
Cannabinoids and Endocannabinoid System Changes in Intestinal Inflammation and Colorectal Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
2. Endocannabinoid System and Intestinal Homeostasis
2.1. Introduction to the Cannabinoid System
2.2. Endocannabinoid System
2.3. Normal Intestinal Cells and Their Functions
2.4. Cannabinoids in the Gastrointestinal Tract
2.4.1. Endocannabinoid System in Intact GIT
2.4.2. Cannabinoid Receptors Expression in Intact GIT
2.4.3. The Microbiome and the Cannabinoid System
Gate Keepers/Openers
Modifiers of CB Expression
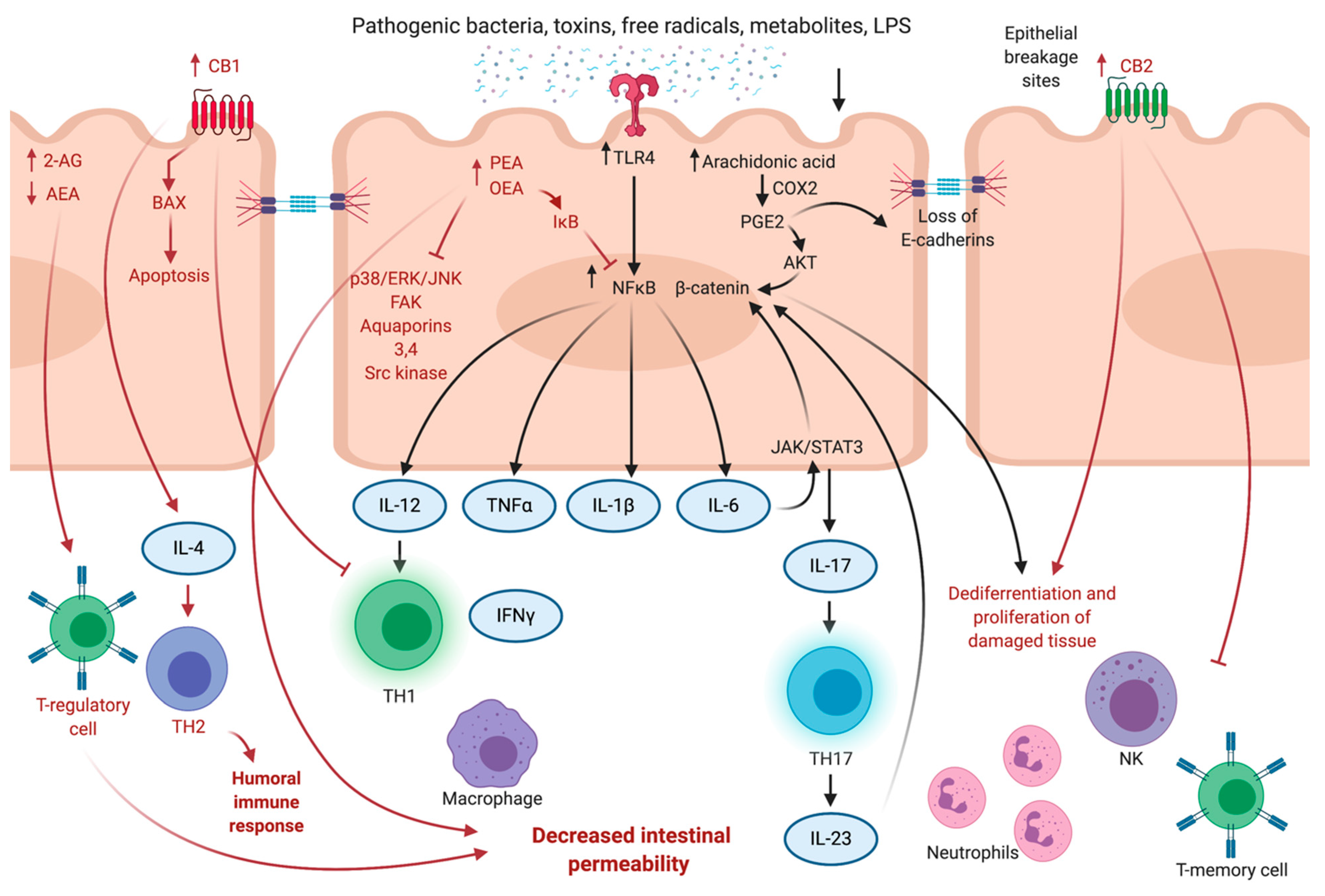
3. Cannabinoids in Intestinal Inflammation
3.1. Colitis and Changes in ECS
3.2. Gatekeeping Mechanisms of ECS
3.3. Anti-Inflammatory Effects of Phytocannabinoids

{kind=link}
{kind=link}
{kind=link}
| Source of Study | Cannabinoid Receptors | Endocannabinoids | Changes in Endocannabinoid Synthesis | Changes in Endocannabinoid Degradation | Methods of Analysis | Effects | References |
|---|---|---|---|---|---|---|---|
| Human intestinal biopsies of CD | CB1, GPR55, GPR119 decreased; PPARδ, TRPV1 increased | OEA elevated | DAGL-α increased | FAAH, NAAA increased | mRNA levels | Correlates with disease severity | [20] |
| Human intestinal biopsies of UC | CB1, CB2, GPR119, PPARα, PPARγ, GPR18, GPR55 decreased; PPARδ, TRPV1 increased | AEA, OEA, and 2-AG elevated | NAPE-PLD decreased | FAAH decreased | mRNA levels | Correlates with disease severity | [20] |
| Human colonic biopsies of UC. Acute mild/moderate colitis | Increased CB2 | - | DAGLα increased, NAPE-PLD decreased | FAAH, MAGL increased | Western blot and immunohistochemistry | CB2 signaling reduces colitis-associated inflammation | [89] |
| Human colonic biopsies of UC Quiescent pancolitis | CB1, CB2 decreased | - | DAGLα decreased, NAPE-PLD elevated | FAAH decreased | Western blot and immunohistochemistry | CB2 signaling reduces colitis-associated inflammation | [89] |
| DNBS, TNBS colitis, human UC biopsies | CB1/CB2 increased | AEA elevated | - | FAAH increased | Chromatography/mass spectrometry | Anti-inflammatory action | [133] |
| DNBS-induced colitis in mice | Increased CB1 expression, and CB1 stimulation | Treatment with CB1 agonist HU210 | - | FAAH experimental genetic ablation | mRNA levels | Alleviates intestinal inflammation | [135] |
| DNBS-induced colitis in mice | TRPV1 and GPR55 downregulation | Increased PEA | NAPE-PLD not changed | NAAA, FAAH not changed | Immunohistochemistry, mRNA, liquid chromatography, and mass spectrometry | Decreased intestinal permeability | [157] |
| DNBS experimental colitis | CB2 stimulation | CBG treatment | - | - | mRNA levels | Anti-inflammatory effect | [166] |
| TNBS-induced colitis in mice | CB2 increased | Addition of CB2 agonists JWH133, AM1241 | - | - | mRNA levels | Protects against inflammation | [136] |
| TNBS- and DSS-induced colitis in mice | Increased PPAR-α | AEA increased; PEA treatment | - | Inhibition of NAAA | HPLC-mass spectrometry, mRNA | Reduction of inflammation | [153] |
| Mustard oil and DSS-induced colitis in mice | CB2 increased expression (higher in mustard oil colitis than in DSS-induced colitis) | CB1, CB2 stimulation with arachidonoyl-chloro-ethanolamide and JWH-133 | - | - | Immunohistochemistry (protein levels) | Alleviates intestinal inflammation | [137] |
| DSS and TNBS-induced colitis in. mice | - | - | - | FAAH inhibition | mRNA levels | Protective on colonic mucosa | [138] |
| DSS-induced colitis in mice | CB1 increased expression | Addition of CB receptor agonists WIN 55,212-2 | - | - | Protein levels | Protective effect on colonic mucosa | [139] |
| TNBS-induced colitis, DSS-induced colitis | - | Addition of AEA | - | Inhibition of FAAH | Microarray analysis, miRNA expression, liquid chromatography/mass spectrometry | Decreased macro- and microscopic signs of colitis | [164,165] |
| TNBS-induced colitis in rats | Inhibition of GPR55, activation of PPAR-γ, TRPV1 | THC, CBD | - | Inhibition of FAAH | - | Anti-inflammatory | [167] |
| Croton oil-induced ileitis in mice | CB1 increased expression | No significant change in AEA and 2-AG levels. Addition of CB receptor agonist CP 55,940 and CBN | - | - | HPLC, protein levels | Reduced intestinal motility | [124] |
| LPS-induced intestinal propulsions | CB2 induction | CB2 induction by JWH-133 | - | - | - | Reduced transit time | [141] |
| LPS-induced colitis and intestinal biopsies from patients with UC | PPAR-γ activation | CBD treatment | - | - | - | Anti-inflammatory, decreased reactive gliosis | [162] |
| Caco-2 CRC cells | OEA acts on TRPV1 and PEA acts on PPAR-α receptor signaling | OEA and PEA treatment | - | Inhibition of FAAH | Liquid chromatography-mass spectrometry | Increased transepithelial electrical resistance and decreased intercellular permeability | [116] |
| Caco-2 CRC cells | CB1 activation | 2-AG treatment | - | Inhibition of FAAH | Liquid chromatography-mass spectrometry | Increased intestinal permeability under inflammation and hypoxia | [163] |
| Human tissue biopsies of IBD patients, HCT-116, HT-29, and Caco-2 CRC cell lines | GPR55 stimulation | High-THCA cannabis extract | - | - | mRNA levels | Anti-inflammatory effect | [34] |
| Human intestinal biopsies from normal mucosa, intestinal adenomas, colorectal carcinomas, CRC cell lines | CB1 and CB2 stimulation | 2-AG, AEA are 2- and 3-fold higher in adenomas and carcinomas | - | Increased FAAH in CRC | Liquid chromatography/mass spectrometry, mRNA levels, western blotting | Anti-cancer effect | [15] |
4. CRC and Colonic Inflammation
5. Changes of the Endocannabinoid System in CRC
5.1. Changes in Cannabinoid Receptors
5.2. Changes in the Level of Endocannabinoids
6. Molecular Mechanisms of Anti-CRC Effects of Cannabinoids
6.1. Ceramide
6.2. Apoptosis
6.3. Extracellular Vesicles
6.4. Autophagy
6.5. CRC Angiogenesis and Metastasis
6.6. Irinotecan and THC
6.7. Cannabis Extracts over Purified Cannabinoids
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoyl glycerol |
| AEA | anandamide |
| ATF4 | activated transcription factor 4 |
| AKT | protein kinase B |
| AMPK | adenosine monophosphate kinase |
| APC mutations | adenomatous polyposis coli mutations |
| APC | antigen-presenting cell |
| cAMP PK | cyclic AMP protein kinase |
| CAC | colitis-associated cancer |
| CB (1 & 2) | cannabinoid receptor (Type 1 & 2) |
| CBD | cannabidiol |
| CBR | cannabinoid receptor |
| CD | Crohn’s disease |
| cdc2 | cell division control 2 |
| CHOP | C/EBP homologous protein |
| CNS | central nervous system |
| COX2 | cyclooxygenase 2 |
| CRC | colorectal cancer |
| CTC | cytotoxic T-cell |
| DGL | diacylglycerol lipase |
| DR5 | death receptor 5 |
| EGFR | epidermal growth factor receptor |
| eIF2α | eukaryotic initiation factor 2α |
| EMT | epithelial-to mesenchymal transition |
| ERK1/2 | extracellular regulated kinase 1/2 |
| FAAH | fatty acid amide hydrolase |
| FGF2 | fibroblast growth factor 2 |
| GIT | gastrointestinal tract |
| GPR55 | G protein coupled receptor 55 |
| HIFα | hypoxia inducible factor α |
| IBDs | inflammatory bowel diseases |
| ICAM-1 | intercellular adhesion molecule 1 |
| IFN-γ | interferon γ |
| IL-(1β, 6, 8, 10, 13, 23) | inerleukin-(1β, 6, 8, 10, 13, 23) |
