
Small Extracellular Vesicles and Metastasis—Blame the Messenger


Abstract
:Simple Summary
Abstract
1. Tumor Metastasis and Nucleic Acid Cargo in Small Extracellular Vesicles
2. sEVs and Their Highly Abundant Exosome-sEV Sub-Population
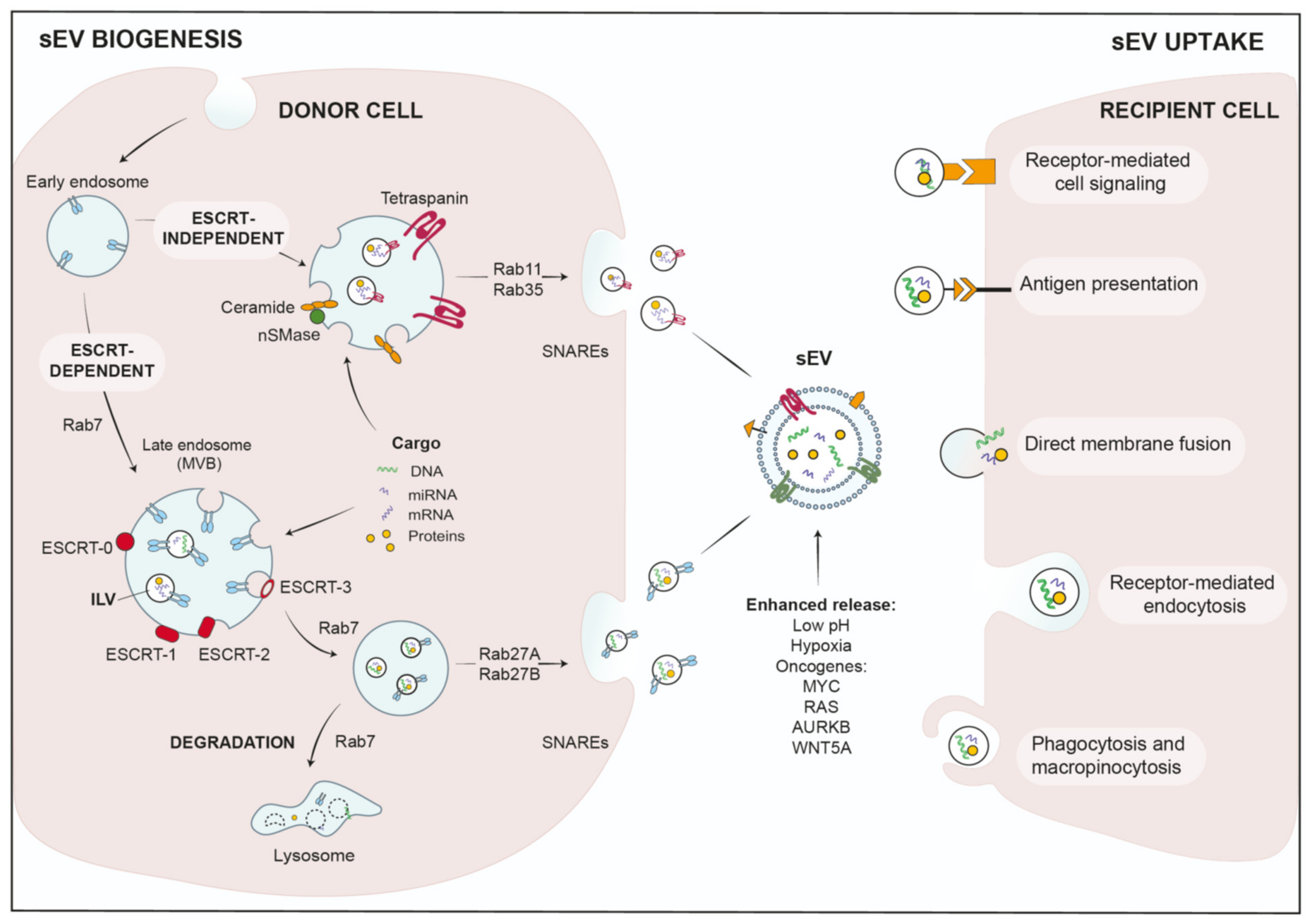
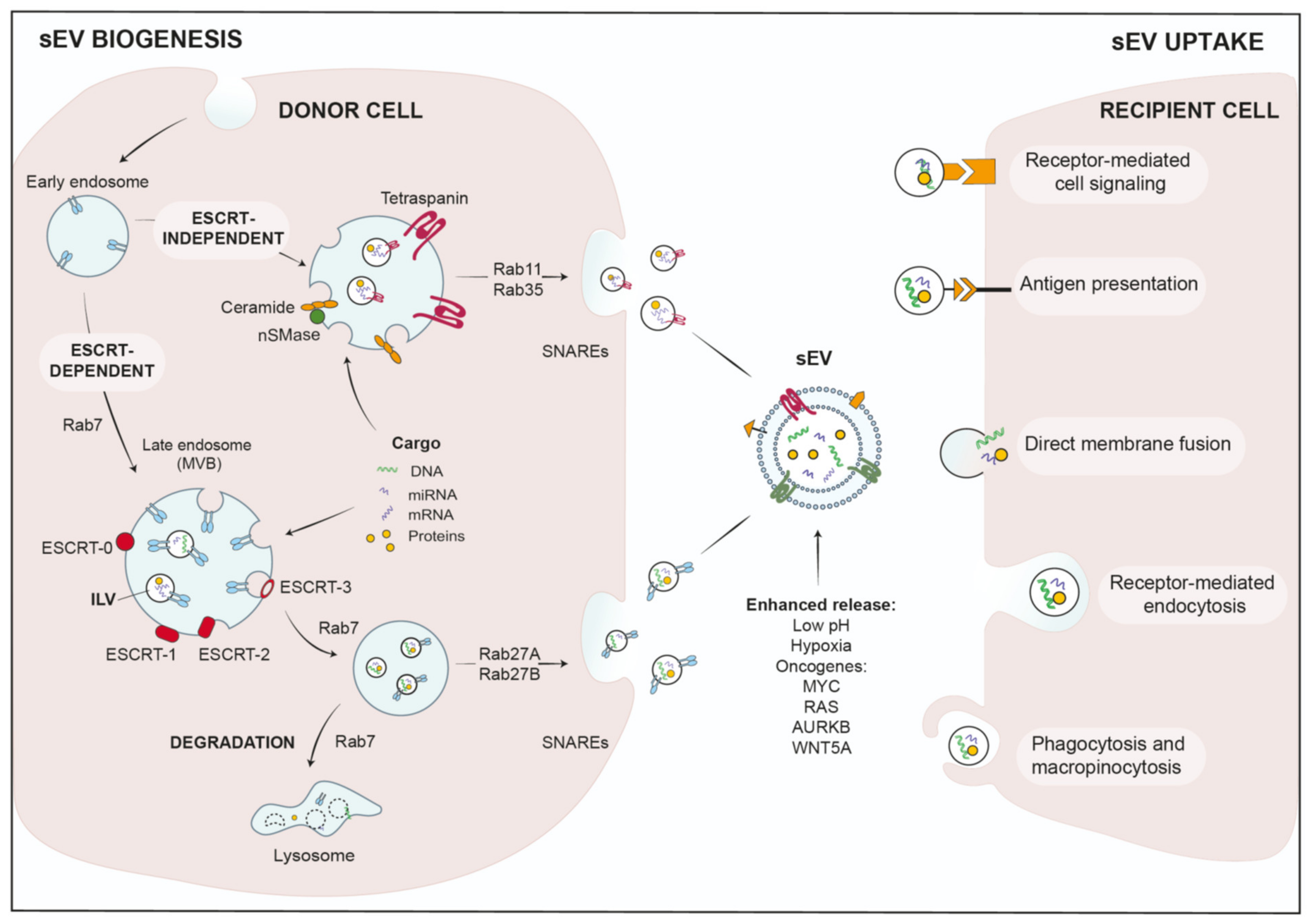
2.1. sEV Biogenesis and Cargo Loading
2.2. sEV-Uptake in Recipient Cells
2.3. Modifiers of sEV-Release in Tumor Cells
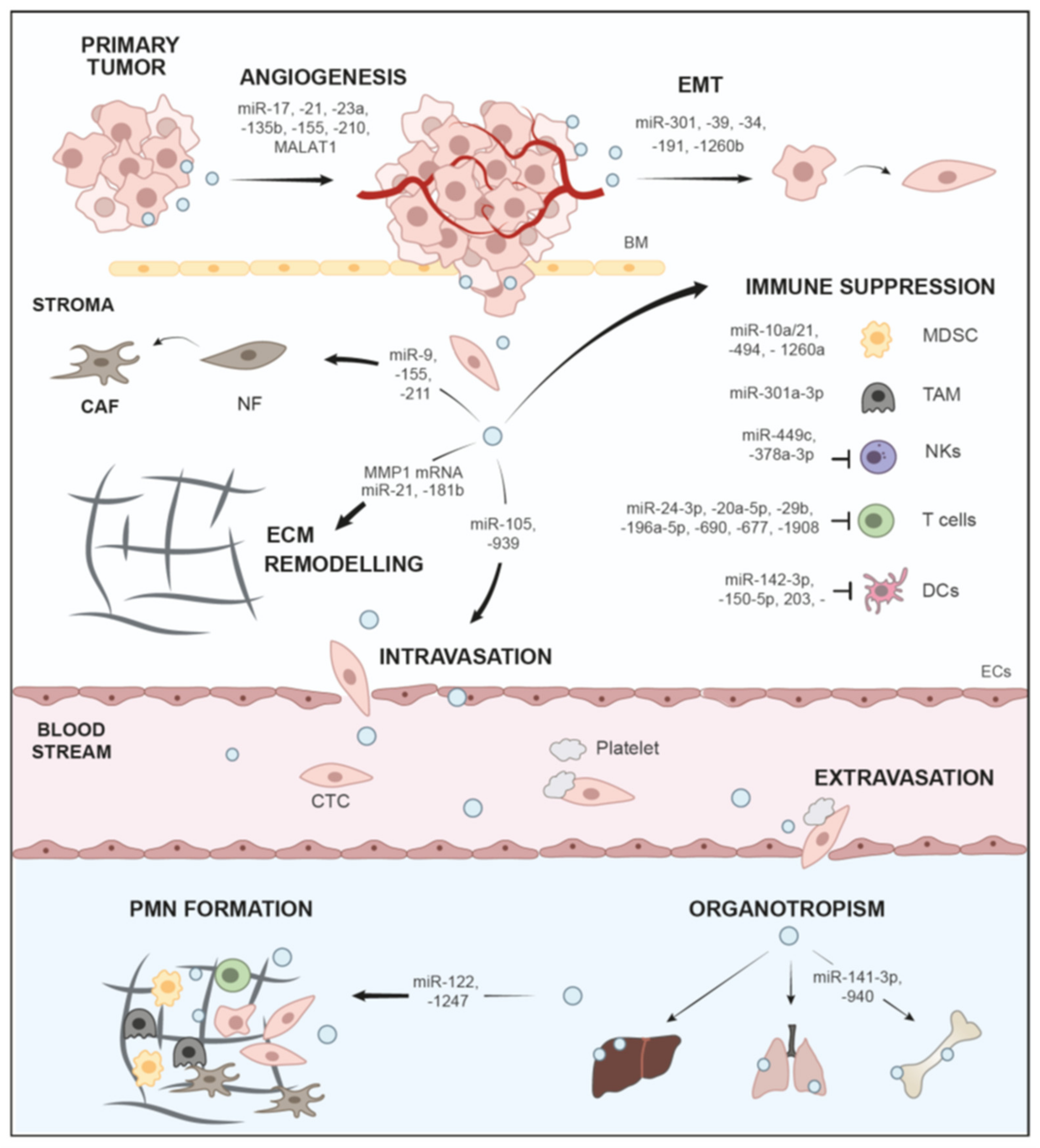
3. Role of sEVs in Local Stroma Invasion
3.1. Epithelial-to-Mesenchymal Transition (EMT)
3.2. ECM Degradation
4. sEV-Based Communication in the Tumor Microenvironment (TME)
4.1. sEV-Based Crosstalk with Cancer-Associated Fibroblasts (CAFs)
4.2. sEVs in Immune Suppression
4.2.1. Innate Antitumor Defense—Natural Killer Cells (NKs)
4.2.2. M2-Polarized Macrophages
4.2.3. Adaptive Antitumor Immunity—Targeting Antigen-Presenting Dendritic Cells (DCs) and Anti-Tumor T-Cells
4.2.4. Myeloid-Derived Suppressor Cells (MDSCs)
4.3. sEVs in Angiogenesis
5. Intravasation of Tumor Cells and Survival in the Circulation
6. Extravasation from the Circulation
7. sEV-Mediated Organotropism and Formation of Pre-Metastatic Niches
8. sEVs as Biomarker Platforms and Therapeutic Vehicles
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Dudas, J. Supportive and rejective functions of tumor stroma on tumor cell growth, survival, and invasivity: The cancer evolution. Front. Oncol. 2015, 5, 44. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Thuma, F.; Zöller, M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin. Cancer Biol. 2014, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D′Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Redis, R.S.; Calin, S.; Yang, Y.; You, M.J.; Calin, G.A. Cell-to-cell miRNA transfer: From body homeostasis to therapy. Pharmacol Ther. 2012, 136, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Dror, S.; Sander, L.; Schwartz, H.; Sheinboim, D.; Barzilai, A.; Dishon, Y.; Apcher, S.; Golan, T.; Greenberger, S.; Barshack, I.; et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 2016, 18, 1006–1017. [Google Scholar] [CrossRef]
- Shang, D.; Xie, C.; Hu, J.; Tan, J.; Yuan, Y.; Liu, Z.; Yang, Z. Pancreatic cancer cell-derived exosomal microRNA-27a promotes angiogenesis of human microvascular endothelial cells in pancreatic cancer via BTG2. J. Cell Mol. Med. 2020, 24, 588–604. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma cell-derived exosomes promote epithelial-mesenchymal transition in primary melanocytes through paracrine/autocrine signaling in the tumor microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Li, S.L.; Ma, Y.Y.; Diao, Y.J.; Yang, L.; Su, M.Q.; Li, Z.; Ji, Y.; Wang, J.; Lei, L.; et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget 2017, 8, 94834–94849. [Google Scholar] [CrossRef] [Green Version]
- Thind, A.; Wilson, C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J. Extracell. Vesicles 2016, 5, 31292. [Google Scholar] [CrossRef]
- Allenson, K.; Castillo, J.; San Lucas, F.A.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol 2017, 28, 741–747. [Google Scholar] [CrossRef]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated with Outcomes of Patients with Pancreatic Cancer. Gastroenterology 2019, 156, 108.e104–118.e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Wolfram, J.; Srivastava, S. Extracellular Vesicles in Cancer Detection: Hopes and Hypes. Trends Cancer 2021, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nature Commun. 2018, 9, 960. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Théry, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer-implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugeratski, F.G.; Hodge, K.; Lilla, S.; McAndrews, K.M.; Zhou, X.; Hwang, R.F.; Zanivan, S.; Kalluri, R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell Biol. 2021, 23, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. Exosomes as a Multicomponent Biomarker Platform in Cancer. Trends Cancer 2020, 6, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.B.; Oestreich, A.J.; Winistorfer, S.; Nguyen, D.; Payne, J.A.; Katzmann, D.J.; Piper, R. ESCRT ubiquitin-binding domains function cooperatively during MVB cargo sorting. J. Cell Biol. 2009, 185, 213–224. [Google Scholar] [CrossRef]
- Meister, M.; Banfer, S.; Gartner, U.; Koskimies, J.; Amaddii, M.; Jacob, R.; Tikkanen, R. Regulation of cargo transfer between ESCRT-0 and ESCRT-I complexes by flotillin-1 during endosomal sorting of ubiquitinated cargo. Oncogenesis 2017, 6, e344. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Babst, M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr. Opin. Cell Biol. 2011, 23, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Tschuschke, M.; Kocherova, I.; Bryja, A.; Mozdziak, P.; Angelova Volponi, A.; Janowicz, K.; Sibiak, R.; Piotrowska-Kempisty, H.; Izycki, D.; Bukowska, D.; et al. Inclusion Biogenesis, Methods of Isolation and Clinical Application of Human Cellular Exosomes. J. Clin. Med. 2020, 9, 436. [Google Scholar] [CrossRef] [Green Version]
- Middleton, R.C.; Rogers, R.G.; De Couto, G.; Tseliou, E.; Luther, K.; Holewinski, R.; Soetkamp, D.; Van Eyk, J.E.; Antes, T.J.; Marbán, E. Newt cells secrete extracellular vesicles with therapeutic bioactivity in mammalian cardiomyocytes. J. Extracell. Vesicles 2018, 7, 1456888. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging. 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Yang, H.; Wang, J.; Ru, W.; Wu, J.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Exosome biogenesis, secretion and function of exosomal miRNAs in skeletal muscle myogenesis. Cell Prolif. 2020, 53, e12857. [Google Scholar] [CrossRef]
