
Epigenetic Changes Affecting the Development of Hepatocellular Carcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
2. HCC Etiology and Prevalence
3. DNA Methylation Pattern in HCC
4. Function of DNA Methyltransferases in HCC
5. Histone Modifications
6. Non-Coding RNAs
7. N6-Methyladenosine mRNA Modification
8. HCC Risk Factors and Epigenetics
9. Liver Cancer Stem Cells
10. Targeted Therapies
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Roayaie, S.; Konstadoulakis, M. Strategies for the management of hepatocellular carcinoma. Nat. Clin. Pract. Oncol. 2007, 4, 424–432. [Google Scholar] [CrossRef] [PubMed]
- El–Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of Viral Hepatitis and Hepatocellular Carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Wolk, A. Overweight, obesity and risk of liver cancer: A meta-analysis of cohort studies. Br. J. Cancer 2007, 97, 1005–1008. [Google Scholar] [CrossRef]
- Kalra, M.; Mayes, J.; Assefa, S.; Kaul, A.K.; Kaul, R. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 5945–5961. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counter-parts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. Mechanisms underlying epigenetically mediated gene silencing in cancer. Semin. Cancer Biol. 2002, 12, 331–337. [Google Scholar] [CrossRef]
- Hama, N.; Totoki, Y.; Miura, F.; Tatsuno, K.; Saito-Adachi, M.; Nakamura, H.; Arai, Y.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Epigenetic landscape influences the liver cancer genome architecture. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.-S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Investig. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.P. Epigenetic mechanisms involved in the pathogenesis of hepatobiliary malignancies. Epigenomics 2010, 2, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, C.T.; Li, H.-M.; Lo, K.W.; Leow, C.K.; Chan, J.Y.; Hin, L.-Y.; Lau, W.Y.; Lai, P.B.-S.; Lim, B.K.; Huang, J.; et al. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene 1999, 18, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Newell, P.; Toffanin, S.; Villanueva, A.; Chiang, D.; Minguez, B.; Cabellos, L.; Savic, R.; Hoshida, Y.; Lim, K.H.; Melgar-Lesmes, P.; et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J. Hepatol. 2009, 51, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Cho, S.B.; Lee, W.S.; Park, C.H.; Joo, Y.E.; Kim, H.S.; Choi, S.K.; Rew, J.S.; Lee, J.H.; Kim, S.J. Methylation pattern of DNA repair genes and microsatellite instability in hepatocelluar carcinoma. Korean J. Gastroenterol. 2006, 48, 327–336. [Google Scholar] [PubMed]
- Liang, Y.; Ma, B.; Jiang, P.; Yang, H.-M. Identification of methylation-regulated differentially expressed genes and related pathways in hepatocellular carcinoma: A study based on tcga database and bioinformatics analysis. Front. Oncol. 2021, 11, 2040. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Tu, Y.; Chen, C.; Sun, H.; Wan, C.; Cai, X. DNA methylation biomarkers for hepatocellular carcinoma. Cancer Cell Int. 2018, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Seo, D.; Choi, K.-J.; Andersen, J.B.; Won, M.-A.; Kitade, M.; Gómez-Quiroz, L.E.; Judge, A.D.; Marquardt, J.; Raggi, C.; et al. Antitumor Effects in Hepatocarcinoma of Isoform-Selective Inhibition of HDAC2. Cancer Res. 2014, 74, 4752–4761. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acet-ylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.F.; Cao, X.Y.; Zhu, Y.J.; Wu, Z.R.; Zhuang, X.; Shao, M.Y.; Xu, Q.; Zhou, Y.-J.; Ji, H.-J.; Lu, Q.-R.; et al. Histone deacetylase 3 promotes liver regeneration and liver cancer cells proliferation through signal transduc-er and activator of transcription 3 signaling pathway. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Wu, H.; Yang, T.Y.; Li, Y.; Ye, W.L.; Liu, F.; He, X.S.; Wang, J.-R.; Gan, W.-J.; Li, X.-M.; Zhang, S.; et al. Tumor necrosis factor receptor-associated factor 6 promotes hepatocarcinogenesis by interacting with histone deacetylase 3 to enhance c-Myc gene expression and protein stability. Hepatology 2020, 71, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Zhou, Y.; Zhuang, X.; Wu, Z.; Lu, Y.; Li, S.; Zeng, Y.; Lu, Q.R.; Huo, Y.; Shi, Y.; et al. HDAC3 deficiency promotes liver cancer through a defect in H3K9ac/H3K9me3 transition. Cancer Res. 2019, 79, 3676–3688. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Nie, Q.; Dai, M.; Chen, F.; Wu, H. Histone Deacetylases Inhibit the Snail2-Mediated EMT During Metastasis of Hepatocellular Carcinoma Cells. Front. Cell Dev. Biol. 2020, 8, 752. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-T.; Chiou, S.-S.; Chai, C.-Y.; Hsi, E.; Wang, S.-N.; Huang, S.-K.; Hsu, S.-H. Aryl hydrocarbon receptor regulates histone deacetylase 8 expression to repress tumor suppressive activity in hepatocellular carcinoma. Oncotarget 2017, 8, 7489–7501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Lou, B.; Chen, W.; Zhang, J.; Lin, S.; Lv, F.-F.; Chen, Y. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumor Biol. 2014, 35, 11523–11532. [Google Scholar] [CrossRef]
- Kanki, K.; Watanabe, R.; Thai, L.N.; Zhao, C.-H.; Naito, K. HDAC9 Is Preferentially Expressed in Dedifferentiated Hepatocellular Carcinoma Cells and Is Involved in an Anchorage-Independent Growth. Cancers 2020, 12, 2734. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.M.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Li, F.; Wang, J.; Hu, J.; Li, Z.; Gu, Y.; Feng, Y. miR-369 inhibits Liver Cancer progression by targeting ZEB1 pathway and predicts the prognosis of HCC patients. J. Cancer 2021, 12, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, X.; Zhou, X.; Wu, B.; Zhu, D.; Jia, W.; Chu, J.; Wang, J.; Wu, J.; Kong, L. MiR-3174 promotes proliferation and inhibits apoptosis by targeting FOXO1 in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2020, 526, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, L.; Luo, Z.; Li, W.; Lu, Y.; Tang, Q.; Pu, J. miR-383 inhibits cell growth and promotes cell apoptosis in hepatocellular carcinoma by targeting IL-17 via STAT3 signaling pathway. Biomed. Pharmacother. 2019, 120, 109551. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-J.; Chen, G.-Y.; Xie, Z.-T. MicroRNA-361-5p Inhibits Cancer Cell Growth by Targeting CXCR6 in Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2016, 38, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Yang, Z.; Lou, Y.; Huang, J.; Yang, P.; Jiang, W.; Chen, S. miR-186 Inhibits Liver Cancer Stem Cells Expansion via Targeting PTPN11. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Cheng, Z.; Liu, H.; Wang, X.; Han, T.; Sun, W.; Li, X.; Yang, W.; Chen, C.; Xia, M.; et al. Shp2 promotes liver cancer stem cell expansion by augmenting beta-catenin signaling and predicts chemo-therapeutic response of patients. Hepatology 2017, 65, 1566–1580. [Google Scholar] [CrossRef] [Green Version]