| JAK | Janus kinase |
| MAGL | monoacylglycerol lipase |
| MAPK | mitogen-activated protein kinase |
| MMP 9 | matrix metalloproteinase 9 |
| MSI | microsatellite instability |
| mTOR | mammalian target of rapamycin |
| NAPE-PLD | N-acyl-phosphatidiyl-ethanolamine-hydrolyzing phosphatase D |
| OEA | oleoylethanolamide |
| PDGFA | platelet-derived growth factor A |
| PEA | palmitoylethanolamide |
| PGE2 | prostaglandin E2 |
| PGF2α | prostaglandin F2α |
| PI3K | phosphoinositide-3 kinase |
| PLC | phospholipase C |
| PPAR | peroxisome proliferator activating receptor |
| STAT (1, 3, 5) | signal transducer and activator of transcription (1, 3, & 5) |
| TH17 | T-helper 17 |
| TLR 4 | Toll-like receptor 4 |
| TNF-α | tumor necrosis factor α |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| TRIB3 | tribbles homolog 3 |
| TXA2 | thromboxane A2 |
| UC | Ulcerative colitis |
| VEGF | vascular endothelial growth facto |
| WNT | Wingless pathway |
References
- Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2019; Canadian Cancer Society: Toronto, ON, Canada, 2019; Available online: cancer.ca/Canadian-Cancer-Statistics-2019-EN (accessed on 10 August 2021).
- WHO Global Status Report on Noncommunicable Diseases. 2020, pp. 1–162. Available online: https://www.who.int/nmh/publications/ncd_report_full_en.pdf (accessed on 10 August 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.E.; Zepp, J.M.; Gilmore, M.J.; Davis, J.V.; Esterberg, E.J.; Muessig, K.R.; Peterson, S.K.; Syngal, S.; Acheson, L.S.; Wiesner, G.L.; et al. Universal tumor screening for Lynch syndrome: Assessment of the perspectives of patients with colorectal cancer regarding benefits and barriers. Cancer 2015, 121, 3281–3289. [Google Scholar] [CrossRef] [Green Version]
- Erdrich, J.; Zhang, X.; Giovannucci, E.; Willett, W. Proportion of colon cancer attributable to lifestyle in a cohort of US women. Cancer Causes Control 2015, 26, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Guraya, S.Y. Association of type 2 diabetes mellitus and the risk of colorectal cancer: A meta-analysis and systematic review. World J. Gastroenterol. 2015, 21, 6026–6031. [Google Scholar] [CrossRef]
- Gerasymchuk, M.; Cherkasova, V.; Kovalchuk, O.; Kovalchuk, I. The Role of microRNAs in Organismal and Skin Aging. Int. J. Mol. Sci. 2020, 21, 5281. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of Colorectal Cancer in Patients With Ulcerative Colitis: A Meta-analysis of Population-Based Cohort Studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Papamichael, D.; Audisio, R.A.; Glimelius, B.; de Gramont, A.; Glynne-Jones, R.; Haller, D.; Köhne, C.H.; Rostoft, S.; Lemmens, V.; Mitry, E.; et al. Treatment of colorectal cancer in older patients: International Society of Geriatric Oncology (SIOG) consensus recommendations 2013. Ann. Oncol. 2015, 26, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor α-mediated ceramide de novo synthesis in colon cancer cells. Clin. Cancer Res. 2008, 14, 7691–7700. [Google Scholar] [CrossRef] [Green Version]
- Nallathambi, R.; Mazuz, M.; Namdar, D.; Shik, M.; Namintzer, D.; Vinayaka, A.C.; Ion, A.; Faigenboim, A.; Nasser, A.; Laish, I.; et al. Identification of Synergistic Interaction Between Cannabis-Derived Compounds for Cytotoxic Activity in Colorectal Cancer Cell Lines and Colon Polyps That Induces Apoptosis-Related Cell Death and Distinct Gene Expression. Cannabis Cannabinoid Res. 2018, 3, 120–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javid, F.A.; Phillips, R.M.; Afshinjavid, S.; Verde, R.; Ligresti, A. Cannabinoid pharmacology in cancer research: A new hope for cancer patients? Eur. J. Pharmacol. 2016, 775, 1–14. [Google Scholar] [CrossRef]
- Raup-Konsavage, W.M.; Johnson, M.; Legare, C.A.; Yochum, G.S.; Morgan, D.J.; Vrana, K.E. Synthetic cannabinoid activity against colorectal cancer cells. Cannabis Cannabinoid Res. 2018, 3, 272–281. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; Dubois, R.N. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2009, 68, 6468–6476. [Google Scholar] [CrossRef] [Green Version]
- Tutino, V.; Caruso, M.G.; De Nunzio, V.; Lorusso, D.; Veronese, N.; Gigante, I.; Notarnicola, M.; Giannelli, G. Down-regulation of cannabinoid type 1 (CB1) receptor and its downstream signaling pathways in metastatic colorectal cancer. Cancers 2019, 11, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, A.; Bisogno, T.; Matias, I.; De Petrocellis, L.; Cascio, M.G.; Cosenza, V.; D’Argenio, G.; Scaglione, G.; Bifulco, M.; Sorrentini, I.; et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 2003, 125, 677–687. [Google Scholar] [CrossRef]
- Anderson, S.P.; Zylla, D.M.; McGriff, D.M.; Arneson, T.J. Impact of Medical Cannabis on Patient-Reported Symptoms for Patients With Cancer Enrolled in Minnesota’s Medical Cannabis Program. J. Oncol. Pract. 2019, 15, e338–e345. [Google Scholar] [CrossRef] [PubMed]
- Grill, M.; Högenauer, C.; Blesl, A.; Haybaeck, J.; Golob-Schwarzl, N.; Ferreirós, N.; Thomas, D.; Gurke, R.; Trötzmüller, M.; Köfeler, H.C.; et al. Members of the endocannabinoid system are distinctly regulated in inflammatory bowel disease and colorectal cancer. Sci. Rep. 2019, 9, 2358. [Google Scholar] [CrossRef] [Green Version]
- Madras, B.K. Update of Cannabis and Its Medical Use; 37th ECDD (2015) Agenda Item 6.2; World Health Organization: Geneva, Switzerland, 2015; pp. 1–41. [Google Scholar]
- Hall, W.; Christie, M.; Currow, D. Cannabinoids and cancer: Causation, remediation, and palliation. Lancet Oncol. 2005, 6, 35–42. [Google Scholar] [CrossRef]
- Hoffenberg, E.J.; McWilliams, S.; Mikulich-Gilbertson, S.; Murphy, B.; Hoffenberg, A.; Hopfer, C.J. Cannabis oil use by adolescents and young adults with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 348–352. [Google Scholar] [CrossRef]
- Couch, D.G.; Cook, H.; Ortori, C.; Barrett, D.; Lund, J.N.; O’Sullivan, S.E. Palmitoylethanolamide and Cannabidiol Prevent Inflammation-induced Hyperpermeability of the Human Gut in Vitro and in Vivo-A Randomized, Placebo-controlled, Double-blind Controlled Trial. Inflamm. Bowel Dis. 2019, 25, 1006–1018. [Google Scholar] [CrossRef]
- Sanger, G.J. Endocannabinoids and the gastrointestinal tract: What are the key questions? Br. J. Pharmacol. 2007, 152, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhough, A.; Patsos, H.A.; Williams, A.C.; Paraskeva, C. The cannabinoid Δ9-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int. J. Cancer 2007, 121, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Pellerito, O.; Notaro, A.; Sabella, S.; De Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARγ down-regulation. Apoptosis 2014, 19, 1029–1042. [Google Scholar] [CrossRef]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Romano, B.; Borrelli, F.; Pagano, E.; Cascio, M.G.; Pertwee, R.G.; Izzo, A.A. Inhibition of colon carcinogenesis by a standardized Cannabis sativa extract with high content of cannabidiol. Phytomedicine 2014, 21, 631–639. [Google Scholar] [CrossRef]