- Andreu, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.; Rossello, C.A.; Fernandez-Garcia, P.; Llado, V.; Kakhlon, O.; Escriba, P.V. The Implications for Cells of the Lipid Switches Driven by Protein-Membrane Interactions and the Development of Membrane Lipid Therapy. Int. J. Mol. Sci. 2020, 21, 2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Ren, Y.; Hu, X.; Mu, J.; Samykutty, A.; Zhuang, X.; Deng, Z.; Kumar, A.; Zhang, L.; Merchant, M.L.; et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat. Commun. 2017, 8, 14448. [Google Scholar] [CrossRef] [PubMed]
- Ni, K.; Wang, C.; Carnino, J.M.; Jin, Y. The Evolving Role of Caveolin-1: A Critical Regulator of Extracellular Vesicles. Med. Sci. 2020, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, S.P.; Lin, Z.X.; Jiang, X.Y.; Yu, X.Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharm. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef] [Green Version]
- Di Giaimo, R.; Penna, E.; Pizzella, A.; Cirillo, R.; Perrone-Capano, C.; Crispino, M. Cross Talk at the Cytoskeleton-Plasma Membrane Interface: Impact on Neuronal Morphology and Functions. Int. J. Mol. Sci. 2020, 21, 9133. [Google Scholar] [CrossRef]
- Jin, H.; Tang, Y.; Yang, L.; Peng, X.; Li, B.; Fan, Q.; Wei, S.; Yang, S.; Li, X.; Wu, B.; et al. Rab GTPases: Central Coordinators of Membrane Trafficking in Cancer. Front. Cell Dev. Biol. 2021, 9, 648384. [Google Scholar] [CrossRef]
- Sinha, S.; Hoshino, D.; Hong, N.H.; Kirkbride, K.C.; Grega-Larson, N.E.; Seiki, M.; Tyska, M.J.; Weaver, A.M. Cortactin promotes exosome secretion by controlling branched actin dynamics. J. Cell Biol. 2016, 214, 197–213. [Google Scholar] [CrossRef]
- Helgeson, L.A.; Nolen, B.J. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. eLife 2013, 2, e00884. [Google Scholar] [CrossRef] [PubMed]
- Armacki, M.; Polaschek, S.; Waldenmaier, M.; Morawe, M.; Ruhland, C.; Schmid, R.; Lechel, A.; Tharehalli, U.; Steup, C.; Bektas, Y.; et al. Protein Kinase D1, Reduced in Human Pancreatic Tumors, Increases Secretion of Small Extracellular Vesicles From Cancer Cells That Promote Metastasis to Lung in Mice. Gastroenterology 2020, 159, 1019.e22–1035.e22. [Google Scholar] [CrossRef]
- Eiseler, T.; Hausser, A.; De Kimpe, L.; Van Lint, J.; Pfizenmaier, K. Protein kinase D controls actin polymerization and cell motility through phosphorylation of cortactin. J. Biol. Chem. 2010, 285, 18672–18683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, S.; Döppler, H.; Perez, E.A.; Andorfer, C.A.; Sun, Z.; Anastasiadis, P.Z.; Thompson, E.; Geiger, X.J.; Storz, P. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013, 15, R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Messenger, S.W.; Woo, S.S.; Sun, Z.; Martin, T.F.J. A Ca(2+)-stimulated exosome release pathway in cancer cells is regulated by Munc13-4. J. Cell Biol. 2018, 217, 2877–2890. [Google Scholar] [CrossRef]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chen, C.K.; Ho, C.M.; Lee, S.S.; Chang, C.Y.; Chen, K.J.; Jou, Y.S. EIF3C-enhanced exosome secretion promotes angiogenesis and tumorigenesis of human hepatocellular carcinoma. Oncotarget 2018, 9, 13193–13205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin. Chim. Acta 2016, 454, 28–32. [Google Scholar] [CrossRef]
- Arbelaiz, A.; Azkargorta, M.; Krawczyk, M.; Santos-Laso, A.; Lapitz, A.; Perugorria, M.J.; Erice, O.; Gonzalez, E.; Jimenez-Agüero, R.; Lacasta, A.; et al. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2017, 66, 1125–1143. [Google Scholar] [CrossRef]
- Cumba Garcia, L.M.; Peterson, T.E.; Cepeda, M.A.; Johnson, A.J.; Parney, I.F. Isolation and Analysis of Plasma-Derived Exosomes in Patients With Glioma. Front. Oncol. 2019, 9, 651. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, E.M.; Schultz, S.W.; Schink, K.O.; Pedersen, N.M.; Nähse, V.; Carlson, A.; Brech, A.; Stenmark, H.; Raiborg, C. Concerted ESCRT and clathrin recruitment waves define the timing and morphology of intraluminal vesicle formation. Nat. Commun. 2018, 9, 2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattissek, C.; Teis, D. The role of the endosomal sorting complexes required for transport (ESCRT) in tumorigenesis. Mol. Membr. Biol. 2014, 31, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Li, X.; Wang, Y.; Dong, M.; Zhan, F.H.; Liu, J. The ceramide pathway is involved in the survival, apoptosis and exosome functions of human multiple myeloma cells in vitro. Acta Pharm. Sin. 2018, 39, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, S.; Paisner, R.; Camarda, R.; Gupta, S.; Momcilovic, O.; Kohnz, R.A.; Avsaroglu, B.; L’Etoile, N.D.; Perera, R.M.; Nomura, D.K.; et al. Oncogene-regulated release of extracellular vesicles. Dev. Cell 2021, 56, 1989.e6–2006.e6. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991–52002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demory Beckler, M.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell Proteom. 2013, 12, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, D.J.; Franklin, J.L.; Dou, Y.; Liu, Q.; Higginbotham, J.N.; Demory Beckler, M.; Weaver, A.M.; Vickers, K.; Prasad, N.; Levy, S.; et al. KRAS-dependent sorting of miRNA to exosomes. eLife 2015, 4, e07197. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Demory Beckler, M.; Poulin, E.J.; Walsh, A.J.; Skala, M.C.; McKinley, E.T.; Manning, H.C.; Hight, M.R.; et al. Mutant KRAS Exosomes Alter the Metabolic State of Recipient Colonic Epithelial Cells. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 627.e6–629.e6. [Google Scholar] [CrossRef] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Ekström, E.J.; Bergenfelz, C.; von Bülow, V.; Serifler, F.; Carlemalm, E.; Jönsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Zavyalova, M.V.; Denisov, E.V.; Tashireva, L.A.; Savelieva, O.E.; Kaigorodova, E.V.; Krakhmal, N.V.; Perelmuter, V.M. Intravasation as a Key Step in Cancer Metastasis. Biochem 2019, 84, 762–772. [Google Scholar] [CrossRef]