- Thakral, S.; Ghoshal, K. miR-122 is a unique molecule with great potential in diagnosis, prognosis of liver disease, and therapy both as miRNA mimic and antimir. Curr. Gene Ther. 2015, 15, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Lanzafame, M.; Bianco, G.; Terracciano, L.M.; Ng, C.K.Y.; Piscuoglio, S. The Role of Long Non-Coding RNAs in Hepatocarcinogenesis. Int. J. Mol. Sci. 2018, 19, 682. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Yang, L.Y.; Xu, Z.P. Long non-coding RNA HOTTIP is able to predict poor prognosis in various neoplasms: A me-ta-analysis. Mol. Clin. Oncol. 2017, 7, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhou, L.; Li, H.; Sun, T.; Wen, X.; Li, X.; Meng, Y.; Li, Y.; Liu, M.; Liu, S.; et al. Nuclear-Encoded lncRNA MALAT1 Epigenetically Controls Metabolic Reprogramming in HCC Cells through the Mitophagy Pathway. Mol. Ther.-Nucleic Acids 2021, 23, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Lin, Z.; Li, X.; Xin, X.; An, J.; Zheng, Q.; Yang, Y.; Lu, D. HULC cooperates with MALAT1 to aggravate liver cancer stem cells growth through telomere repeat-binding factor 2. Sci. Rep. 2016, 6, 36045. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, Y.; Ma, L. Long noncoding RNA highly upregulated in liver cancer promotes the progression of hepatocellular carcinoma and attenuates the chemosensitivity of oxaliplatin by regulating miR-383-5p/vesicle-associated membrane pro-tein-2 axis. Pharmacol. Res. Perspect. 2021, 9, e00815. [Google Scholar] [CrossRef]
- Yan, C.; Wei, S.; Han, D.; Wu, L.; Tan, L.; Wang, H.; Dong, Y.; Hua, J.; Yang, W. LncRNA HULC shRNA disinhibits miR-377-5p to suppress the growth and invasion of hepatocellular carci-noma in vitro and hepatocarcinogenesis in vivo. Ann. Transl. Med. 2020, 8, 1294. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yang, T.; He, W.; Jiang, S.; Zhong, D.; Xu, Z.; Wei, Q.; Zhang, Y.; Shi, C. Liver X receptor inhibits the growth of hepatocellular carcinoma cells via regulating HULC/miR-134-5p/FOXM1 axis. Cell. Signal. 2020, 74, 109720. [Google Scholar] [CrossRef] [PubMed]
- Matsukura, S.; Soejima, H.; Nakagawachi, T.; Yakushiji, H.; Ogawa, A.; Fukuhara, M.; Miyazaki, K.; Nakabeppu, Y.; Sekiguchi, M.; Mukai, T. CpG methylation of MGMT and hMLH1 promoter in hepatocellular carcinoma associated with hepatitis viral infection. Br. J. Cancer 2003, 88, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischoff, I.; Tannapfe, A. DNA methylation in hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Peng, B.; Tang, Y.; Qian, Y.; Guo, P.; Li, M.; Luo, J.; Chen, B.; Tang, H.; Lu, C.; et al. CpG methylation signature predicts recurrence in early-stage hepatocellular carcinoma: Results from a multicenter study. J. Clin. Oncol. 2017, 35, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Tong, W.; Xie, F.; Zhu, L.; Wu, H.; Shi, R.; Wang, L.; Yang, L.; Liu, Z.; Miao, F.; et al. DNA methylation biomarkers for diagnosis of primary liver cancer and distinguishing hepatocellular carcinoma from intrahepatic cholangiocarcinoma. Aging 2021, 13, 17592–17606. [Google Scholar] [CrossRef]