- Pagano, E.; Capasso, R.; Piscitelli, F.; Romano, B.; Parisi, O.A.; Finizio, S.; Lauritano, A.; Di Marzo, V.; Izzo, A.A.; Borrelli, F. An orally active Cannabis extract with high content in cannabidiol attenuates chemically-induced intestinal inflammation and hypermotility in the mouse. Front. Pharmacol. 2016, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Orrego-González, E.; Londoño-Tobón, L.; Ardila-González, J.; Polania-Tovar, D.; Valencia-Cárdenas, A.; Velez-Van Meerbeke, A. Cannabinoid Effects on Experimental Colorectal Cancer Models Reduce Aberrant Crypt Foci (ACF) and Tumor Volume: A Systematic Review. Evid.-Based Complement. Altern. Med. 2020, 2020, 2371527. [Google Scholar] [CrossRef]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [Green Version]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallathambi, R.; Mazuz, M.; Ion, A.; Selvaraj, G.; Weininger, S.; Fridlender, M.; Nasser, A.; Sagee, O.; Kumari, P.; Nemichenizer, D.; et al. Anti-Inflammatory Activity in Colon Models Is Derived from Δ9-Tetrahydrocannabinolic Acid That Interacts with Additional Compounds in Cannabis Extracts. Cannabis Cannabinoid Res. 2017, 2, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Hasenoehrl, C.; Feuersinger, D.; Sturm, E.M.; Bärnthaler, T.; Graf, R.; Grill, M.; Pichler, M.; Beck, S.; Butcher, L.; Thomas, D.; et al. G protein-coupled receptor GPR55 promotes colorectal cancer and has opposing effects to cannabinoid receptor 1. Int. J. Cancer 2018, 142, 121–132. [Google Scholar] [CrossRef]
- Dall’Stella, P.B.; Docema, M.F.L.; Maldaun, M.V.C.; Feher, O.; Lancellotti, C.L.P. Case report: Clinical outcome and image response of two patients with secondary high-grade glioma treated with chemoradiation, PCV, and cannabidiol. Front. Oncol. 2019, 8, 643. [Google Scholar] [CrossRef] [Green Version]
- Sulé-Suso, J.; Watson, N.A.; van Pittius, D.G.; Jegannathen, A. Striking lung cancer response to self-administration of cannabidiol: A case report and literature review. SAGE Open Med. Case Rep. 2019, 7, 2050313X1983216. [Google Scholar] [CrossRef] [PubMed]
- Erices, J.I.; Torres, Á.; Niechi, I.; Bernales, I.; Quezada, C. Current natural therapies in the treatment against glioblastoma. Phyther. Res. 2018, 32, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.W.; Ho, R.C.M. The Cannabis Dilemma: A Review of Its Associated Risks and Clinical Efficacy. J. Addict. 2015, 2015, 707596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, A.C. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 619–631. [Google Scholar] [CrossRef]
- Lutz, B. Molecular biology of cannabinoid receptors. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 123–142. [Google Scholar] [CrossRef]
- Glass, M.; Northup, J.K. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1999, 56, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Brown, S.; Roche, J.P.; Hsieh, C.; Celver, J.P.; Kovoor, A.; Chavkin, C.; Mackie, K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J. Neurosci. 1999, 19, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K.; Hille, B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3825–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caulfield, M.P.; Brown, D.A. Cannabinoid receptor agonists inhibit Ca current in NG108–15 neuroblastoma cells via a Pertussis toxin-sensitive mechanism. Br. J. Pharmacol. 1992, 106, 231–232. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K.; Lai, Y.; Westenbroek, R.; Mitchell, R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J. Neurosci. 1995, 15, 6552–6561. [Google Scholar] [CrossRef] [Green Version]
- Henry, D.J.; Chavkin, C. Activation of inwardly rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci. Lett. 1995, 186, 91–94. [Google Scholar] [CrossRef]
- Watkins, A.R. Cannabinoid interactions with ion channels and receptors. Channels 2019, 13, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Bennett, A. Cannabinoids: A new group of agonists of PPARs. PPAR Res. 2007, 2007, 023513. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.C.; Reddy, P.V.; Natarajan, V.; Schmid, H.H. Metabolism of N-acylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. J. Biol. Chem. 1983, 258, 9302–9306. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef] [Green Version]
- Simon, G.M.; Cravatt, B.F. Characterization of mice lacking candidate N-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol. Biosyst. 2010, 6, 1411–1418. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, K.; Ikematsu, N.; Uyama, T.; G. Deutsch, D.; Tokumura, A.; Ueda, N. Biosynthetic Pathways of Bioactive N-Acylethanolamines in Brain. CNS Neurol. Disord. Drug Targets 2013, 12, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Capasso, R.; Aviello, G.; Borrelli, F.; Romano, B.; Piscitelli, F.; Gallo, L.; Capasso, F.; Orlando, P.; Di Marzo, V. Inhibitory effect of cannabichromene, a major non-psychotropic cannabinoid extracted from Cannabis sativa, on inflammation-induced hypermotility in mice. Br. J. Pharmacol. 2012, 166, 1444–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, V.; Daeninck, P.J. A user’s guide to cannabinoid therapies in oncology. Curr. Oncol. 2016, 23, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Woodward, D.F.; Liang, Y.; Krauss, A.H.P. Prostamides (prostaglandin-ethanolamides) and their pharmacology. Br. J. Pharmacol. 2008, 153, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, K.; Sun, Y.X.; Okamoto, Y.; Araki, N.; Tonai, T.; Ueda, N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J. Biol. Chem. 2005, 280, 11082–11092. [Google Scholar] [CrossRef] [Green Version]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef]
- Luk, T.; Jin, W.; Zvonok, A.; Lu, D.; Lin, X.Z.; Chavkin, C.; Makriyannis, A.; Mackie, K. Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br. J. Pharmacol. 2004, 142, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauckner, J.E.; Hille, B.; Mackie, K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, M.G.; Ruiz-Llorente, L.; Sánchez, A.M.; Díaz-Laviada, I. Activation of phosphoinositide 3-kinase/PKB pathway by CB1 and CB2 cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell Signal. 2003, 15, 851–859. [Google Scholar] [CrossRef]
- Johnson, D.E.; Heald, S.L.; Dally, R.D.; Janis, R.A. Isolation, identification and synthesis of an endogenous arachidonic amide that inhibits calcium channel antagonist 1,4-dihydropyridine binding. Prostaglandins Leukot. Essent. Fat. Acids 1993, 48, 429–437. [Google Scholar] [CrossRef]
- Shimasue, K.; Urushidani, T.; Hagiwara, M.; Nagao, T. Effects of anandamide and arachidonic acid on specific binding of (+)-PN200-110, diltiazem and (−)-desmethoxyverapamil to L-type Ca2+ channel. Eur. J. Pharmacol. 1996, 296, 347–350. [Google Scholar] [CrossRef]
- Sugiura, T.; Kobayashi, Y.; Oka, S.; Waku, K. Biosynthesis and degradation of anandamide and 2-arachidonoylglycerol and their possible physiological significance. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 173–192. [Google Scholar] [CrossRef]