- Knights, A.J.; Funnell, A.P.; Crossley, M.; Pearson, R.C. Holding Tight: Cell Junctions and Cancer Spread. Trends Cancer Res. 2012, 8, 61–69. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Nam, R.K.; Benatar, T.; Wallis, C.J.; Amemiya, Y.; Yang, W.; Garbens, A.; Naeim, M.; Sherman, C.; Sugar, L.; Seth, A. MiR-301a regulates E-cadherin expression and is predictive of prostate cancer recurrence. Prostate 2016, 76, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shan, Z.; Hong, J.; Yang, L. MicroRNA-92a promotes epithelial-mesenchymal transition through activation of PTEN/PI3K/AKT signaling pathway in non-small cell lung cancer metastasis. Int. J. Oncol. 2017, 51, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Wei, K.; Hu, L.Q.; Zhou, C.R.; Lu, Z.B.; Zhan, G.S.; Pan, X.L.; Pan, C.F.; Wang, J.; Wen, W.; et al. Exosome-mediated transfer of miR-1260b promotes cell invasion through Wnt/β-catenin signaling pathway in lung adenocarcinoma. J. Cell Physiol. 2020, 235, 6843–6853. [Google Scholar] [CrossRef]
- Bigagli, E.; Luceri, C.; Guasti, D.; Cinci, L. Exosomes secreted from human colon cancer cells influence the adhesion of neighboring metastatic cells: Role of microRNA-210. Cancer Biol. Ther. 2016, 17, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Ma, C.; Zhou, T.; Dong, X.; Luo, Q.; Geng, L.; Ding, L.; Zhang, Y.; Zhang, L.; Li, N.; et al. Exosomes derived from gemcitabine-resistant cells transfer malignant phenotypic traits via delivery of miRNA-222-3p. Mol. Cancer 2017, 16, 132. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell. Biochem. 2008, 105, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Shan, Y.; Chen, J.; Yue, H.; You, B.; Shi, S.; Li, X.; Cao, X. Matrix metalloproteinase 13-containing exosomes promote nasopharyngeal carcinoma metastasis. Cancer Sci. 2015, 106, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.Y.; Dugas-Ford, J.; Seiki, M.; Chang, J.H.; Azar, D.T. Evidence for the Involvement of MMP14 in MMP2 Processing and Recruitment in Exosomes of Corneal Fibroblasts. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5323–5329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, A.; Yoshioka, Y.; Yamamoto, Y.; Ishikawa, M.; Ikeda, S.I.; Kato, T.; Kiyono, T.; Takeshita, F.; Kajiyama, H.; Kikkawa, F.; et al. Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 2017, 8, 14470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Liu, R.; Shi, Y.J.; Yin, L.H.; Pu, Y.P. Exosome-shuttling microRNA-21 promotes cell migration and invasion-targeting PDCD4 in esophageal cancer. Int. J. Oncol. 2016, 48, 2567–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J. Considering Exosomal miR-21 as a Biomarker for Cancer. J. Clin. Med. 2016, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.J.; Wang, Z.Y.; Chen, R.; Xiong, J.; Yao, Y.L.; Wu, J.H.; Li, G.X. Macrophage-secreted Exosomes Delivering miRNA-21 Inhibitor can Regulate BGC-823 Cell Proliferation. Asian Pac. J. Cancer Prev. 2015, 16, 4203–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.H.; Tian, D.; Yang, Z.C.; Li, J.L. Exosomal miR-21 promotes proliferation, invasion and therapy resistance of colon adenocarcinoma cells through its target PDCD4. Sci. Rep. 2020, 10, 8271. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lu, J.; Chen, L.; Bian, H.; Hu, J.; Li, D.; Xia, C.; Xu, H. Tumor-Derived EV-Encapsulated miR-181b-5p Induces Angiogenesis to Foster Tumorigenesis and Metastasis of ESCC. Mol. Ther. Nucleic. Acids 2020, 20, 421–437. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, N.; Caetano, M.S.; Cumpian, A.M.; Unver, N.; De la Garza Ramos, C.; Noble, O.; Daliri, S.; Hernandez, B.J.; Gutierrez, B.A.; Evans, S.E.; et al. IL22 Promotes Kras-Mutant Lung Cancer by Induction of a Protumor Immune Response and Protection of Stemness Properties. Cancer Immunol. Res. 2018, 6, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Todoric, J.; Karin, M. The Fire within: Cell-Autonomous Mechanisms in Inflammation-Driven Cancer. Cancer Cell 2019, 35, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of Exosomes in Crosstalk Between Cancer-Associated Fibroblasts and Cancer Cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. Febs. j. 2021, 288, 10–35. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef]
- Duan, B.; Shi, S.; Yue, H.; You, B.; Shan, Y.; Zhu, Z.; Bao, L.; You, Y. Exosomal miR-17-5p promotes angiogenesis in nasopharyngeal carcinoma via targeting BAMBI. J. Cancer 2019, 10, 6681–6692. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.S.; Kanugula, S.S.; Sudhir, S.; Pereira, M.P.; Jain, S.; Aghi, M.K. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012, 40, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef]
- Gu, J.; Qian, H.; Shen, L.; Zhang, X.; Zhu, W.; Huang, L.; Yan, Y.; Mao, F.; Zhao, C.; Shi, Y.; et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-β/Smad pathway. PLoS ONE 2012, 7, e52465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015, 106, 1362–1369. [Google Scholar] [CrossRef]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther Nucleic Acids 2020, 19, 654–667. [Google Scholar] [CrossRef]
- Yue, X.; Lan, F.; Xia, T. Hypoxic Glioma Cell-Secreted Exosomal miR-301a Activates Wnt/β-catenin Signaling and Promotes Radiation Resistance by Targeting TCEAL7. Mol. Ther. 2019, 27, 1939–1949. [Google Scholar] [CrossRef]
- Cao, M.; Seike, M.; Soeno, C.; Mizutani, H.; Kitamura, K.; Minegishi, Y.; Noro, R.; Yoshimura, A.; Cai, L.; Gemma, A. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. Int. J. Oncol. 2012, 41, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhu, W.; Chen, Q.; Yuan, Y.; Wang, Y.; Wang, J.; Wu, X. Ovarian cancer cell-secreted exosomal miR-205 promotes metastasis by inducing angiogenesis. Theranostics 2019, 9, 8206–8220. [Google Scholar] [CrossRef]