- Zopf, S.; Ocker, M.; Neureiter, D.; Alinger, B.; Gahr, S.; Neurath, M.F.; Di Fazio, P. Inhibition of DNA methyltransferase activity and expression by treatment with the pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma cell lines. BMC Cancer 2012, 12, 386. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kanai, Y.; Nakagawa, T.; Sakamoto, M.; Saito, H.; Ishii, H.; Hirohashi, S. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malig-nant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer 2003, 105, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.-K.; Kim, H.; Park, H.-J.; Shim, Y.-H.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 2007, 20, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturelli, S.; Berger, A.; Weiland, T.; Zimmermann, M.; Häcker, S.; Peter, C.; Wesselborg, S.; Königsrainer, A.; Weiss, T.; Gregor, M.; et al. Dual antitumour effect of 5-azacytidine by inducing a breakdown of resistance-mediating factors and epigenetic modulation. Gut 2011, 60, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous Activation of Ras and Jak/Stat Pathways in Human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Li, Y.; Pandit, H.; Li, S.; Pulliam, Z.; Zheng, Q.; Yu, Y.; Martin, R.C. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell. Immunol. 2019, 336, 66–74. [Google Scholar] [CrossRef]
- Bannister, A.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Ler, S.Y.; LEuNG, C.H.W.; Khin, L.W.; Lu, G.D.; Salto-Tellez, M.; Hartman, M.; Iau, P.T.C.; Yap, C.T.; Hooi, S.C. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk re-gression model in a Southeast Asian population. Oncol. Rep. 2015, 34, 2238–2250. [Google Scholar] [CrossRef] [Green Version]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, L.; Tao, S.; Dai, M.; Wang, Y.; Li, Y.; Wu, J. Clinical significance of HDAC9 in hepatocellular carcinoma. Cell. Mol. Biol. 2019, 65, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Han, H.D.; Lopez-Berestein, G.; Sood, A.K. MicroRNA therapeutics: Principles, expectations, and challenges. Chin. J. Cancer 2011, 30, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Li, Y.; Xu, K.; Chen, G.; Lu, X.; Duan, Q.; Kang, Z. RETRACTED: miR-361-5p inhibits hepatocellular carcinoma cell proliferation and invasion by targeting VEGFA. Biochem. Biophys. Res. Commun. 2016, 479, 901–906. [Google Scholar] [CrossRef]
- Ren, Q.; Xiao, X.; Leng, X.; Zhang, Q.; Zhou, X.; Ren, Z.; Xiao, H. MicroRNA-361-5p induces hepatocellular carcinoma cell apoptosis and enhances drug sensitivity by targeting MAP3K9. Exp. Ther. Med. 2021, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Kogure, T.; Nuovo, G.J.; Jiang, J.; He, L.; Kim, J.H.; Phelps, M.A.; Papenfuss, T.L.; Croce, C.M.; Patel, T.; et al. miR-221 Silencing Blocks Hepatocellular Carcinoma and Promotes Survival. Cancer Res. 2011, 71, 7608–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, S.; Hu, Q.; Wu, W.; Wang, M.; Huang, J.; Zhao, X.; Tang, G.; Liang, T. Tumor-triggered personalized microRNA cocktail therapy for hepatocellular carcinoma. Biomater. Sci. 2020, 8, 6579–6591. [Google Scholar] [CrossRef]
- Stiuso, P.; Potenza, N.; Lombardi, A.; Ferrandino, I.; Monaco, A.; Zappavigna, S.; Vanacore, D.; Mosca, N.; Castiello, F.; Porto, S.; et al. MicroRNA-423-5p Promotes Autophagy in Cancer Cells and Is Increased in Serum From Hepatocarcinoma Patients Treated With Sorafenib. Mol. Ther.-Nucleic Acids 2015, 4, e233. [Google Scholar] [CrossRef]