- Navia-Paldanius, D.; Aaltonen, N.; Lehtonen, M.; Savinainen, J.R.; Taschler, U.; Radner, F.P.W.; Zimmermann, R.; Laitinen, J.T. Increased tonic cannabinoid CB1R activity and brain region-specific desensitization of CB1R Gi/o signaling axis in mice with global genetic knockout of monoacylglycerol lipase. Eur. J. Pharm. Sci. 2015, 77, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Imperatore, R.; Morello, G.; Luongo, L.; Taschler, U.; Romano, R.; De Gregorio, D.; Belardo, C.; Maione, S.; Di Marzo, V.; Cristino, L. Genetic deletion of monoacylglycerol lipase leads to impaired cannabinoid receptor CB1R signaling and anxiety-like behavior. J. Neurochem. 2015, 135, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Goggolidou, P. Ulcerative colitis: Understanding its cellular pathology could provide insights into novel therapies. J. Inflamm. 2020, 17, 15. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.M.; Calabrese, P.P.; Miller, A.J.; Munoz, N.M.; Grady, W.M.; Shibata, D.; Liskay, R.M. Single cell lineage tracing reveals a role for TgfβR2 in intestinal stem cell dynamics and differentiation. Proc. Natl. Acad. Sci. USA 2016, 113, 12192–12197. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.C.; Van Den Berg, D.; Kang, H.; Hsieh, C.L.; Lieber, M.R. Large chromosome deletions, duplications, and gene conversion events accumulate with age in normal human colon crypts. Aging Cell 2013, 12, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Argaw, A.; Duff, G.; Zabouri, N.; Cecyre, B.; Chaine, N.; Cherif, H.; Tea, N.; Lutz, B.; Ptito, M.; Bouchard, J.-F. Concerted Action of CB1 Cannabinoid Receptor and Deleted in Colorectal Cancer in Axon Guidance. J. Neurosci. 2011, 31, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, K.A.; Wiley, J.W. Getting into the weed: The role of the endocannabinoid system in the brain-gut axis. Gastroenterology 2016, 151, 252–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storr, M.A.; Sharkey, K.A. The endocannabinoid system and gut-brain signalling. Curr. Opin. Pharmacol. 2007, 7, 575–582. [Google Scholar] [CrossRef]
- Hasenoehrl, C.; Taschler, U.; Storr, M. The gastrointestinal tract—A central organ of cannabinoid signaling in health and disease. Neurogastroenterol. Motil. 2016, 28, 1765–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids-at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.; Davison, J.S.; Sharkey, K.A. Review article: Endocannabinoids and their receptors in the enteric nervous system. Aliment. Pharmacol. Ther. 2005, 22, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Amaya, F.; Shimosato, G.; Kawasaki, Y.; Hashimoto, S.; Tanaka, Y.; Ji, R.R.; Tanaka, M. Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain 2006, 124, 175–183. [Google Scholar] [CrossRef]
- Kulkarni-Narla, A.; Brown, D.R. Localization of CB1-cannabinoid receptor immunoreactivity in the porcine enteric nervous system. Cell Tissue Res. 2000, 302, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Marquéz, L.; Suárez, J.; Iglesias, M.; Bermudez-Silva, F.J.; de Fonseca, F.R.; Andreu, M. Ulcerative colitis induces changes on the expression of the endocannabinoid system in the human colonic tissue. PLoS ONE 2009, 4, e6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyires, K.; S. Zádori, Z. Role of Cannabinoids in Gastrointestinal Mucosal Defense and Inflammation. Curr. Neuropharmacol. 2016, 14, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; Rooney, N.; Feeney, M.; Tate, J.; Robertson, D.; Welham, M.; Ward, S. Differential expression of cannabinoid receptors in the human colon: Cannabinoids promote epithelial wound healing. Gastroenterology 2005, 129, 437–453. [Google Scholar] [CrossRef]
- Wright, K.L.; Duncan, M.; Sharkey, K.A. Cannabinoid CB2 receptors in the gastrointestinal tract: A regulatory system in states of inflammation. Br. J. Pharmacol. 2008, 153, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Sharir, H.; Console-Bram, L.; Mundy, C.; Popoff, S.N.; Kapur, A.; Abood, M.E. The endocannabinoids anandamide and virodhamine modulate the activity of the candidate cannabinoid receptor GPR55. J. Neuroimmune Pharmacol. 2012, 7, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Lo Verme, J.; Gaetani, S.; Fu, J.; Oveisi, F.; Burton, K.; Piomelli, D. Regulation of food intake by oleoylethanolamide. Cell. Mol. Life Sci. 2005, 62, 708–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thabuis, C.; Tissot-Favre, D.; Bezelgues, J.B.; Martin, J.C.; Cruz-Hernandez, C.; Dionisi, F.; Destaillats, F. Biological functions and metabolism of oleoylethanolamide. Lipids 2008, 43, 887–894. [Google Scholar] [CrossRef]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and pharmacology of endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar] [CrossRef]
- Bisogno, T.; Mahadevan, A.; Coccurello, R.; Chang, J.W.; Allarà, M.; Chen, Y.; Giacovazzo, G.; Lichtman, A.; Cravatt, B.; Moles, A.; et al. A novel fluorophosphonate inhibitor of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol with potential anti-obesity effects. Br. J. Pharmacol. 2013, 169, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Janssen, F.J.; van der Stelt, M. Inhibitors of diacylglycerol lipases in neurodegenerative and metabolic disorders. Bioorg. Med. Chem. Lett. 2016, 26, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, J.L.; Ghosh, S.; Bagdas, D.; Mason, B.L.; Crowe, M.S.; Hsu, K.L.; Wise, L.E.; Kinsey, S.G.; Damaj, M.I.; Cravatt, B.F.; et al. Diacylglycerol lipase β inhibition reverses nociceptive behaviour in mouse models of inflammatory and neuropathic pain. Br. J. Pharmacol. 2016, 173, 1678–1692. [Google Scholar] [CrossRef]
- Ward, S.M.; Bayguinov, J.; Won, K.J.; Grundy, D.; Berthoud, H.R. Distribution of the vanilloid receptor (VR1) in the gastrointestinal tract. J. Comp. Neurol. 2003, 465, 121–135. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Nabissi, M.; Santoni, G.; Ligresti, A. Actions and Regulation of Ionotropic Cannabinoid Receptors, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 80. [Google Scholar]
- Liberati, S.; Morelli, M.; Amantini, C.; Farfariello, V.; Santoni, M.; Conti, A.; Nabissi, M.; Cascinu, S.; Santoni, G. Loss of TRPV2 Homeostatic Control of Cell Proliferation Drives Tumor Progression. Cells 2014, 3, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Caprodossi, S.; Arcella, A.; Santoni, M.; Giangaspero, F.; De Maria, R.; et al. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis 2010, 31, 794–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Kato, I. Gut microbiota, inflammation and colorectal cancer. Genes Dis. 2016, 3, 130–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Cohen, L.J.; Esterhazy, D.; Kim, S.H.; Lemetre, C.; Aguilar, R.R.; Gordon, E.A.; Pickard, A.J.; Cross, J.R.; Emiliano, A.B.; Han, S.M.; et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 2017, 549, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Montalban-Arques, A.; Scharl, M. Intestinal microbiota and colorectal carcinoma: Implications for pathogenesis, diagnosis, and therapy. EBioMedicine 2019, 48, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Everard, A. Keeping gut lining at bay: Impact of emulsifiers. Trends Endocrinol. Metab. 2015, 26, 273–274. [Google Scholar] [CrossRef]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Gueimonde, M.; Ouwehand, A.; Huhtinen, H.; Salminen, E.; Salminen, S. Qualitative and quantitative analyses of the bifidobacterial microbiota in the colonic mucosa of patients with colorectal cancer, diverticulitis and inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 3985–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Everard, A.; Geurts, L.; Caesar, R.; Van Hul, M.; Matamoros, S.; Duparc, T.; Denis, R.G.P.; Cochez, P.; Pierard, F.; Castel, J.; et al. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat. Commun. 2014, 5, 5648. [Google Scholar] [CrossRef] [Green Version]