- Sun, L.P.; Xu, K.; Cui, J.; Yuan, D.Y.; Zou, B.; Li, J.; Liu, J.L.; Li, K.Y.; Meng, Z.; Zhang, B. Cancer-associated fibroblast-derived exosomal miR-382-5p promotes the migration and invasion of oral squamous cell carcinoma. Oncol. Rep. 2019, 42, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.B.; Li, Z.L.; Luo, D.H.; Huang, B.J.; Chen, Y.S.; Zhang, X.S.; Cui, J.; Zeng, Y.X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4(+) T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Gao, C.; Huang, X.Y.; Lu, J.C.; Guo, X.J.; Shi, G.M.; Cai, J.B.; Ke, A.W. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol. Cancer 2020, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briand, J.; Garnier, D.; Nadaradjane, A.; Clément-Colmou, K.; Potiron, V.; Supiot, S.; Bougras-Cartron, G.; Frenel, J.S.; Heymann, D.; Vallette, F.M.; et al. Radiotherapy-induced overexpression of exosomal miRNA-378a-3p in cancer cells limits natural killer cells cytotoxicity. Epigenomics 2020, 12, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Zhou, L.; Qian, Y.; Fu, M.; Chen, J.; Chen, J.; Xiang, J.; Wu, Z.; Jiang, G.; Cao, L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 2015, 6, 29877–29888. [Google Scholar] [CrossRef] [Green Version]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.M.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 6065. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yu, J.; Chen, L.; Tao, T.; Yi, S.; Hanley, S.J.B.; Yue, J.; Watari, H.; Sakuragi, N. Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene 2018, 37, 5257–5268. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef]
- Karimi, N.; Ali Hosseinpour Feizi, M.; Safaralizadeh, R.; Hashemzadeh, S.; Baradaran, B.; Shokouhi, B.; Teimourian, S. Serum overexpression of miR-301a and miR-23a in patients with colorectal cancer. J. Chin. Med. Assoc. 2019, 82, 215–220. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerloff, D.; Lützkendorf, J.; Moritz, R.K.C.; Wersig, T.; Mäder, K.; Müller, L.P.; Sunderkötter, C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J. Cell Mol. Med. 2020, 24, 9560–9573. [Google Scholar] [CrossRef]
- Di Modica, M.; Regondi, V.; Sandri, M.; Iorio, M.V.; Zanetti, A.; Tagliabue, E.; Casalini, P.; Triulzi, T. Breast cancer-secreted miR-939 downregulates VE-cadherin and destroys the barrier function of endothelial monolayers. Cancer Lett. 2017, 384, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, H.; Deng, T.; Ning, T.; Liu, R.; Liu, D.; Bai, M.; Ying, G.; Ba, Y. Exosomes Carrying MicroRNA-155 Target Forkhead Box O3 of Endothelial Cells and Promote Angiogenesis in Gastric Cancer. Mol. Ther. Oncolytics 2019, 15, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Wang, Y.; Dong, L.; Zhang, Y.; Zhang, Y.; Wang, C.; Zhang, Q.; Yang, S.; Cao, L.; Zhang, X.; et al. Hypoxic exosomes facilitate angiogenesis and metastasis in esophageal squamous cell carcinoma through altering the phenotype and transcriptome of endothelial cells. J. Exp. Clin. Cancer Res. 2019, 38, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Dang, W.; Zhang, S.; Yue, W.; Yang, L.; Zhai, X.; Yan, Q.; Lu, J. The role of exosomal noncoding RNAs in cancer. Mol. Cancer 2019, 18, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.J.; Lin, X.J.; Tang, X.Y.; Zheng, T.T.; Lin, Y.Y.; Hua, K.Q. Exosomal Metastasis-Associated Lung Adenocarcinoma Transcript 1 Promotes Angiogenesis and Predicts Poor Prognosis in Epithelial Ovarian Cancer. Int. J. Biol. Sci. 2018, 14, 1960–1973. [Google Scholar] [CrossRef] [Green Version]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar]
- Zhou, C.F.; Ma, J.; Huang, L.; Yi, H.Y.; Zhang, Y.M.; Wu, X.G.; Yan, R.M.; Liang, L.; Zhong, M.; Yu, Y.H.; et al. Cervical squamous cell carcinoma-secreted exosomal miR-221-3p promotes lymphangiogenesis and lymphatic metastasis by targeting VASH1. Oncogene 2019, 38, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Luo, Y.; He, W.; Zhao, Y.; Kong, Y.; Liu, H.; Zhong, G.; Li, Y.; Li, J.; Huang, J.; et al. Exosomal long noncoding RNA LNMAT2 promotes lymphatic metastasis in bladder cancer. J. Clin. Invest. 2020, 130, 404–421. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazaei, S.; Nouraee, N.; Moradi, A.; Mowla, S.J. A novel signaling role for miR-451 in esophageal tumor microenvironment and its contribution to tumor progression. Clin. Transl. Oncol. 2017, 19, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [Green Version]
- Egen, J.G.; Ouyang, W.; Wu, L.C. Human Anti-tumor Immunity: Insights from Immunotherapy Clinical Trials. Immunity 2020, 52, 36–54. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S.; et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 2019, 10, 254. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [Green Version]
- Batista, I.A.; Melo, S.A. Exosomes and the Future of Immunotherapy in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 567. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The effect of tumor-derived exosomes on immune regulation and cancer immunotherapy. Future Oncol. 2017, 13, 2583–2592. [Google Scholar] [CrossRef] [PubMed]
