Quedenau, C.; et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548, 456–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peikon, I.D.; Gizatullina, D.I.; Zador, A.M. In vivo generation of DNA sequence diversity for cellular barcoding. Nucleic. Acids Res. 2014, 42, e127. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, W.; Yum, S.-Y.; Jeon, Y.; Cho, N.; Jang, G.; Bang, D. Lineage tracing using a Cas9-deaminase barcoding system targeting endogenous L1 elements. Nat. Commun. 2019, 10, 1234. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Findlay, G.M.; Gagnon, J.A.; Horwitz, M.S.; Schier, A.F.; Shendure, J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 2016, 353, aaf7907. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.E.; Weinreb, C.; Collins, Z.M.; Briggs, J.A.; Megason, S.G.; Klein, A.M. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 2018, 360, 981. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.-J.; Klein, A.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef]
- Gerlach, C.; Van Heijst, J.W.J.; Swart, E.; Sie, D.; Armstrong, N.; Kerkhoven, R.M.; Zehn, D.; Bevan, M.J.; Schepers, K.; Schumacher, T.N.M. One naive T cell, multiple fates in CD8+ T cell differentiation. J. Exp. Med. 2010, 207, 1235–1246. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.M.; Gerlach, C.; Van Heijst, J.W.J. Mapping the life histories of T cells. Nat. Rev. Immunol. 2010, 10, 621–631. [Google Scholar] [CrossRef]
- Alemany, A.; Florescu, M.; Baron, C.S.; Peterson-Maduro, J.; Van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 2018, 556, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kalhor, R.; Mali, P.; Church, G.M. Rapidly evolving homing CRISPR barcodes. Nat. Methods 2017, 14, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieda, K.L.; Linton, J.M.; Hormoz, S.; Choi, J.; Chow, K.-H.K.; Singer, Z.S.; Budde, M.W.; Elowitz, M.B.; Cai, L. Synthetic recording and in situ readout of lineage information in single cells. Nature 2017, 541, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, B.; Wagner, D.E.; McKenna, A.; Pandey, S.; Klein, A.M.; Shendure, J.; Gagnon, J.A.; Schier, A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018, 36, 442–450. [Google Scholar] [CrossRef]
- Umkehrer, C.; Holstein, F.; Formenti, L.; Jude, J.; Froussios, K.; Neumann, T.; Cronin, S.M.; Haas, L.; Lipp, J.J.; Burkard, T.R.; et al. Isolating live cell clones from barcoded populations using CRISPRa-inducible reporters. Nat. Biotechnol. 2021, 39, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.N.; Baker, L.C.; Mittelman, D.; Porteus, M.H. Lentiviral and targeted cellular barcoding reveals ongoing clonal dynamics of cell lines in vitro and in vivo. Genome Biol. 2014, 15, R75. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [Green Version]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Roh, V.; Abramowski, P.; Hiou-Feige, A.; Cornils, K.; Rivals, J.-P.; Zougman, A.; Aranyossy, T.; Thielecke, L.; Truan, Z.; Mermod, M.; et al. Cellular Barcoding Identifies Clonal Substitution as a Hallmark of Local Recurrence in a Surgical Model of Head and Neck Squamous Cell Carcinoma. Cell Rep. 2018, 25, 2208–2222.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, C.H.; Greenside, P.G.; Rogers, Z.N.; Brady, J.J.; Yang, D.; Ma, R.K.; Caswell, D.R.; Chiou, S.H.; Winters, A.F.; Grüner, B.M.; et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat. Med. 2017, 23, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Compound of Interest | Putative Molecular Target | Subtype Specificity | Pubmed-ID & Year of Publication |
|---|---|---|---|
| Metformin | HNF4G via AMPK COX6B2 | SMAD4-deficiency High levels of COX6B2 | 32737864; 2021 [36] 32415061; 2020 [38] |
| Phenformin | Mitochondrial Complex I | High OXPHOS | 33294863; 2020 [45] |
| CB-839+-ß-lapachone | GLS 1/NQO1 | Mutant KRAS + NQO1 overexpression | 26462257; 2015 [50] |
| CD40 ligand | CD40 | Immune-poor/high levels of M2 macrophages | 27906162; 2017 [58] |
| Chimeric antigen receptor-engineered T cells | Human epidermal growth factor receptor 2 (HER2) CEACAM7 (Carcinoembryonic antigen-related cell adhesion molecule 7) Immune Checkpoint Inhibitors PD-1/PD-L1 Mesothelin | HER-2 positive tumors Exclusive expression in PDAC tumors PD-1/PD-L1 overexpression Mesothelin high PDAC | 33742099; 2021 [59] 30121627; 2019 [60] 33479048; 2021 [61] 32637575; 2020 [62] 29859625; 2018 [63] 28929447; 2017 [64] |
| Compound | Treatment Regime | Subtype Specific Patient Stratification | Clinical Phase | NCT-Number |
|---|---|---|---|---|
| CPI-613 (Devimistat) | FOLFIRINOX | No | Phase 1/2 Recruiting | NCT03699319 |
| Mitazalimab (CD40 agonist) | mFOLFIRINOX | No | Phase1b/2 Not yet recruiting | NCT04888312 |
| LOAd703 (oncolytic adenovirus encoding TMZ-CD40L) | Gemcitabine+nab-paclitaxel+/-atezolizumab (anti PD-L1) | No | Phase 1/2a Recruiting | NCT02705196 |
| CART-meso cells | / | No | Not applicable | NCT03638193 |
| / | No | Phase 1 | NCT03323944 | |
| Anti-CEA CAR-T cells | Systemic chemotherapy regimens | CEA-expressing PDAC with livermet | Phase 2b Recruiting | NCT04037241 |
| / | CEA expressing tumors | Phase1/2 Recruiting | NCT04348643 | |
| GVAX | Nivolumab, Ipilimumab, Cyclophosphamide, CRS-207 | No | Phase 2 Recruiting | NCT03190265 |
| Epacadostat, Pembrolizumab, CRS-207, Cyclophosphamide | No | Phase 2 Recruiting | NCT03006302 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dujardin, P.; Baginska, A.K.; Urban, S.; Grüner, B.M. Unraveling Tumor Heterogeneity by Using DNA Barcoding Technologies to Develop Personalized Treatment Strategies in Advanced-Stage PDAC. Cancers 2021, 13, 4187. https://doi.org/10.3390/cancers13164187
Dujardin P, Baginska AK, Urban S, Grüner BM. Unraveling Tumor Heterogeneity by Using DNA Barcoding Technologies to Develop Personalized Treatment Strategies in Advanced-Stage PDAC. Cancers. 2021; 13(16):4187. https://doi.org/10.3390/cancers13164187
Chicago/Turabian StyleDujardin, Philip, Anna K. Baginska, Sebastian Urban, and Barbara M. Grüner. 2021. "Unraveling Tumor Heterogeneity by Using DNA Barcoding Technologies to Develop Personalized Treatment Strategies in Advanced-Stage PDAC" Cancers 13, no. 16: 4187. https://doi.org/10.3390/cancers13164187
APA StyleDujardin, P., Baginska, A. K., Urban, S., & Grüner, B. M. (2021). Unraveling Tumor Heterogeneity by Using DNA Barcoding Technologies to Develop Personalized Treatment Strategies in Advanced-Stage PDAC. Cancers, 13(16), 4187. https://doi.org/10.3390/cancers13164187