
Targeting DNA Damage Repair Mechanisms in Pancreas Cancer

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
Aim and Scope
2. Materials and Methods
Composition of the Expert Panel and Recommendations
3. Results
3.1. The Incidence of Germline BRCA1/2 Mutations
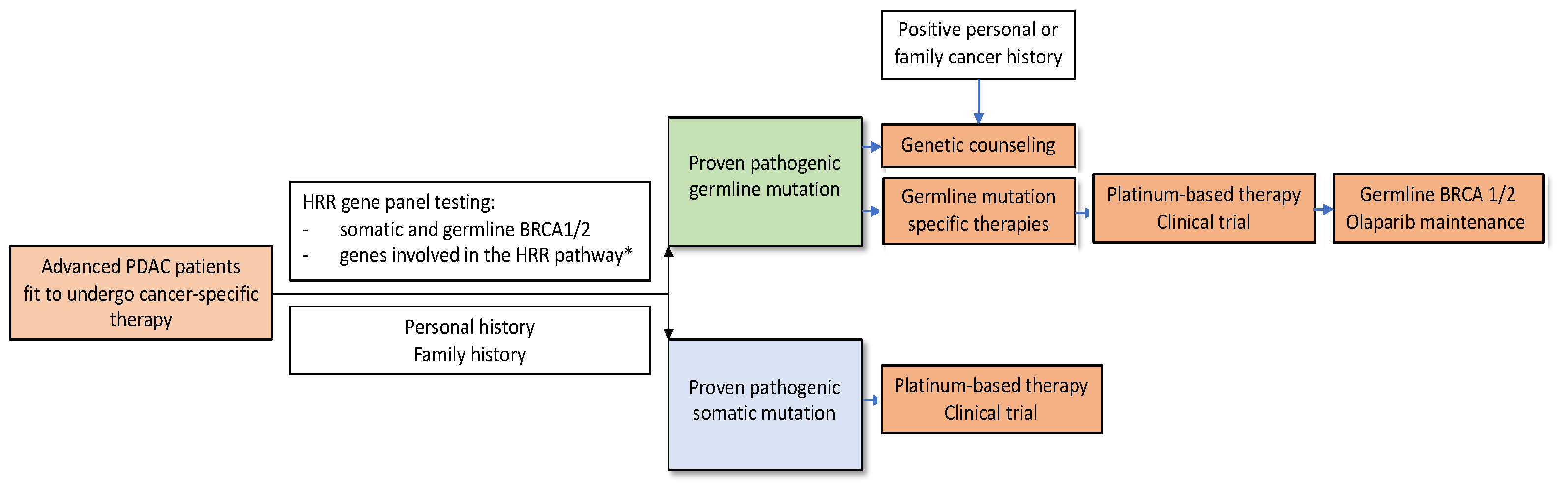
3.2. BRCA1/2 Testing Recommendations
3.3. Genetic Counselling for Patients with a Germline BRCA1/2 Mutation
3.4. Analysing Homologous Recombination Repair Pathways
3.5. Treatment of PDAC Patients with BRCA1/2 Mutations
3.6. Treatment of PDAC Patients with Other Mutations in DDR Genes
3.7. Acquired Resistance to PARP Inhibitor Therapy
3.8. Future Directions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef]
- Rainone, M.; Singh, I.; Salo-Mullen, E.E.; Stadler, Z.K.; O’Reilly, E.M. An Emerging Paradigm for Germline Testing in Pancreatic Ductal Adenocarcinoma and Immediate Implications for Clinical Practice: A Review. JAMA Oncol. 2020, 6, 764–771. [Google Scholar] [CrossRef]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; O‘Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021, 160, 2119–2132.e2119. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.; Perkhofer, L.; Morawe, M.; Arnold, F.; Ihle, M.; Biber, S.; Lange, S.; Roger, E.; Kraus, J.M.; Stifter, K.; et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2021, 70, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Wang, H.; Parenti, S.; He, A.R.; Hwang, J.J.; Ley, L.; Difebo, H.; Smaglo, B.G.; Kim, S.S.; Weinberg, B.A.; et al. Final report of a phase I/II study of veliparib (Vel) in combination with 5-FU and oxaliplatin (FOLFOX) in patients (pts) with metastatic pancreatic cancer (mPDAC). J. Clin. Oncol. 2019, 37, 4015. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis Oncol. 2018, 2018. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer. Res. 2014, 34, 471–476. [Google Scholar]
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer With DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol. 2021, 10.1001/jamaoncol.2021.0006. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Kindler, H.L.; Park, J.O.; Reni, M.; Macarulla, T.; Hammel, P.; Van Cutsem, E.; Arnold, D.; Hochhauser, D.; McGuinness, D.; et al. Geographic and Ethnic Heterogeneity of Germline BRCA1 or BRCA2 Mutation Prevalence among Patients with Metastatic Pancreatic Cancer Screened for Entry Into the POLO Trial. J. Clin. Oncol. 2020, 38, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.B.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet. Med. 2015, 17, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients with Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef]