- O′Brien, D.I.; Nally, K.; Kelly, R.G.; O’Connor, T.M.; Shanahan, F.; O’Connell, J. Targeting the Fas/Fas ligand pathway in cancer. Expert Opin. Ther. Targets 2005, 9, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yang, F.; Yu, L.; Yu, Z.; Jiang, L.; Wang, Q.; Yang, Y.; Wang, L.; Cao, X.; Wang, J. Activated T cell exosomes promote tumor invasion via Fas signaling pathway. J. Immunol. 2012, 188, 5954–5961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Sharma, P.; Theodoraki, M.N.; Pietrowska, M.; Yerneni, S.S.; Lang, S.; Ferrone, S.; Whiteside, T.L. Molecular and Functional Profiles of Exosomes From HPV(+) and HPV(-) Head and Neck Cancer Cell Lines. Front. Oncol. 2018, 8, 445. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Sung, Y.H.; Pack, C.G.; Jung, M.K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fan, Y.; Che, X.; Hou, K.; Zhang, C.; Li, C.; Wen, T.; Wang, S.; Cheng, Y.; Liu, Y.; et al. 5-FU-Induced Upregulation of Exosomal PD-L1 Causes Immunosuppression in Advanced Gastric Cancer Patients. Front. Oncol. 2020, 10, 492. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, D.; Gnatta, E.; Padoan, A.; Fogar, P.; Furlanello, S.; Aita, A.; Bozzato, D.; Zambon, C.F.; Arrigoni, G.; Frasson, C.; et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget 2017, 8, 84928–84944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotelli, M.R.; Reinhart-King, C.A. Mechanical Forces in Tumor Angiogenesis. Adv. Exp. Med. Biol. 2018, 1092, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Jászai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1989–2000. [Google Scholar] [CrossRef]
- Sruthi, T.V.; Edatt, L.; Raji, G.R.; Kunhiraman, H.; Shankar, S.S.; Shankar, V.; Ramachandran, V.; Poyyakkara, A.; Kumar, S.V.B. Horizontal transfer of miR-23a from hypoxic tumor cell colonies can induce angiogenesis. J. Cell Physiol. 2018, 233, 3498–3514. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Langheinrich, M.C.; Schellerer, V.; Perrakis, A.; Lohmüller, C.; Schildberg, C.; Naschberger, E.; Stürzl, M.; Hohenberger, W.; Croner, R.S. Molecular mechanisms of lymphatic metastasis in solid tumors of the gastrointestinal tract. Int. J. Clin. Exp. Pathol. 2012, 5, 614–623. [Google Scholar] [PubMed]
- Rahman, M.; Mohammed, S. Breast cancer metastasis and the lymphatic system. Oncol. Lett. 2015, 10, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Pak, K.H.; Park, K.C.; Cheong, J.H. VEGF-C induced by TGF- β1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis. BMC Cancer 2019, 19, 799. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.; Shaker, H.; Morris, K.; Tugwood, J.D.; Kirwan, C.C. The significance of circulating tumour cells in breast cancer: A review. Breast 2014, 23, 552–560. [Google Scholar] [CrossRef]
- Hou, J.M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.J.; Greystoke, A.; Zhou, C.; Morris, K.; et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef]
- Brandt, B.; Junker, R.; Griwatz, C.; Heidl, S.; Brinkmann, O.; Semjonow, A.; Assmann, G.; Zänker, K.S. Isolation of prostate-derived single cells and cell clusters from human peripheral blood. Cancer Res. 1996, 56, 4556–4561. [Google Scholar] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Zhang, Q.; Lou, Y.; Yang, J.; Nie, G.; Chen, Q.; Chen, Y.; Zhang, J.; Wang, J.; Wei, T.; et al. Primary tumor-derived exosomes facilitate metastasis by regulating adhesion of circulating tumor cells via SMAD3 in liver cancer. Oncogene 2018, 37, 6105–6118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial-mesenchymal plasticity. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.t.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, P.; Martínez-Pena, I.; Piñeiro, R. Dangerous Liaisons: Circulating Tumor Cells (CTCs) and Cancer-Associated Fibroblasts (CAFs). Cancers 2020, 12, 2861. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Karpatkin, S.; Pearlstein, E.; Ambrogio, C.; Coller, B.S. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J. Clin. Investig. 1988, 81, 1012–1019. [Google Scholar] [CrossRef]
- Gil-Bernabé, A.M.; Lucotti, S.; Muschel, R.J. Coagulation and metastasis: What does the experimental literature tell us? Br. J. Haematol. 2013, 162, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Hisada, Y.; Mackman, N. Cancer-associated pathways and biomarkers of venous thrombosis. Blood 2017, 130, 1499–1506. [Google Scholar] [CrossRef]
- Khorana, A.A. Venous thromboembolism and prognosis in cancer. Thromb. Res. 2010, 125, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, D.; Magnus, N.; Lee, T.H.; Bentley, V.; Meehan, B.; Milsom, C.; Montermini, L.; Kislinger, T.; Rak, J. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J. Biol. Chem. 2012, 287, 43565–43572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Dean, W.L.; Lee, M.J.; Cummins, T.D.; Schultz, D.J.; Powell, D.W. Proteomic and functional characterisation of platelet microparticle size classes. Thromb. Haemost. 2009, 102, 711–718. [Google Scholar] [CrossRef]
- Dovizio, M.; Alberti, S.; Sacco, A.; Guillem-Llobat, P.; Schiavone, S.; Maier, T.J.; Steinhilber, D.; Patrignani, P. Novel insights into the regulation of cyclooxygenase-2 expression by platelet-cancer cell cross-talk. Biochem. Soc. Trans. 2015, 43, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Reymond, N.; d’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Köhler, S.; Ullrich, S.; Richter, U.; Schumacher, U. E-/P-selectins and colon carcinoma metastasis: First in vivo evidence for their crucial role in a clinically relevant model of spontaneous metastasis formation in the lung. Br. J. Cancer 2010, 102, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Chan, Y.K.; Zhang, H.; Liu, P.; Tsao, S.W.; Lung, M.L.; Mak, N.K.; Ngok-Shun Wong, R.; Ying-Kit Yue, P. Proteomic analysis of exosomes from nasopharyngeal carcinoma cell identifies intercellular transfer of angiogenic proteins. Int. J. Cancer 2015, 137, 1830–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Castellana, D.; Klingbeil, P.; Cuesta Hernández, I.; Vitacolonna, M.; Orlicky, D.J.; Roffler, S.R.; Brodt, P.; Zöller, M. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 2009, 11, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; von Au, A.; Schnölzer, M.; Hackert, T.; Zöller, M. CD44v6-competent tumor exosomes promote motility, invasion and cancer-initiating cell marker expression in pancreatic and colorectal cancer cells. Oncotarget 2016, 7, 55409–55436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, S.; Pucci, M.; Giallombardo, M.; Di Bella, M.A.; Santarpia, M.; Reclusa, P.; Gil-Bazo, I.; Rolfo, C.; Alessandro, R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep. 2017, 7, 3170. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Donoso-Quezada, J.; Ayala-Mar, S.; González-Valdez, J. The role of lipids in exosome biology and intercellular communication: Function, analytics and applications. Traffic 2021, 22, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhou, J.; Yuan, C.; Zhang, L.; Li, D.; Si, D.; Xiu, D.; Zhong, L. Metabolomics identifies serum and exosomes metabolite markers of pancreatic cancer. Metab. Off. J. Metab. Soc. 2019, 15, 86. [Google Scholar] [CrossRef]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-Y.; Sung, C.W.-H.; Chen, C.; Cheng, C.-M.; Lin, D.P.-C.; Huang, C.-T.; Hsu, M.-Y. Advances in exosomes technology. Clin. Chim. Acta 2019, 493, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef]