- Hsu, S.H.; Wang, B.; Kutay, H.; Bid, H.; Shreve, J.; Zhang, X.; Costinean, S.; Bratasz, A.; Houghton, P.; Ghoshal, K. Hepatic loss of miR-122 predisposes mice to hepatobiliary cyst and hepatocellular carcinoma upon diethyl-nitrosamine exposure. Am. J. Pathol. 2013, 183, 1719–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Tsang, F.H.; Au, S.L.; Wei, L.; Fan, D.N.; Lee, J.M.; Wong, C.C.L.; Ng, I.O.; Wong, C.-M. Long non-coding RNA HOTTIP is frequently up-regulated in hepatocellular carcinoma and is targeted by tumour suppressive miR-125b. Liver Int. 2015, 35, 1597–1606. [Google Scholar] [CrossRef]
- Luo, F.; Sun, B.; Li, H.; Xu, Y.; Liu, Y.; Liu, X.; Lu, L.; Li, J.; Wang, Q.; Wei, S.; et al. A MALAT1/HIF-2α feedback loop contributes to arsenite carcinogenesis. Oncotarget 2016, 7, 5769–5787. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Yang, Z.; Zhou, L.; Zhu, Q.Q.; Xie, H.Y.; Zhang, F.; Wu, L.-M.; Chen, L.-M.; Zheng, S.-S. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med. Oncol. 2012, 29, 1810–1816. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Gholipour, M.; Hussen, B.M.; Taheri, M. The Impact of Long Non-Coding RNAs in the Pathogenesis of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 1150. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.I.; Lin, Y.-Y.; Macaes, B.; Meneshian, A.; Hung, C.-F.; Wu, T.-C. Prospects of RNA interference therapy for cancer. Gene Ther. 2006, 13, 464–477. [Google Scholar] [CrossRef]
- Shen, S.; Yan, J.; Zhang, Y.; Dong, Z.; Xing, J.; He, Y. N6-methyladenosine (m6A)-mediated messenger RNA signatures and the tumor immune microenvironment can predict the prognosis of hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Lan, Y.; Zhao, Y.; Shi, Y.; Jin, J.; Xie, W. The emerging roles of N6-methyladenosine RNA methylation in human cancers. Biomark. Res. 2020, 8, 1–16. [Google Scholar] [CrossRef]
- Park, I.Y.; Sohn, B.H.; Yu, E.; Suh, D.J.; Chung, Y.; Lee, J.; Surzycki, S.J.; Lee, Y.I. Aberrant Epigenetic Modifications in Hepatocarcinogenesis Induced by Hepatitis B Virus X Protein. Gastroenterology 2007, 132, 1476–1494. [Google Scholar] [CrossRef]
- Zhao, P.; Malik, S.; Xing, S. Epigenetic Mechanisms Involved in HCV-Induced Hepatocellular Carcinoma (HCC). Front. Oncol. 2021, 11, 2752. [Google Scholar] [CrossRef]
- Domovitz, T.; Gal-Tanamy, M. Tracking Down the Epigenetic Footprint of HCV-Induced Hepatocarcinogenesis. J. Clin. Med. 2021, 10, 551. [Google Scholar] [CrossRef]
- Quan, H.; Zhou, F.; Nie, D.; Chen, Q.; Cai, X.; Shan, X.; Zhou, Z.; Chen, K.; Huang, A.; Li, S.; et al. Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial-mesenchymal transition. Oncogene 2014, 33, 2826–2835. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, N.N.; ul Haq, A.; Siddiqui, O.A.; Khan, R. DNA methyltransferase 1, 3a, and 3b expression in hepatitis C associated human hepatocellular carci-noma and their clinicopathological association. Tumour Biol. 2016, 37, 10487–10497. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Kim, E.O.; Jung, J.K.; Jang, K.L. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransfer-ase 1 and 3b. Cancer Lett. 2008, 261, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.L.; Heo, S.; Jang, K.L. Hepatitis C virus core protein overcomes H2O2-induced apoptosis by downregulating p14 expression via DNA methylation. J. Gen. Virol. 2015, 96, 822–832. [Google Scholar] [CrossRef]