- De Winter, B.Y.; De Man, J.G. Interplay between inflammation, immune system and neuronal pathways: Effect on gastrointestinal motility. World J. Gastroenterol. 2010, 16, 5523–5535. [Google Scholar] [CrossRef]
- Karwad, M.A.; Macpherson, T.; Wang, B.; Theophilidou, E.; Sarmad, S.; Barrett, D.A.; Larvin, M.; Wright, K.L.; Lund, J.N.; O’Sullivan, S.E. Oleoylethanolamine and palmitoylethanolamine modulate intestinal permeability in vitro via TRPV1 and PPARα. FASEB J. 2017, 31, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrpouya-Bahrami, P.; Chitrala, K.N.; Ganewatta, M.S.; Tang, C.; Murphy, E.A.; Enos, R.T.; Velazquez, K.T.; McCellan, J.; Nagarkatti, M.; Nagarkatti, P. Blockade of CB1 cannabinoid receptor alters gut microbiota and attenuates inflammation and diet-induced obesity. Sci. Rep. 2017, 7, 15645. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van De Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluny, N.L.; Keenan, C.M.; Reimer, R.A.; Foll, B.L.; Sharkey, K.A. Prevention of Diet-Induced Obesity Effects on Body Weight and Gut Microbiota in Mice Treated Chronically with Δ9-Tetrahydrocannabinol. PLoS ONE 2015, 10, e0144270. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, C.; Di Marzo, V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef]
- Izzo, A.A.; Fezza, F.; Capasso, R.; Bisogno, T.; Pinto, L.; Iuvone, T.; Esposito, G.; Mascolo, N.; Di Marzo, V.; Capasso, F. Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br. J. Pharmacol. 2001, 134, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdú, E.F.; Bercik, P.; Verma-Gandhu, M.; Huang, X.X.; Blennerhassett, P.; Jackson, W.; Mao, Y.; Wang, L.; Rochat, F.; Collins, S.M. Specific probiotic therapy attenuates antibiotic induced visceral hypersensitivity in mice. Gut 2006, 55, 182–190. [Google Scholar] [CrossRef]
- Rousseaux, C.; Thuru, X.; Gelot, A.; Barnich, N.; Neut, C.; Dubuquoy, L.; Dubuquoy, C.; Merour, E.; Geboes, K.; Chamaillard, M.; et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 2007, 13, 35–37. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.D. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.C.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.Z.; Wright, N.A. Clonality, Founder Mutations, and Field Cancerization in Human Ulcerative Colitis-Associated Neoplasia. Gastroenterology 2009, 136, 542–550.e6. [Google Scholar] [CrossRef]
- Kern, S.E.; Redston, M.; Seymour, A.B.; Caldas, C.; Powell, S.M.; Kornacki, S.; Kinzler, K.W. Molecular genetic profiles of colitis-associated neoplasms. Gastroenterology 1994, 107, 420–428. [Google Scholar] [CrossRef]
- Galandiuk, S.; Rodriguezjusto, M.; Jeffery, R.; Nicholson, A.M.; Cheng, Y.; Oukrif, D.; Elia, G.; Leedham, S.J.; McDonald, S.A.C.; Wright, N.A.; et al. Field cancerization in the intestinal epithelium of patients with Crohn’s ileocolitis. Gastroenterology 2012, 142, 855–864.e8. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.W.; Newton, C.; Larsen, K.; Lu, L.; Perkins, I.; Nong, L.; Friedman, H. The cannabinoid system and immune modulation. J. Leukoc. Biol. 2003, 74, 486–496. [Google Scholar] [CrossRef] [Green Version]
- D’Argenio, G.; Valenti, M.; Scaglione, G.; Cosenza, V.; Sorrentini, I.; Di Marzo, V. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006, 20, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Camilleri, M. Cannabinoids in intestinal inflammation and cancer. Pharmacol. Res. 2009, 60, 117–125. [Google Scholar] [CrossRef]
- Massa, F.; Marsicano, G.; Hermana, H.; Cannich, A.; Monory, K.; Cravatt, B.F.; Ferri, G.L.; Sibaev, A.; Storr, M.; Lutz, B. The endogenous cannabinoid system protects against colonic inflammation. J. Clin. Investig. 2004, 113, 1202–1209. [Google Scholar] [CrossRef] [Green Version]
- Storr, M.A.; Keenan, C.M.; Zhang, H.; Patel, K.D.; Makriyannis, A.; Sharkey, K.A. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm. Bowel Dis. 2009, 15, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Kimball, E.S.; Schneider, C.R.; Wallace, N.H.; Hornby, P.J. Agonists of cannabinoid receptor 1 and 2 inhibit experimental colitis induced by oil of mustard and by dextran sulfate sodium. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 291, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storr, M.A.; Keenan, C.M.; Emmerdinger, D.; Zhang, H.; Yüce, B.; Sibaev, A.; Massa, F.; Buckley, N.E.; Lutz, B.; Göke, B.; et al. Targeting endocannabinoid degradation protects against experimental colitis in mice: Involvement of CB1 and CB2 receptors. J. Mol. Med. 2008, 86, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.J.; Li, Y.Y.; Lin, X.H.; Li, K.; Cao, M.H. Anti-inflammatory effect of cannabinoid agonist win55, 212 on mouse experimental colitis is related to inhibition of p38MAPK. World J. Gastroenterol. 2016, 22, 9515–9524. [Google Scholar] [CrossRef]
- Di Marzo, V.; Izzo, A.A. Endocannabinoid overactivity and intestinal inflammation. Gut 2006, 55, 1373–1376. [Google Scholar] [CrossRef] [Green Version]
- Mathison, R.; Ho, W.; Pittman, Q.J.; Davison, J.S.; Sharkey, K.A. Effects of cannabinoid receptor-2 activation on accelerated gastrointestinal transit in lipopolysaccharide-treated rats. Br. J. Pharmacol. 2004, 142, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Sanson, M.; Bueno, L.; Fioramonti, J. Involvement of cannabinoid receptors in inflammatory hypersensitivity to colonic distension in rats. Neurogastroenterol. Motil. 2006, 18, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Camilleri, M. Emerging role of cannabinoids in gastrointestinal and liver diseases: Basic and clinical aspects. Gut 2008, 57, 1140–1155. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Kiertscher, S.M.; Cheng, Q.; Zoumalan, R.; Tashkin, D.P.; Roth, M.D. Δ9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. J. Neuroimmunol. 2002, 133, 124–131. [Google Scholar] [CrossRef]
- Ziring, D.; Wei, B.; Velazquez, P.; Schrage, M.; Buckley, N.E.; Braun, J. Formation of B and T cell subsets require the cannabinoid receptor CB2. Immunogenetics 2006, 58, 714–725. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Romero, J.; Velasco, G.; Tolón, R.M.; Ramos, J.A.; Guzmán, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef]
- Ihenetu, K.; Molleman, A.; Parsons, M.E.; Whelan, C.J. Inhibition of interleukin-8 release in the human colonic epithelial cell line HT-29 by cannabinoids. Eur. J. Pharmacol. 2003, 458, 207–215. [Google Scholar] [CrossRef]
- Acharya, N.; Penukonda, S.; Shcheglova, T.; Hagymasi, A.T.; Basu, S.; Srivastava, P.K. Endocannabinoid system acts as a regulator of immune homeostasis in the gut. Proc. Natl. Acad. Sci. USA 2017, 114, 5005–5010. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.W.; Cabral, G.A. Cannabinoid-Induced Immune Suppression and Modulation of Antigen-Presenting Cells. J. Neuroimmune Pharmacol. 2006, 1, 50. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARa-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. Harnessing the anti-inflammatory potential of palmitoylethanolamide. Drug Discov. Today 2014, 19, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Izzo, A.A. Role of acylethanolamides in the gastrointestinal tract with special reference to food intake and energy balance. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 33–49. [Google Scholar] [CrossRef]