- Buscail, E.; Chauvet, A.; Quincy, P.; Degrandi, O.; Buscail, C.; Lamrissi, I.; Moranvillier, I.; Caumont, C.; Verdon, S.; Brisson, A.; et al. CD63-GPC1-Positive Exosomes Coupled with CA19-9 Offer Good Diagnostic Potential for Resectable Pancreatic Ductal Adenocarcinoma. Transl. Oncol. 2019, 12, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Frampton, A.E.; Prado, M.M.; Lopez-Jimenez, E.; Fajardo-Puerta, A.B.; Jawad, Z.A.R.; Lawton, P.; Giovannetti, E.; Habib, N.A.; Castellano, L.; Stebbing, J.; et al. Glypican-1 is enriched in circulating-exosomes in pancreatic cancer and correlates with tumor burden. Oncotarget 2018, 9, 19006–19013. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, S.; Iinuma, H.; Wada, K.; Takahashi, K.; Minezaki, S.; Kainuma, M.; Shibuya, M.; Miura, F.; Sano, K. Exosome-encapsulated microRNA-4525, microRNA-451a and microRNA-21 in portal vein blood is a high-sensitive liquid biomarker for the selection of high-risk pancreatic ductal adenocarcinoma patients. J. Hepatobiliary Pancreat. Sci. 2019, 26, 63–72. [Google Scholar] [CrossRef]
- Yuan, X.; Qian, N.; Ling, S.; Li, Y.; Sun, W.; Li, J.; Du, R.; Zhong, G.; Liu, C.; Yu, G.; et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics 2021, 11, 1429–1445. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Wang, P.Y.; Li, X.Y.; Chen, J.X.; Li, Y.; Zhang, X.Z.; Zhang, C.G.; Jiang, T.; Li, W.B.; Ding, W.; et al. Exosomal levels of miRNA-21 from cerebrospinal fluids associated with poor prognosis and tumor recurrence of glioma patients. Oncotarget 2015, 6, 26971–26981. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, Y.; Lu, J.; Sun, Y.; Xiao, H.; Liu, M.; Tian, L. Combined detection of serum exosomal miR-21 and HOTAIR as diagnostic and prognostic biomarkers for laryngeal squamous cell carcinoma. Med. Oncol. 2014, 31, 148. [Google Scholar] [CrossRef] [PubMed]
- Ohzawa, H.; Kumagai, Y.; Yamaguchi, H.; Miyato, H.; Sakuma, Y.; Horie, H.; Hosoya, Y.; Kawarai Lefor, A.; Sata, N.; Kitayama, J. Exosomal microRNA in peritoneal fluid as a biomarker of peritoneal metastases from gastric cancer. Ann. Gastroenterol. Surg. 2020, 4, 84–93. [Google Scholar] [CrossRef]
- Shin, S.; Park, Y.H.; Jung, S.H.; Jang, S.H.; Kim, M.Y.; Lee, J.Y.; Chung, Y.J. Urinary exosome microRNA signatures as a noninvasive prognostic biomarker for prostate cancer. NPJ Genom. Med. 2021, 6, 45. [Google Scholar] [CrossRef]
- De Miguel Perez, D.; Rodriguez Martinez, A.; Ortigosa Palomo, A.; Delgado Urena, M.; Garcia Puche, J.L.; Robles Remacho, A.; Exposito Hernandez, J.; Lorente Acosta, J.A.; Ortega Sanchez, F.G.; Serrano, M.J. Extracellular vesicle-miRNAs as liquid biopsy biomarkers for disease identification and prognosis in metastatic colorectal cancer patients. Sci. Rep. 2020, 10, 3974. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer*. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möhrmann, L.; Huang, H.J.; Hong, D.S.; Tsimberidou, A.M.; Fu, S.; Piha-Paul, S.A.; Subbiah, V.; Karp, D.D.; Naing, A.; Krug, A.; et al. Liquid Biopsies Using Plasma Exosomal Nucleic Acids and Plasma Cell-Free DNA Compared with Clinical Outcomes of Patients with Advanced Cancers. Clin. Cancer Res. 2018, 24, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.S.; Hoff, D.V.; Cridebring, D.; Goel, A. Extracellular Vesicles in Diagnosis and Treatment of Pancreatic Cancer: Current State and Future Perspectives. Cancers 2020, 12, 1530. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Bernard, V.; San Lucas, F.A.; Allenson, K.; Capello, M.; Kim, D.U.; Gascoyne, P.; Mulu, F.C.; Stephens, B.M.; Huang, J.; et al. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann. Oncol. 2018, 29, 223–229. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Wang, F.; Li, L.; Piontek, K.; Sakaguchi, M.; Selaru, F.M. Exosome miR-335 as a novel therapeutic strategy in hepatocellular carcinoma. Hepatology 2018, 67, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Raimondi, L.; Costa, V.; De Luca, A.; Carina, V.; Maglio, M.; Fini, M.; Alessandro, R.; Giavaresi, G. Engineered exosomes: A new promise for the management of musculoskeletal diseases. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Gonzalo, D.H.; Feely, M.; Rinaldi, C.; Belsare, S.; Zhai, H.; Kalra, K.; Gerber, M.H.; Forsmark, C.E.; Hughes, S.J. Stroma-derived extracellular vesicles deliver tumor-suppressive miRNAs to pancreatic cancer cells. Oncotarget 2018, 9, 5764–5777. [Google Scholar] [CrossRef] [Green Version]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.L.; Yao, J.L.; Wang, K.; Ai, H. Human Umbilical Cord Mesenchymal Stem Cell-Derived Extracellular Vesicles Inhibit Endometrial Cancer Cell Proliferation and Migration through Delivery of Exogenous miR-302a. Stem. Cells Int. 2019, 2019, 8108576. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D. Exosomal miRNA Cargo as Mediator of Immune Escape Mechanisms in Neuroblastoma. Cancer Res. 2019, 79, 1293–1294. [Google Scholar] [CrossRef] [Green Version]