- Park, S.H.; Lim, J.S.; Lim, S.Y.; Tiwari, I.; Jang, K.L. Hepatitis C virus core protein stimulates cell growth by down-regulating p16 expression via DNA methyl-ation. Cancer Lett. 2011, 310, 61–68. [Google Scholar]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Downregulation of Gadd45beta expression by hepatitis C virus leads to defective cell cycle arrest. Cancer Res. 2010, 70, 4901–4911. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Holmes, J.A.; Dai, C.-Y.; Huang, C.-F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meroni, M.; Longo, M.; Rametta, R.; Dongiovanni, P. Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. Int. J. Mol. Sci. 2018, 19, 3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stem-ness and miR-122-mediated HIF-1alpha activation. Sci. Rep. 2016, 6, 21340. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Johnson, N.D.; Wu, X.; Still, C.D.; Chu, X.; Petrick, A.T.; Gerhard, G.S.; Conneely, K.N.; DiStefano, J.K. Differential DNA methylation and changing cell-type proportions as fibrotic stage progresses in NAFLD. Clin. Epigenetics 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Gerhard, G.S.; Malenica, I.; Llaci, L.; Chu, X.; Petrick, A.T.; Still, C.D.; Distefano, J.K. Differentially methylated loci in NAFLD cirrhosis are associated with key signaling pathways. Clin. Epigenetics 2018, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef]
- Haraguchi, N.; Utsunomiya, T.; Inoue, H.; Tanaka, F.; Mimori, K.; Barnard, G.F.; Mori, M. Characterization of a Side Population of Cancer Cells from Human Gastrointestinal System. Stem Cells 2006, 24, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar] [CrossRef]
- Ma, S.; Tang, K.H.; Chan, Y.P.; Lee, K.W.; Kwan, P.S.; Castilho, A.; Ng, I.O.-L.; Man, K.; Wong, N.; To, K.-F.; et al. miR-130b Promotes CD133+ Liver Tumor-Initiating Cell Growth and Self-Renewal via Tumor Protein 53-Induced Nuclear Protein 1. Cell Stem Cell 2010, 7, 694–707. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.S.; Hur, W.; Kim, T.-K.; Hong, S.W.; Kim, S.W.; Choi, J.E.; Sung, P.S.; Song, M.J.; Lee, B.-C.; Hwang, D.; et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012, 315, 129–137. [Google Scholar] [CrossRef]
- Lan, X.; Wu, Y.Z.; Wang, Y.; Wu, F.R.; Zang, C.B.; Tang, C.; Cao, S.; Li, S.-L. CD133 silencing inhibits stemness properties and enhances chemoradiosensitivity in CD133-positive liver can-cer stem cells. Int. J. Mol. Med. 2013, 31, 315–324. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, J.; Tsui, Y.M.; Shi, C.; Wang, Y.; Zhang, X.; Yan, Q.; Chen, M.; Jiang, C.; Yuan, Y.-F.; et al. RALYL increases hepatocellular carcinoma stemness by sustaining the mRNA stability of TGF-beta2. Nat. Commun. 2021, 12, 1–14. [Google Scholar]
- De Mattia, E.; Cecchin, E.; Guardascione, M.; Foltran, L.; Di Raimo, T.; Angelini, F.; D’Andrea, M.; Toffoli, G. Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3870–3896. [Google Scholar] [CrossRef]
- Thillai, K.; Srikandarajah, K.; Ross, P. Regorafenib as treatment for patients with advanced hepatocellular cancer. Futur. Oncol. 2017, 13, 2223–2232. [Google Scholar] [CrossRef]