- Eldridge, R.C.; Gapstur, S.M.; Newton, C.C.; Goodman, M.; Patel, A.V.; Jacobs, E.J. Jewish ethnicity and pancreatic cancer mortality in a large U.S. cohort. Cancer Epidemiol. Biomark. Prev. 2011, 20, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Salo-Mullen, E.E.; O’Reilly, E.M.; Kelsen, D.P.; Ashraf, A.M.; Lowery, M.A.; Yu, K.H.; Reidy, D.L.; Epstein, A.S.; Lincoln, A.; Saldia, A.; et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 2015, 121, 4382–4388. [Google Scholar] [CrossRef]
- Stadler, Z.K.; Salo-Mullen, E.; Patil, S.M.; Pietanza, M.C.; Vijai, J.; Saloustros, E.; Hansen, N.A.; Kauff, N.D.; Kurtz, R.C.; Kelsen, D.P.; et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer 2012, 118, 493–499. [Google Scholar] [CrossRef]
- Iqbal, J.; Ragone, A.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; Neuhausen, S.L.; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goere, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs. Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef] [PubMed]
- NCT04348045 Phase II Study to Evaluate Maintenance Therapy With Olaparib or Selumetinib Plus Durvalumab According to BRCAness and KRAS Somatic Status Personalized in Metastatic Pancreatic Adenocarcinoma Patients (D19-02). Available online: https://clinicaltrials.gov/ct2/show/NCT04348045 (accessed on 11 September 2020).
- Golan, T.; Barenboim, A.; Lahat, G.; Nachmany, I.; Goykhman, Y.; Shacham-Shmueli, E.; Halpern, N.; Brazowski, E.; Geva, R.; Wolf, I.; et al. Increased Rate of Complete Pathologic Response After Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 3963–3970. [Google Scholar] [CrossRef]
- Okur, V.; Chung, W.K. The impact of hereditary cancer gene panels on clinical care and lessons learned. Cold Spring Harb. Mol. Case Stud. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, S.E.; Nussbaum, R.L.; Kurian, A.W.; Nielsen, S.M.; Das, K.; Michalski, S.; Yang, S.; Ngo, N.; Blanco, A.; Esplin, E.D. Yield and Utility of Germline Testing Following Tumor Sequencing in Patients With Cancer. JAMA Netw. Open 2020, 3, e2019452. [Google Scholar] [CrossRef] [PubMed]
- Fda Approves Liquid Biopsy Ngs Companion Diagnostic Test for Multiple Cancers and Biomarkers. Available online: https://www.esmo.org/oncology-news/fda-approves-liquid-biopsy-ngs-companion-diagnostic-test-for-multiple-cancers-and-biomarkers?hit=mail-snews&utm_campaign=Scientific&utm_medium=email&_hsmi=99701691&_hsenc=p2ANqtz--42fHKGds8l-q3_YSxhBlDVQ5YR6MmMmOWHr9IYpORVqtKmrv_T1-_YowWzoiKZwYnAk1l4sAeN8R71XG_LjCw_i7kXQ&utm_content=99701691&utm_source=hs_email (accessed on 10 November 2020).