- Lunavat, T.R.; Jang, S.C.; Nilsson, L.; Park, H.T.; Repiska, G.; Lässer, C.; Nilsson, J.A.; Gho, Y.S.; Lötvall, J. RNAi delivery by exosome-mimetic nanovesicles-Implications for targeting c-Myc in cancer. Biomaterials 2016, 102, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, S.; Bjornetro, T.; Lyckander, L.G.; Flatmark, K.; Dueland, S.; Samiappan, R.; Johansen, C.; Kalanxhi, E.; Ree, A.H.; Redalen, K.R. Circulating Exosomal miR-141-3p and miR-375 in Metastatic Progression of Rectal Cancer. Transl. Oncol. 2019, 12, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, T.; Zheng, X.; Yang, S.; Xu, K.; Chen, X.; Xu, F.; Wang, L.; Shen, Y.; Wang, T.; et al. Colorectal cancer-derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis 2018, 39, 1368–1379. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, X.; Gao, S.; Jing, F.; Yang, Y.; Du, L.; Zheng, G.; Li, P.; Li, C.; Wang, C. Exosomal long noncoding RNA CRNDE-h as a novel serum-based biomarker for diagnosis and prognosis of colorectal cancer. Oncotarget 2016, 7, 85551–85563. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.Y.; Gu, R.H.; Yan, B. Downregulation of exosome-encapsulated miR-548c-5p is associated with poor prognosis in colorectal cancer. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microRNA levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Ma, Y.Y.; Wang, J.; Zeng, X.F.; Li, R.; Kang, W.; Hao, X.K. Exosomal microRNA-141 is upregulated in the serum of prostate cancer patients. Onco Targets Ther. 2016, 9, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagirath, D.; Yang, T.L.; Bucay, N.; Sekhon, K.; Majid, S.; Shahryari, V.; Dahiya, R.; Tanaka, Y.; Saini, S. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res. 2018, 78, 1833–1844. [Google Scholar] [CrossRef] [Green Version]
- Tokuhisa, M.; Ichikawa, Y.; Kosaka, N.; Ochiya, T.; Yashiro, M.; Hirakawa, K.; Kosaka, T.; Makino, H.; Akiyama, H.; Kunisaki, C.; et al. Exosomal miRNAs from Peritoneum Lavage Fluid as Potential Prognostic Biomarkers of Peritoneal Metastasis in Gastric Cancer. PLoS ONE 2015, 10, e0130472. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fu, H.; Wang, B.; Zhang, X.; Mao, J.; Li, X.; Wang, M.; Sun, Z.; Qian, H.; Xu, W. Exosomal miR-423-5p targets SUFU to promote cancer growth and metastasis and serves as a novel marker for gastric cancer. Mol. Carcinog. 2018, 57, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, T.; Feng, D.; Li, J.; Wu, M.; Peng, X.; Wang, B.; Zhan, X.; Fu, P. Screening of non-invasive miRNA biomarker candidates for metastasis of gastric cancer by small RNA sequencing of plasma exosomes. Carcinogenesis 2020, 41, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Muller, V.; Milde-Langosch, K.; Trillsch, F.; Pantel, K.; Schwarzenbach, H. Diagnostic and prognostic relevance of circulating exosomal miR-373, miR-200a, miR-200b and miR-200c in patients with epithelial ovarian cancer. Oncotarget 2016, 7, 16923–16935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Kamohara, H.; Kinoshita, K.; Kurashige, J.; Ishimoto, T.; Iwatsuki, M.; Watanabe, M.; Baba, H. Clinical impact of serum exosomal microRNA-21 as a clinical biomarker in human esophageal squamous cell carcinoma. Cancer 2013, 119, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Wu, J.; Wu, J.; Ji, A.; Qiang, G.; Jiang, Y.; Jiang, C.; Ding, Y. Exosomal miR-665 as a novel minimally invasive biomarker for hepatocellular carcinoma diagnosis and prognosis. Oncotarget 2017, 8, 80666–80678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Nan, A.; Chen, L.; Li, X.; Jia, Y.; Qiu, M.; Dai, X.; Zhou, H.; Zhu, J.; Zhang, H.; et al. Circular RNA circSATB2 promotes progression of non-small cell lung cancer cells. Mol. Cancer 2020, 19, 101. [Google Scholar] [CrossRef]
- Que, R.; Ding, G.; Chen, J.; Cao, L. Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J. Surg. Oncol. 2013, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, Z.; Jiang, P.; Peng, M.; Zhang, X.; Chen, K.; Liu, H.; Bi, H.; Liu, X.; Li, X. Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.R.; Grossmann, K.F.; Cassidy, P.B.; Yang, C.H.; Fan, M.; Kopelovich, L.; Leachman, S.A.; Pfeffer, L.M. Detection of Exosomal miRNAs in the Plasma of Melanoma Patients. J. Clin. Med. 2015, 4, 2012–2027. [Google Scholar] [CrossRef]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release Off. Control. Release Soc. 2015, 220, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Delivery of Exogenous miR-124 to Glioblastoma Multiform Cells by Wharton’s Jelly Mesenchymal Stem Cells Decreases Cell Proliferation and Migration, and Confers Chemosensitivity. Stem Cell Rev. Rep. 2018, 14, 236–246. [Google Scholar] [CrossRef]
- De la Fuente, A.; Alonso-Alconada, L.; Costa, C.; Cueva, J.; Garcia-Caballero, T.; Lopez-Lopez, R.; Abal, M. M-Trap: Exosome-Based Capture of Tumor Cells as a New Technology in Peritoneal Metastasis. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Step | Molecule | Cell of Origin | Effect | Ref. |
|---|---|---|---|---|
| EMT | miR-301a | GBM, PC | Activation of Wnt/β-catenin signaling, suppression of TCEAL7, p63 and E-cadherin | [89,124] |
| miR-92a | NSCLC | Activation of PTEN/PI3K/AKT | [90] | |
| miR-191 | Melanoma | Activation of MAPK signaling | [14] | |
| miR-1260b | LC | Activation of Wnt/β-catenin signaling via inhibition of sFRP1 and Smad4 | [91] | |
| miR-181d-5p | CAFs in BC | Suppression of CDC2 and HOXA5 | ||
| miR-23a | NSCLC | Induction of TGFβ signaling and inhibition of E-cadherin | [125] | |
| miR-21 miR-143 miR-378e | CAFs in BC | Upregulation of NANOG, SOX2, SNAIL and ZEB | [126] | |
| miR-499a-5p | LC | Activation of mTOR signaling | [127] | |
| miR-19b-3p | CCRCC | Downregulation of PTEN | [127] | |
| miR-382-5p | CAFs in OSCC | Upregulation of ß-catenin and N-cadherin | [128] | |
| ECM degradation | miR-21 | CRC, EC | MMP activation via inhibition of PDCD4 and TIMP-3 | [101,102,104] |