- Alhouayek, M.; Bottemanne, P.; Subramanian, K.V.; Lambert, D.M.; Makriyannis, A.; Cani, P.D.; Muccioli, G.G. N-acylethanolamine-hydrolyzing acid amidase inhibition increases colon N-palmitoylethanolamine levels and counteracts murine colitis. FASEB J. 2015, 29, 650–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut 2013, 63, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Maurelli, S.; Melck, D.; De Petrocellis, L.; Di Marzo, V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997, 272, 3315–3323. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Melck, D.; Orlando, P.; Bisogno, T.; Zagoory, O.; Bifulco, M.; Vogel, Z.; de Petrocellis, L. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem. J. 2001, 358, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [Green Version]
- De Petrocellis, L.; Bisogno, T.; Ligresti, A.; Bifulco, M.; Melck, D.; Di Marzo, V. Effect on cancer cell proliferation of palmitoylethanolamide, a fatty acid amide interacting with both the cannabinoid and vanilloid signalling systems. Fundam. Clin. Pharmacol. 2002, 16, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Smart, D.; Jonsson, K.O.; Vandevoorde, S.; Lambert, D.M.; Fowler, C.J. “Entourage” effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br. J. Pharmacol. 2002, 136, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Riquer, Z.P.; Ibarra-Sánchez, A.; Vibhushan, S.; Bratti, M.; Charles, N.; Blank, U.; Rodríguez-Manzo, G.; González-Espinosa, C. TLR4 Receptor Induces 2-AG–Dependent Tolerance to Lipopolysaccharide and Trafficking of CB2 Receptor in Mast Cells. J. Immunol. 2019, 202, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Li, Y.N.; Ni, J.B.; Chen, C.J.; Lv, S.; Chai, S.Y.; Wu, R.H.; Yüce, B.; Storr, M. Involvement of cannabinoid-1 and cannabinoid-2 receptors in septic ileus. Neurogastroenterol. Motil. 2010, 22, 350–358. [Google Scholar] [CrossRef]
- de Filippis, D.; Esposito, G.; Cirillo, C.; Cipriano, M.; de Winter, B.Y.; Scuderi, C.; Sarnelli, G.; Cuomo, R.; Steardo, L.; de Man, J.G.; et al. Cannabidiol reduces intestinal inflammation through the control of neuroimmune axis. PLoS ONE 2011, 6, e28159. [Google Scholar] [CrossRef]
- Karwad, M.A.; Couch, D.G.; Theophilidou, E.; Sarmad, S.; Barrett, D.A.; Larvin, M.; Wright, K.L.; Lund, J.N.; O’Sullivan, S.E. The role of CB1 in intestinal permeability and inflammation. FASEB J. 2017, 31, 3267–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamran, H.; Singh, N.P.; Zumbrun, E.E.; Murphy, A.; Taub, D.D.; Mishra, M.K.; Price, R.L.; Chatterjee, S.; Nagarkatti, M.; Nagarkatti, P.S.; et al. Fatty acid amide hydrolase (FAAH) blockade ameliorates experimental colitis by altering microRNA expression and suppressing inflammation. Brain Behav. Immun. 2017, 59, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Sałaga, M.; Mokrowiecka, A.; Zakrzewski, P.K.; Cygankiewicz, A.; Leishman, E.; Sobczak, M.; Zatorski, H.; Małecka-Panas, E.; Kordek, R.; Storr, M.; et al. Experimental colitis in mice is attenuated by changes in the levels of endocannabinoid metabolites induced by selective inhibition of fatty acid amide hydrolase (FAAH). J. Crohn’s Colitis 2014, 8, 998–1009. [Google Scholar] [CrossRef]
- Borrelli, F.; Fasolino, I.; Romano, B.; Capasso, R.; Maiello, F.; Coppola, D.; Orlando, P.; Battista, G.; Pagano, E.; Di, V.; et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem. Pharmacol. 2013, 85, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Jamontt, J.M.; Molleman, A.; Pertwee, R.G.; Parsons, M.E. The effects of Δ9-tetrahydrocannabinol and cannabidiol alone and in combination on damage, inflammation and in vitro motility disturbances in rat colitis. Br. J. Pharmacol. 2010, 160, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, T.; Hanuš, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snipper, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in the mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Kharusi, M.R.A.; Smartt, H.J.M.; Greenhough, A.; Collard, T.J.; Emery, E.D.; Williams, A.C.; Paraskeva, C. LGR5 promotes survival in human colorectal adenoma cells and is upregulated by PGE2: Implications for targeting adenoma stem cells with nsaids. Carcinogenesis 2013, 34, 1150–1157. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Maeda, S.; Hikiba, Y.; Nakagawa, H.; Hayakawa, Y.; Shibata, W.; Yanai, A.; Ogura, K.; Omata, M. Constitutive NF-κB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin. Cancer Res. 2009, 15, 2248–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1Β stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [Green Version]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in Apc(Δ716) knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Atreya, I.; Neurath, M.F. Immune cells in colorectal cancer: Prognostic relevance and therapeutic strategies. Expert Rev. Anticancer Ther. 2008, 8, 561–572. [Google Scholar] [CrossRef]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebs, A.; Herrero San Juan, M.; Schmidt, K.G.; Wiercinska, E.; Anlauf, M.; Ottenlinger, F.; Thomas, D.; Elwakeel, E.; Weigert, A.; Farin, H.F.; et al. Cancer-induced inflammation and inflammation-induced cancer in colon: A role for S1P lyase. Oncogene 2019, 38, 4788–4803. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer Find the latest version: Review series A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Pallone, F.; Monteleone, G.; Fantini, M.C. Intestinal inflammation and colorectal cancer: A double-edged sword? World J. Gastroenterol. 2011, 17, 3092–3100. [Google Scholar] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Cui, G.; Yao, Y.L.; Wang, Q.C.; Gu, H.G.; Li, X.N.; Zhang, H.; Feng, W.M.; Shi, Q.L.; Cui, W. Value of CNRIP1 promoter methylation in colorectal cancer screening and prognosis assessment and its influence on the activity of cancer cells. Arch. Med. Sci. 2017, 13, 1281–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.K.; Kang, W.K.; Park, J.M.; Ahn, H.J.; Kim, S.W.; Oh, S.T.; Choi, K.Y. Expression of the cannabinoid type I receptor and prognosis following surgery in colorectal cancer. Oncol. Lett. 2013, 5, 870–876. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Gomez, I.; Martin, P.; Sanchez, A.; Roman, L.; Tejerina, E.; Bonilla, F.; Garcia-Merino, A.; de Herreros, A.G.; Provencio, M.; et al. Cannabinoids receptor type 2, CB2, expression correlates with human colon cancer progression and predicts patient survival. Oncoscience 2015, 2, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, S.B.; Palmqvist, R.; Henriksson, M.L.; Dahlin, A.M.; Edin, S.; Jacobsson, S.O.P.; Öberg, Å.; Fowler, C.J. High tumour cannabinoid CB 1 receptor immunoreactivity negatively impacts disease-specific survival in stage II microsatellite stable colorectal cancer. PLoS ONE 2011, 6, e23003. [Google Scholar] [CrossRef]