- Bakouny, Z.; Assi, T.; El Rassy, E.; Nasr, F. Second-line treatments of advanced hepatocellular carcinoma: Systematic review and network meta-analysis of randomized controlled trials. J. Clin. Gastroenterol. 2019, 53, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anti-cancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar] [PubMed]
- Bitzer, M.; Horger, M.; Giannini, E.; Ganten, T.M.; Wörns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Soukupova, J.; Bertran, E.; Peñuelas-Haro, I.; Urdiroz-Urricelqui, U.; Borgman, M.; Kohlhof, H.; Fabregat, I. Resminostat induces changes in epithelial plasticity of hepatocellular carcinoma cells and sensitizes them to sorafenib-induced apoptosis. Oncotarget 2017, 8, 110367–110379. [Google Scholar] [CrossRef] [Green Version]
- Streubel, G.; Schrepfer, S.; Kallus, H.; Parnitzke, U.; Wulff, T.; Hermann, F.; Borgmann, M.; Hamm, S. Histone deacetylase inhibitor resminostat in combination with sorafenib counteracts platelet-mediated pro-tumoral effects in hepatocellular carcinoma. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Tak, W.Y.; Ryoo, B.-Y.; Lim, H.Y.; Kim, D.-Y.; Okusaka, T.; Ikeda, M.; Hidaka, H.; Yeon, J.-E.; Mizukoshi, E.; Morimoto, M.; et al. Phase I/II study of first-line combination therapy with sorafenib plus resminostat, an oral HDAC inhibitor, versus sorafenib monotherapy for advanced hepatocellular carcinoma in east Asian patients. Investig. New Drugs 2018, 36, 1072–1084. [Google Scholar] [CrossRef]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.-W.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhib-itor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Yeo, W.; Chung, H.; Chan, S.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y.; et al. Epigenetic Therapy Using Belinostat for Patients With Unresectable Hepatocellular Carcinoma: A Multicenter Phase I/II Study With Biomarker and Pharmacokinetic Analysis of Tumors From Patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, J.; Rao, S. Epigenetics and immunotherapy: The current state of play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef]
- Shin, S.; Kim, M.; Lee, S.-J.; Park, K.-S. Trichostatin A Sensitizes Hepatocellular Carcinoma Cells to Enhanced NK Cell-mediated Killing by Regulating Immune-related Genes. Cancer Genom. Proteom. 2017, 14, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Mufti, G.J.; Hellström-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.; Gore, S.; Seymour, J.F.; et al. Azacitidine Prolongs Overall Survival Compared With Conventional Care Regimens in Elderly Patients With Low Bone Marrow Blast Count Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Li, H.; Hinoue, T.; Zhou, W.; Ohtani, H.; El-Khoueiry, A.; Daniels, J.; O’Connell, C.; Dorff, T.B.; et al. Integrative epigenetic analysis reveals therapeutic targets to the dna methyltransferase inhibitor guadecitabine (SGI-110) in hepatocellular carcinoma. Hepatology 2018, 68, 1412–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, Y.; El-Khoueiry, A.; Taverna, P.; Ljungman, M.; Neamati, N. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol. Oncol. 2015, 9, 1799–1814. [Google Scholar] [CrossRef] [PubMed]
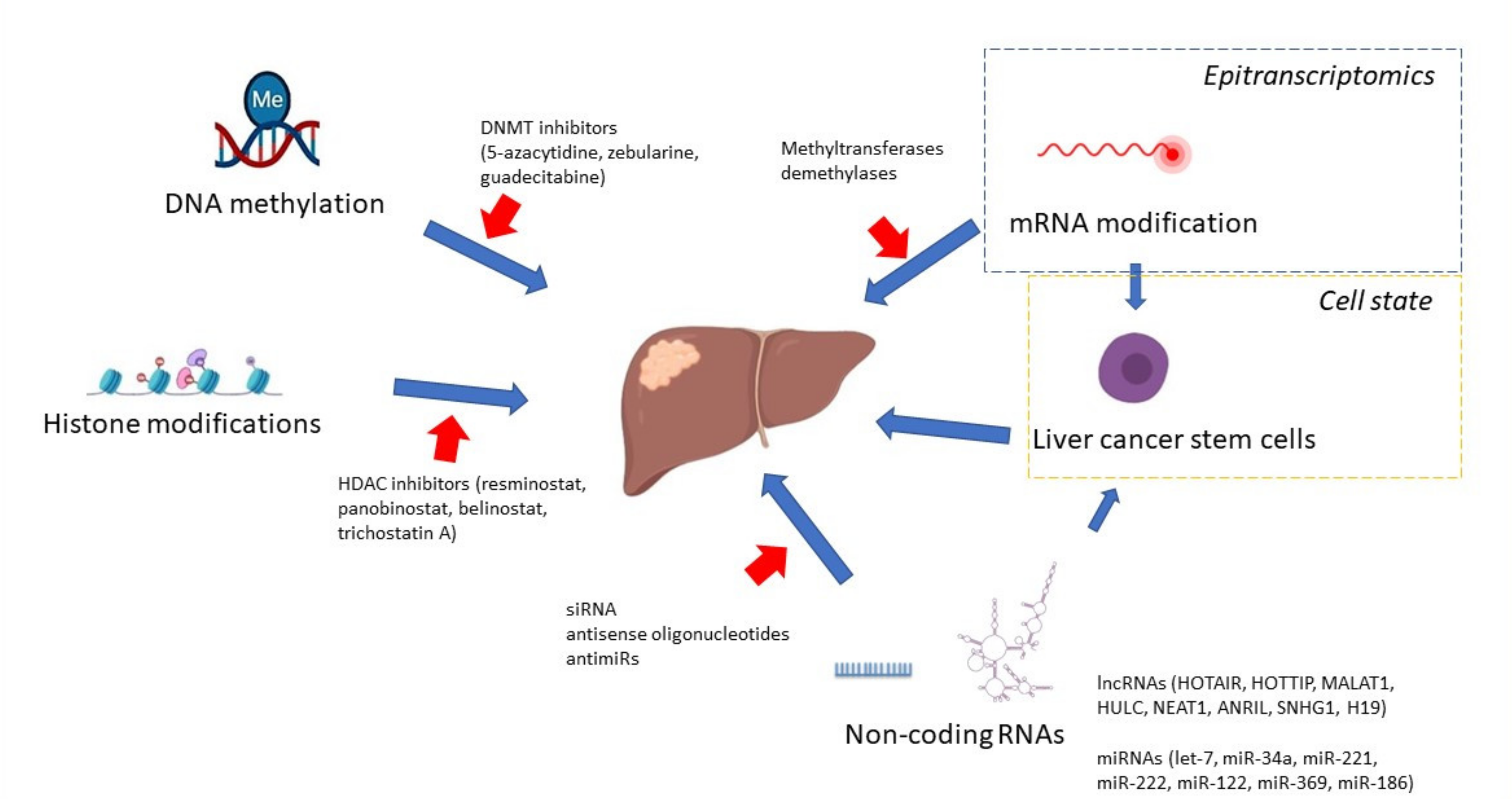

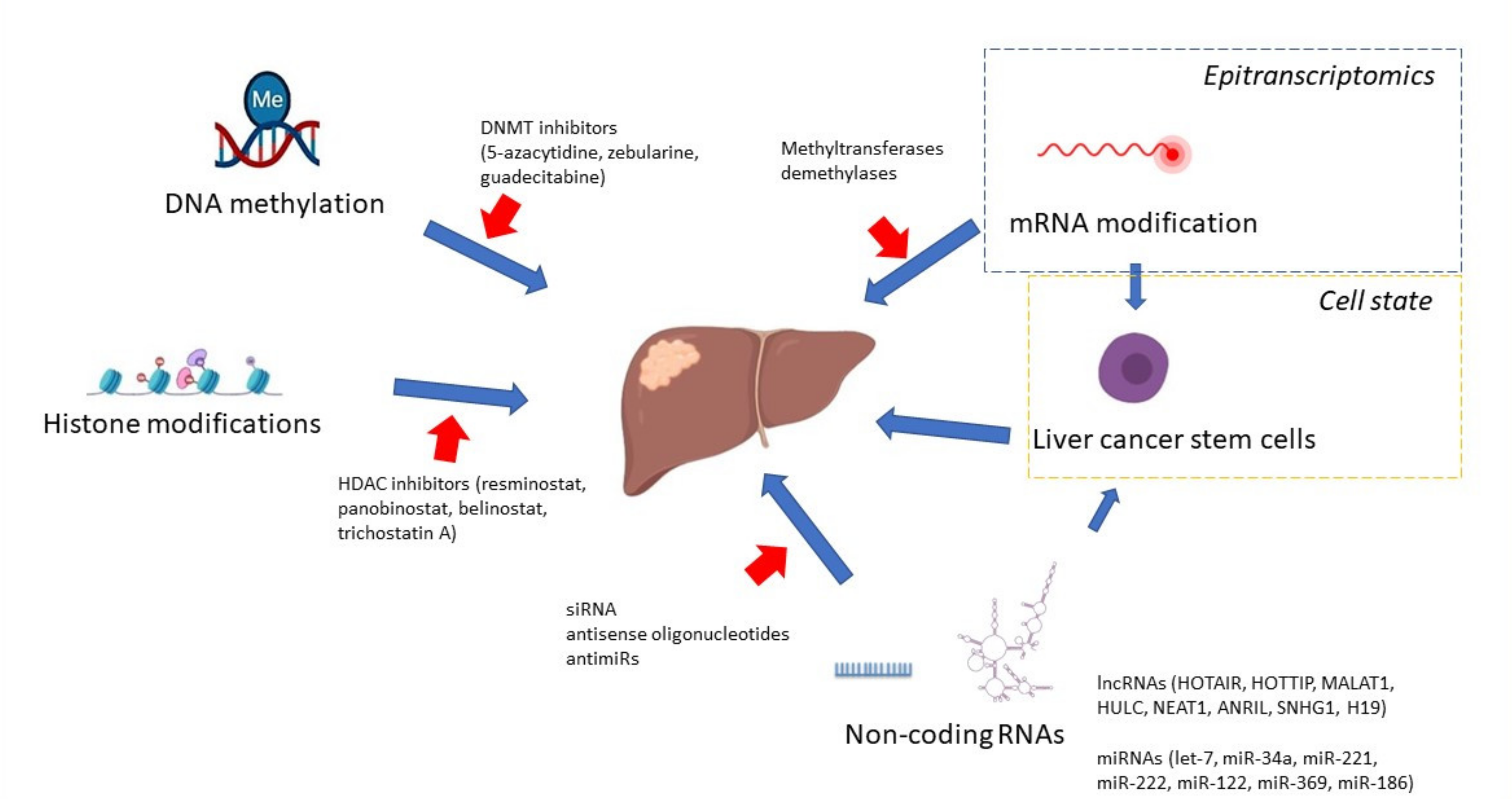{kind=link}
| Epigenetic Changes | Mechanism Affected | Reference |
|---|---|---|
| DNA methylation | ||