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Rahib, L.; Lyons, E.; Arbeloa, P.D.; Hendifar, A.; Mikhail, S.; Chung, V.; Sohal, D.P.S.; et al. Outcomes in Patients With Pancreatic Adenocarcinoma With Genetic Mutations in DNA Damage Response Pathways: Results From the Know Your Tumor Program. JCO Precis. Oncol. 2019, 1–10. [Google Scholar] [CrossRef]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Cutsem, E.V.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. J. Clin. Oncol. 2021, 39, 378. [Google Scholar] [CrossRef]
- FDA Approves Olaparib for gBRCAm Metastatic Pancreatic Adenocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-gbrcam-metastatic-pancreatic-adenocarcinoma (accessed on 27 December 2019).
- Lynparza. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lynparza (accessed on 6 October 2020).
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Guthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, T.; Pentheroudakis, G.; Mishima, S.; Overman, M.J.; Yeh, K.H.; Baba, E.; Naito, Y.; Calvo, F.; Saxena, A.; Chen, L.T.; et al. JSCO-ESMO-ASCO-JSMO-TOS: International expert consensus recommendations for tumour-agnostic treatments in patients with solid tumours with microsatellite instability or NTRK fusions. Ann. Oncol. 2020, 31, 861–872. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Chu, C.; Badve, S.S.; Vinayak, S.; Silver, D.P.; Isakoff, S.J.; Kaklamani, V.; Gradishar, W.; Stearns, V.; Connolly, R.M.; et al. Association of Tumor-Infiltrating Lymphocytes with Homologous Recombination Deficiency and BRCA1/2 Status in Patients with Early Triple-Negative Breast Cancer: A Pooled Analysis. Clin. Cancer Res. 2020, 26, 2704–2710. [Google Scholar] [CrossRef]
- Ricciuti, B.; Recondo, G.; Spurr, L.F.; Li, Y.Y.; Lamberti, G.; Venkatraman, D.; Umeton, R.; Cherniack, A.D.; Nishino, M.; Sholl, L.M.; et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 4135–4142. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. Molecular pathways: How can BRCA-mutated tumors become resistant to PARP inhibitors? Clin. Cancer Res. 2014, 20, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Sella, T.; O’Reilly, E.M.; Katz, M.H.; Epelbaum, R.; Kelsen, D.P.; Borgida, A.; Maynard, H.; Kindler, H.; Friedmen, E.; et al. Overall survival and clinical characteristics of BRCA mutation carriers with stage I/II pancreatic cancer. Br. J. Cancer 2017, 116, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Agarwal, P.; Mamtani, R.; Symecko, H.; Spielman, K.; O’Hara, M.; O’Dwyer, P.J.; Schneider, C.; Teitelbaum, U.; Nathanson, K.L.; et al. Retrospective Survival Analysis of Patients With Resected Pancreatic Ductal Adenocarcinoma and a Germline BRCA or PALB2 Mutation. JCO Precis. Oncol. 2019, 1–11. [Google Scholar] [CrossRef]


{kind=link}
| # | Question |
|---|---|
| Q1 | Do you know the proportion of gBRCA1/2 mutation in PDAC in your area/country? |
| Q2 | Do you regularly determine the BRCA mutation status in patients with PDAC? |
| Q2-1 | If no, explain why not. |
| Q3 | In which situation do you search for BRCA mutation? |
| Q4 | Is your approach different to patients with a suspicion of genetic syndrome and those without any suspicion? |
| Q4-1 | If yes, what is the difference? |
| Q5 | Which material do you use for BRCA analysis? |