- Cudaback, E.; Marrs, W.; Moeller, T.; Stella, N. The expression level of CB1 and CB2 receptors determines their efficacy at inducing apoptosis in astrocytomas. PLoS ONE 2010, 5, e8702. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G.; Cascio, M.G. Known Pharmacological Actions of Delta-9-Tetrahydrocannabinol and of Four Other Chemical Constituents of Cannabis that Activate Cannabinoid Receptors. Handb. Cannabis 2015, 115–136. [Google Scholar]
- Henstridge, C.M.; Balenga, N.A.B.; Kargl, J.; Andradas, C.; Brown, A.J.; Irving, A.; Sanchez, C.; Waldhoer, M. Minireview: Recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol. Endocrinol. 2011, 25, 1835–1848. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.A. L-α-Lysophosphatidylinositol meets GPR55: A deadly relationship. Trends Pharmacol. Sci. 2011, 32, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Henstidge, C.M.; Balenga, N.A.B.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-α-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Andradas, C.; Caffarel, M.M.; Pérez-Gómez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2011, 30, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stančic, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br. J. Pharmacol. 2016, 173, 142–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stančić, A.; Jandl, K.; Hasenöhrl, C.; Reichmann, F.; Marsche, G.; Schuligoi, R.; Heinemann, A.; Storr, M.; Schicho, R. The GPR55 antagonist CID16020046 protects against intestinal inflammation. Neurogastroenterol. Motil. 2015, 27, 1432–1445. [Google Scholar] [CrossRef] [Green Version]
- Balenga, N.A.B.; Aflaki, E.; Kargl, J.; Platzer, W.; Schröder, R.; Blättermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, H.; Li, Y.; Li, L.; Qiu, Y.; Ren, J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol. Rep. 2015, 34, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Zhang, B.; Seviour, E.G.; Tao, K.X.; Liu, X.H.; Ling, Y.; Chen, J.Y.; Wang, G. bin Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011, 307, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Di Popolo, A.; Memoli, A.; Apicella, A.; Tuccillo, C.; Di Palma, A.; Ricchi, P.; Acquaviva, A.M.; Zarrilli, R. IGF-II/IGF-I receptor pathway up-regulates COX-2 mRNA expression and PGE2 synthesis in Caco-2 human colon carcinoma cells. Oncogene 2000, 19, 5517–5524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Kuwano, T.; Nakao, S.; Yamamoto, H.; Tsuneyoshi, M.; Yamamoto, T.; Kuwano, M.; Ono, M. Cyclooxygenase 2 is a key enzyme for inflammatory cytokine-induced angiogenesis. FASEB J. 2004, 18, 300–310. [Google Scholar] [CrossRef]
- Qiao, L.; Kozoni, V.; Tsioulias, G.J.; Koutsos, M.I.; Hanif, R.; Shiff, S.J.; Rigas, B. Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1995, 1258, 215–223. [Google Scholar] [CrossRef]
- Kozak, K.R.; Crews, B.C.; Morrow, J.D.; Wang, L.H.; Ma, Y.H.; Weinander, R.; Jakobsson, P.J.; Marnett, L.J. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 2002, 277, 44877–44885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsos, H.A.; Hicks, D.J.; Dobson, R.R.H.; Greenhough, A.; Woodman, N.; Lane, J.D.; Williams, A.C.; Paraskeva, C. The endogenous cannabinoid, anandamide, induces cell death in colorectal carcinoma cells: A possible role for cyclooxygenase 2. Gut 2005, 54, 1741–1750. [Google Scholar] [CrossRef] [Green Version]
- Galon, J. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Izzo, A.A.; Muccioli, G.G.; Ruggieri, M.R.; Schicho, R. Endocannabinoids and the Digestive Tract and Bladder in Health and Disease. Handb. Exp. Pharmacol. 2015, 231, 423–447. [Google Scholar] [PubMed]
- Sreevalsan, S.; Joseph, S.; Jutooru, I.; Chadalapaka, G.; Safe, S.H. Induction of Apoptosis by Cannabinoids in Prostate and Colon Cancer Cells Is Phosphatase Dependent. Anticancer Res. 2011, 31, 3799–3807. [Google Scholar]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidhar, R.; Ben-Dor, S.; Wang, E.; Kelly, S.; Merrill, A.H.; Futerman, A.H. Acyl chain specificity of ceramide synthases is determined within a region of 150 residues in the tram-lag-CLN8 (TLC) domain. J. Biol. Chem. 2012, 287, 3197–3206. [Google Scholar] [CrossRef] [Green Version]
- Morad, S.A.F.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Sánchez, C.; Galve-Roperh, I.; Rueda, D.; Guzmán, M. Involvement of sphingomyelin hydrolysis and the mitogen-activated protein kinase cascade in the Δ9-Tetrahydrocannabinol-induced stimulation of glucose metabolism in primary astrocytes. Mol. Pharmacol. 1998, 54, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Adam-Klages, S.; Adam, D.; Wiegmann, K.; Struve, S.; Kolanus, W.; Schneider-Mergener, J.; Krönke, M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell 1996, 86, 937–947. [Google Scholar] [CrossRef] [Green Version]
- White-Gilbertson, S.; Mullen, T.; Senkal, C.; Lu, P.; Ogretmen, B.; Obeid, L.; Voelkel-Johnson, C. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 2009, 28, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Osawa, Y.; Uchinami, H.; Bielawski, J.; Schwabe, R.F.; Hannun, Y.A.; Brenner, D.A. Roles for C16-ceramide and sphingosine 1-phosphate in regulating hepatocyte apoptosis in response to tumor necrosis factor-α. J. Biol. Chem. 2005, 280, 27879–27887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Verlaan–de Vries, M.; Van Der Eb, A.J.; Fearon, E.R.; Vogelstein, B.; Hamilton, S.R.; Van Boom, J.H. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Parsons, D.W.; Wang, T.L.; Samuels, Y.; Bardelli, A.; Cummins, J.M.; DeLong, L.; Silliman, N.; Ptak, J.; Szabo, S.; Willson, J.K.V.; et al. Colorectal cancer: Mutations in a signalling pathway. Nature 2005, 436, 792. [Google Scholar] [CrossRef]
- Hart, S.; Fischer, O.M.; Ullrich, A. Cannabinoids Induce Cancer Cell Proliferation via Tumor Necrosis Factor α-Converting Enzyme (TACE/ADAM17)-Mediated Transactivation of the Epidermal Growth Factor Receptor. Cancer Res. 2004, 64, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Premoli, M.; Aria, F.; Bonini, S.A.; Maccarinelli, G.; Gianoncelli, A.; Pina, S.D.; Tambaro, S.; Memo, M.; Mastinu, A. Cannabidiol: Recent advances and new insights for neuropsychiatric disorders treatment. Life Sci. 2019, 224, 120–127. [Google Scholar] [CrossRef] [PubMed]
- McCarberg, B.H.; Barkin, R.L. The future of cannabinoids as analgesic agents: A pharmacologic, pharmacokinetic, and pharmacodynamic overview. Am. J. Ther. 2007, 14, 475–483. [Google Scholar] [CrossRef]
- Johansson, E.; Ohlsson, A.; Lindgren, J.-E.; Agurell, S.; Gillespie, H.; Hollister, L.E. Single-dose kinetics of deuterium-labelled cannabinol in man after intravenous administration and smoking. Biomed. Environ. Mass Spectrom. 1987, 14, 495–499. [Google Scholar] [CrossRef]