| Hypomethylation | Protooncogene c-Jun and c-myc activation | [12] |
| Mitotic recombination/genomic instability | [12] | |
| Hypermethylation | WNT/β-catenin signaling activation | [13] |
| APC inactivation | [13] | |
| p16INK4A activation | [14] | |
| RASSF1A and NORE1A activation | [15] | |
| Mismatch repair system genes (hMLH1, hMSH2, and hMSH3) inactivation | [16] | |
| Cardiotrophin-1 (CTF1), FZD8, pyruvate dehydrogenase kinase 4 (PDK4), and ZNF334 activity | [17] | |
| MAD2L1, CDC20, CCNB1, CCND1, AR, and ESR1 | [18] | |
| p53 and MAPK signaling regulation | [18] | |
| Histone modification | ||
| Upregulated HDAC2 | Dysregulation of cell cycle, apoptosis, and differentiation via p27, p53, BCL-2, or PPAR γ | [19] |
| Downregulated HDAC3 | An increase in p21WAF1/cip1 expression; G1-phase arrest | [20] |
| Downregulated HDAC3 | STAT3-dependent cell proliferation | [21] |
| Downregulated HDAC3 | c-Myc protein synthesis and stability | [22] |
| Downregulated HDAC3 | Defective double-strand breaks repair | [23] |
| HDAC3 and HDAC1 | Cell migration, epithelial-mesenchymal transition (EMT), and tumor metastasis regulation | [24] |
| Upregulated HDAC8 | Downregulation of RB1 | [25] |
| Upregulated HDAC5 | Increased cell proliferation | [26] |
| Downregulation of HDAC5 | Cell apoptosis via antiapoptotic proteins (p53, bax, bcl-2, cyto C, and caspase 3), G1-phase cell cycle arrest via cell cycle regulators (cyclin D1 and CDK2/4/6) | [26] |
| Upregulated HDAC9 | Epithelial–mesenchymal transition process activation; cellular stemness properties regulation | [27] |
| Non-coding RNAs | ||
| miR-221/222 | Enhanced cell growth via p27 regulation mTOR kinase regulation | [28] |
| miR-369 | Zinc finger E-box binding homeobox 1 regulation | [29] |
| miR-3174 | FOXO1 regulation | [30] |
| miR-383 | IL-17 via STAT3 signaling pathway regulation | [31] |
| miR-361-5p | CXCR6, VEGFA, or MAP3K9 regulation | [32] |
| miR-186 | CSCs self-renewal | [33] |
| miR-186 | Protein tyrosine phosphatase non-receptor type 11 regulation | [34] |
| miR-122 | Tumor growth regulation | [35] |
| HOTAIR | Proliferation, regulation of pluripotency, metastasis, and sensitivity to chemotherapeutics | [36] |
| HOTTIP | Survival, tumor grade, and prognosis | [37] |
| MALAT1 | Regulation of mitochondrial metabolism | [38] |
| HULC | Growth of liver cancer stem cells | [39] |
| Chemosensitivity of anti-cancer drug oxaliplatin inhibition | [40] | |
| Regulation of miR-383-5p/vesicle-associated membrane protein-2 pathway; miR-377-5p/HIF-1α pathway and miR-134-5p/FOXM1 pathway | [41,42] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolinska, E.; Skrzypczak, M. Epigenetic Changes Affecting the Development of Hepatocellular Carcinoma. Cancers 2021, 13, 4237. https://doi.org/10.3390/cancers13164237
Wolinska E, Skrzypczak M. Epigenetic Changes Affecting the Development of Hepatocellular Carcinoma. Cancers. 2021; 13(16):4237. https://doi.org/10.3390/cancers13164237
Chicago/Turabian StyleWolinska, Ewa, and Maciej Skrzypczak. 2021. "Epigenetic Changes Affecting the Development of Hepatocellular Carcinoma" Cancers 13, no. 16: 4237. https://doi.org/10.3390/cancers13164237
APA StyleWolinska, E., & Skrzypczak, M. (2021). Epigenetic Changes Affecting the Development of Hepatocellular Carcinoma. Cancers, 13(16), 4237. https://doi.org/10.3390/cancers13164237