| Q6 | Who performs the test? |
| Q7 | What is your acceptable/desirable period of waiting for the results? |
| Q8 | In your opinion, does family history play a role in identifying patients with PDAC and gBRCA1/2 mutation? |
| Q9 | Are patients with gBRCA1/2 mutation regularly referred to a human geneticist in your country? |
| Q10 | For your testing, do you send the patient to the geneticist before or after checking the results positively? |
| Q11 | Do the panels you use comprise other DDR-related genes? |
| Q12 | Do you determine genomic signatures for HRDness in a given PDAC? |
| Q12-1 | If yes, what is the reason? |
| Q13 | Do you pay attention to differences in the BRCA mutational status? |
| Q13-1 | If yes, explain why. Do you think it influences prognosis? |
| Q14 | In the case of a known BRCA mutation, is there a preference for a specific chemotherapy combination? |
| Q15 | Is prolongation of PFS compared to placebo a clinically meaningful endpoint for you? |
| Q16 | Do you treat patients with germline BRCA1/2 mutations with PARP inhibitors as a maintenance treatment? |
| Q17 | Should patients with somatic BRCA1/2 mutations be treated as patients with gBRCA1/2 mutations? |
| Q18 | Do somatic (or germline) mutations in other DDR genes have a therapeutic consequence (e.g., ATM)? |
| Q18-1 | If yes, what is the proposed treatment? |
| Q19 | Do mutations in DDR genes sensitise genes to checkpoint inhibitors? |
| Q20 | Which developments do you foresee in the area of DNA damage repair deficiency in PDAC without gBRCA mutation? |
| Q21 | Do you think we need to consider this BRCA status in a (neo)adjuvant setting for a possible application/trial? |
| “APC” “ATM” “BAP1” “BARD1” “BLM” “BMPR1A” “BRCA1” “BRCA2” “BRIP1” “CBL” “CDC73” “CDH1” “CDK4” “CDKN1B” “CDKN2A” “CHEK2” “CTNNA1” “DICER1” “EPCAM” “ERCC2” “ERCC3” “ERCC4” “ERCC5” “FAM175A” “FANCA” “FANCC” “FANCD2” “FANCE” “FANCF” “FANCG” “FANCI” “FANCL” “FH” “FLCN” “FLT3” “GREM1” “HOXB13” “IDH1” “IDH2” “LZTR1” “MAP2K1” “MAPK1” “MAX” “MEN1” “MITF” “MLH1” “MRE11” “MSH2” “MSH6” “MUTYH” “NBN” “NF1” “NF2” “PALB2” “PMS2” “POLD1” “POLE” “PRKAR1A” “PTCH1” “PTEN” “RAD50” “RAD51C” “RAD51D” “RAF1” “RB1” “RECQL4” “RNF43” “RUNX1” “SDHA” “SDHAF2” “SDHB” “SDHC” “SDHD” “SLX4” “SMAD3” “SMAD4” “SMARCA4” “SMARCB1” “STK11” “SUFU” “TERT” “TGFBR1” “TGFBR2” “TMEM127” “TP53” “TSC1” “TSC2” “VHL” “WT1” “XRCC2” |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perkhofer, L.; Golan, T.; Cuyle, P.-J.; Matysiak-Budnik, T.; Van Laethem, J.-L.; Macarulla, T.; Cauchin, E.; Kleger, A.; Beutel, A.K.; Gout, J.; et al. Targeting DNA Damage Repair Mechanisms in Pancreas Cancer. Cancers 2021, 13, 4259. https://doi.org/10.3390/cancers13174259
Perkhofer L, Golan T, Cuyle P-J, Matysiak-Budnik T, Van Laethem J-L, Macarulla T, Cauchin E, Kleger A, Beutel AK, Gout J, et al. Targeting DNA Damage Repair Mechanisms in Pancreas Cancer. Cancers. 2021; 13(17):4259. https://doi.org/10.3390/cancers13174259
Chicago/Turabian StylePerkhofer, Lukas, Talia Golan, Pieter-Jan Cuyle, Tamara Matysiak-Budnik, Jean-Luc Van Laethem, Teresa Macarulla, Estelle Cauchin, Alexander Kleger, Alica K. Beutel, Johann Gout, and et al. 2021. "Targeting DNA Damage Repair Mechanisms in Pancreas Cancer" Cancers 13, no. 17: 4259. https://doi.org/10.3390/cancers13174259