- Yuan, Y.; Xu, X.; Zhao, C.; Zhao, M.; Wang, H.; Zhang, B.; Wang, N.; Mao, H.; Zhang, A.; Xing, C. The roles of oxidative stress, endoplasmic reticulum stress, and autophagy in aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab. Investig. 2015, 95, 1374–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Ri, M.; Masaki, A.; Mori, F.; Ito, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; Iida, S. Lower expression of activating transcription factors 3 and 4 correlates with shorter progression-free survival in multiple myeloma patients receiving bortezomib plus dexamethasone therapy. Blood Cancer J. 2015, 5, e373. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.L.; Kim, B.R.; Kim, D.Y.; Jeong, Y.A.; Jeong, S.; Na, Y.J.; Park, S.H.; Yun, H.K.; Jo, M.J.; Kim, B.G.; et al. Cannabidiol enhances the therapeutic effects of TRAIL by upregulating DR5 in colorectal cancer. Cancers 2019, 11, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Cascio, M.G.; Gauson, L.A.; Stevenson, L.A.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid cannabigerol is a highly potent α 2-adrenoceptor agonist and moderately potent 5HT 1A receptor antagonist. Br. J. Pharmacol. 2010, 159, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrelli, F.; Pagano, E.; Romano, B.; Panzera, S.; Maiello, F.; Coppola, D.; De Petrocellis, L.; Buono, L.; Orlando, P.; Izzo, A.A. Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-derived non-psychotropic cannabinoid. Carcinogenesis 2014, 35, 2787–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [Green Version]
- Ansa-Addo, E.A.; Lange, S.; Stratton, D.; Antwi-Baffour, S.; Cestari, I.; Ramirez, M.I.; McCrossan, M.V.; Inal, J.M. Human Plasma Membrane-Derived Vesicles Halt Proliferation and Induce Differentiation of THP-1 Acute Monocytic Leukemia Cells. J. Immunol. 2010, 185, 5236–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turola, E.; Furlan, R.; Bianco, F.; Matteoli, M.; Verderio, C. Microglial microvesicle secretion and intercellular signaling. Front. Physiol. 2012, 3, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, M.; Pizzirani, C.; Pinotti, M.; Ferrari, D.; Adinolfi, E.; Calzavarini, S.; Caruso, P.; Bernardi, F.; Di Virgilio, F. Stimulation of P2 (P2X 7) receptors in human dendritic cells induces the release of tissue factor-bearing microparticles. FASEB J. 2007, 21, 1926–1933. [Google Scholar] [CrossRef]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kosgodage, U.S.; Mould, R.; Henley, A.B.; Nunn, A.V.; Guy, G.W.; Thomas, E.L.; Inal, J.M.; Bell, J.D.; Lange, S. Cannabidiol (CBD) is a novel inhibitor for exosome and microvesicle (EMV) release in cancer. Front. Pharmacol. 2018, 9, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Mechanisms of disease: Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Hua, Y.; Zhu, Y.; Zhang, J.; Zhu, Z.; Ning, Z.; Chen, H.; Liu, L.; Chen, Z.; Meng, Z. MiR-122 targets X-linked inhibitor of apoptosis protein to sensitize oxaliplatin-resistant colorectal cancer cells to oxaliplatin-mediated cytotoxicity. Cell. Physiol. Biochem. 2018, 51, 2148–2159. [Google Scholar] [CrossRef]
- Tan, S.; Peng, X.; Peng, W.; Zhao, Y.; Wei, Y. Enhancement of oxaliplatin-induced cell apoptosis and tumor suppression by 3-methyladenine in colon cancer. Oncol. Lett. 2015, 9, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Kim, B.G.; Kim, D.Y.; Kim, B.R.; Kim, J.L.; Park, S.H.; Na, Y.J.; Jo, M.J.; Yun, H.K.; Jeong, Y.A.; et al. Cannabidiol overcomes oxaliplatin resistance by enhancing NOS3- and SOD2-Induced autophagy in human colorectal cancer cells. Cancers 2019, 11, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schläfli, A.M.; Berezowska, S.; Adams, O.; Langer, R.; Tschan, M.P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 2015, 59, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Tournier, N.; Chevillard, L.; Megarbane, B.; Pirnay, S.; Scherrmann, J.M.; Declèves, X. Interaction of drugs of abuse and maintenance treatments with human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2). Int. J. Neuropsychopharmacol. 2010, 13, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, M.L.; Allen, J.D.; Arnold, J.C. Interaction of plant cannabinoids with the multidrug transporter ABCC1 (MRP1). Eur. J. Pharmacol. 2008, 591, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.L.; Lau, D.T.T.; Allen, J.D.; Arnold, J.C. The multidrug transporter ABCG2 (BCRP) is inhibited by plant-derived cannabinoids. Br. J. Pharmacol. 2007, 152, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Stout, S.M.; Cimino, N.M. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: A systematic review. Drug Metab. Rev. 2014, 46, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Notarnicola, M.; Messa, C.; Orlando, A.; Bifulco, M.; Laezza, C.; Gazzerro, P.; Caruso, M.G. Estrogenic induction of cannabinoid CB1 receptor in human colon cancer cell lines. Scand. J. Gastroenterol. 2008, 43, 66–72. [Google Scholar] [CrossRef]
- Thapa, D.; Kang, Y.; Park, P.H.; Noh, S.K.; Lee, Y.R.; Han, S.S.; Ku, S.K.; Jung, Y.; Kim, J.A. Anti-tumor activity of the novel hexahydrocannabinol analog LYR-8 in human colorectal tumor xenograft is mediated through the inhibition of Akt and hypoxia-inducible factor-1α activation. Biol. Pharm. Bull. 2012, 35, 924–932. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.; Kogan, N.M. HU-331: A cannabinoid quinone, with uncommon cytotoxic properties and low toxicity. Expert Opin. Investig. Drugs 2007, 16, 1405–1413. [Google Scholar] [CrossRef]
- Solinas, M.; Massi, P.; Cinquina, V.; Valenti, M.; Bolognini, D.; Gariboldi, M.; Monti, E.; Rubino, T.; Parolaro, D. Cannabidiol, a Non-Psychoactive Cannabinoid Compound, Inhibits Proliferation and Invasion in U87-MG and T98G Glioma Cells through a Multitarget Effect. PLoS ONE 2013, 8, e76918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinas, M.; Massi, P.; Cantelmo, A.R.; Cattaneo, M.G.; Cammarota, R.; Bartolini, D.; Cinquina, V.; Valenti, M.; Vicentini, L.M.; Noonan, D.M.; et al. Cannabidiol inhibits angiogenesis by multiple mechanisms. Br. J. Pharmacol. 2012, 167, 1218–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, C.P.; Hind, W.H.; Tufarelli, C.; O’Sullivan, S.E. Cannabidiol causes endothelium-dependent vasorelaxation of human mesenteric arteries via CB1 activation. Cardiovasc. Res. 2015, 107, 568–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol. Immunother. 2004, 53, 723–728. [Google Scholar] [CrossRef]
- Prester, L.; Mikolić, A.; Jurič, A.; Fuchs, N.; Neuberg, M.; Lucić Vrdoljak, A.; Brčić Karačonji, I. Effects of Δ9-tetrahydrocannabinol on irinotecan-induced clinical effects in rats. Chem. Biol. Interact. 2018, 294, 128–134. [Google Scholar] [CrossRef]
- Raup-Konsavage, W.M.; Carkaci-Salli, N.; Greenland, K.; Gearhart, R.; Vrana, K.E. Cannabidiol (CBD) Oil Does Not Display an Entourage Effect in Reducing Cancer Cell Viability in vitro. Med. Cannabis Cannabinoids 2020, 3, 95–102. [Google Scholar] [CrossRef]
- Booth, J.K.; Bohlmann, J. Terpenes in Cannabis sativa—From plant genome to humans. Plant Sci. 2019, 284, 67–72. [Google Scholar] [CrossRef]
- Russo, E.B. History of cannabis and its preparations in saga, science, and sobriquet. Chem. Biodivers. 2007, 4, 1614–1648. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherkasova, V.; Kovalchuk, O.; Kovalchuk, I. Cannabinoids and Endocannabinoid System Changes in Intestinal Inflammation and Colorectal Cancer. Cancers 2021, 13, 4353. https://doi.org/10.3390/cancers13174353
Cherkasova V, Kovalchuk O, Kovalchuk I. Cannabinoids and Endocannabinoid System Changes in Intestinal Inflammation and Colorectal Cancer. Cancers. 2021; 13(17):4353. https://doi.org/10.3390/cancers13174353
Chicago/Turabian StyleCherkasova, Viktoriia, Olga Kovalchuk, and Igor Kovalchuk. 2021. "Cannabinoids and Endocannabinoid System Changes in Intestinal Inflammation and Colorectal Cancer" Cancers 13, no. 17: 4353. https://doi.org/10.3390/cancers13174353