| miR-181b | HCC, ESCC | MMP activation via inhibition of TIMP-3 | [105,106] | |
| MMP1 mRNA | OC | Enhanced MMP1 expression in recipient cells | [100] | |
| miR-382-5p | CAFs in OSCC | Upregulation of MMP-9 and MMP-3 | [128] | |
| Anoikis resistance | miR-210 | CRC | [92] | |
| miR-222-3p | NSCLC | Inhibition of SOCS3 | [93] | |
| Stroma | miR-9 | BC | CAF formation | [121] |
| miR-211 | Melanoma | CAF formation via MAPK activation | [12] | |
| miR-155 | PDAC | CAF formation via downregulation of TP53INP1 | [122] | |
| Immune modulation | miR-24-3p, miR-891a, miR-106a-5p, miR-20a-5p, miR-1908 | NPC | T-cell exhaustion via downregulation of the MARK1 signaling pathway | [129] |
| miR-690, miR-677, miR-29b | Melanoma | CD4+ T-cell apoptosis, increase of caspase-3, caspase-7 and caspase-9, downregulation of BCL-2 | [130] | |
| circUHRF1 | HCC | NK dysfunction via inhibition of IFN-γ and TNF-α secretion | [131] | |
| miR-301a-3p | PDAC | M2 macrophage polarization via activation of PTEN/PI3Kγ signaling | [132] | |
| miR-378a-3p | Various cell lines | Decreased NK cytotoxicity via inhibition of granzyme B | [133] | |
| miR-212-3p | PDAC | DC dysfunction via PFXAP inhibition and compromised MHCII expression | [134] | |
| miR-150-5p, miR-142-3p | Tregs | DC exhaustion | [135] | |
| miR-203 | PDAC | DC dysfunction via TLR4 inhibition | [136] | |
| miR-18a | CeC | T-cell exhaustion, suppression of PTEN, WNK2 and SOX6, and enhanced PD-L1 levels | [137] | |
| miR-23a | HCC | T-cell exhaustion, suppression of PTEN and enhanced PD-L1 levels | [138,139] | |
| miR-10a, miR-21 | Glioma | MDSC expansion by targeting RAR and PTEN | [140] | |
| miR-1246 | Mutant p53 cancer cell lines | M2 macrophage polarization via targeting TERF2IP | [141,142] | |
| Let7 | Hypoxic tumor cells | M2 polarization and metabolic reprogramming in macrophages via suppression of AKT-mTOR signaling | [143] | |
| miR-29a-3p and miR-21-5p | TAMs in OC | Suppression of STAT3 in CD4 T-cells and corresponding Treg induction | [144] | |
| miR-125b-5p | Melanoma | M2 macrophage polarization via downregulation of LIPA | [145] | |
| miR-27a-3p | BC | PD-L1-mediated immune evasion via targeting MAGI2/PTEN/PI3K signaling | [146] | |
| Intra- vasation | miR-939 | BC | Downregulation of VE-Cadherin in ECs | [147] |
| miR-105 | BC | Downregulation of ZO-1 in ECs | [148] | |
| miR-181c | BC | Destruction of blood–brain barrier and promotion of brain metastasis via downregulation of PDPK1 | [149] | |
| Angio- genesis | miR-17-5p | NPC | Promoting angiogenic activity in ECs via AKT/VEGF-A expression | [115] |
| Mir-155 | GC | Suppression of FOXO3a and c-MYC to enhance the expression of VEGF in ECs | [150] | |
| miR-27a | PDAC | Proliferation of ECs via inhibition of BTG2 | [13] | |
| miR-135b, miR-210, miR-21, miR-23a | Hypoxic tumor cells | Induction of blood vessel formation | [151,152,153] | |
| MALAT1 | OC | Pro-angiogenic gene expression in HUVECs | [154] | |
| miR-205 | OC | Promotes angiogenesis in ECs via PTEN-AKT signaling | [127] | |
| lncRNA-Ccat2 | Glioma | Proliferation of ECs via upregulation of VEGF-A and TGFβ. Inhibition of apoptosis by targeting Bax and caspase-3 | [155] | |
| lncRNA-Pouf3 | Glioma | Pro-angiogenic gene expression in ECs | [155] | |
| Lymphangiogenesis | miR-221-3p | CeC | Expansion of lymphatic vessels via downregulation of VASH | [156] |
| LNMAT2 | Bladder cancer | Growth of lymphatic vessels via upregulation of PROX1 | [157] | |
| Organo- tropism | miR-141-3p | PC | Bone metastasis via increased osteoblast activity | [15] |
| miR-940 | PC, BC | Bone metastasis via increased osteoblast activity | [158] | |
| PMN | miR-122 | BC | Suppression of glycolytic enzymes in non-tumor cells | [116] |
| miR-1247-3p | HCC | CAF formation | [159] | |
| miR-451 | CAFs in ESCC | Pro-tumor PMN formation | [160] | |
| miR-25-3p | CRC | Enhanced vascular permeability and angiogenesis in PMN via targeting KLF2 and KLF4 | [161] |
| Cancer | sEV Cargo | Source | Diagnostic/Prognostic Value | Reference |
|---|---|---|---|---|
| CRC | miR-92a-3p | Serum | Liver and lung metastasis | [262] |
| miR-193a | Plasma | Liver metastasis | [47] | |
| miR-25-3p | Serum | Metastasis (Liver and lung metastasis in mice; involved PMN formation) | [161] | |
| miR-141-3p miR-375 | Plasma | Liver metastasis | [263] | |
| miR-21-5p | Plasma | Liver metastasis | [264] | |
| lncRNA CRNDE-h | Serum | Regional lymph node and distant metastasis | [265] | |
| miR-19b, miR-21, miR-222, miR-92a | Serum | Metastasis | [246] | |
| Low miR-548c-5p | Serum | Liver metastasis | [266] | |
| PC | miR-141 miR-375 | Serum | Metastasis | [267,268] |
| Low miR-636 High miR-21 High miR-16 High miR-142-3p High miR-451 | Urine | Metastasis | [245] | |
| miR-1246 | Serum | Metastasis | [269] | |
| GC | miR-21 miR-1225-5p | PLF | Peritoneal metastasis | [270] |
| miR-21-5p miR-92a-3p miR-223-3p miR-342-3p | PLF | Peritoneal metastasis | [244] | |
| miR-423-5p | Serum | Lymph node metastasis | [271] | |
| miR-10b-5p | Plasma | Lymph node metastasis | [272] | |
| miR-101-3p | Plasma | Ovarian metastasis | [272] | |
| miR-143-5p | Plasma | Liver metastasis | [272] | |
| OC | miR-200b miR-200c | Serum | Lymph node metastasis | [273] |
| ESCC | miR-21 | Serum | Metastasis | [274] |
| HCC | miR-665 | Serum | Metastasis | [275] |
| miR-1247-3p | Serum | Lung metastasis | [159] | |
| LSCC | miR-21 | Serum | Lymph node metastasis | [243] |
| Glioma | miR-21 | CSF | Tumor spinal/ventricle metastasis | [242] |
| BC | miR-105 | serum | Distant metastasis | [148] |
| miR-21 | Serum | Bone metastasis | [241] | |
| NSCLC | circSATB2 | Serum | Lymphatic metastasis | [276] |
| PDAC | miR-17-5p | Serum | Metastasis | [277] |
| Circ-IARS | Plasma | Tumor-node metastasis and liver metastasis | [278] | |
| miR-21 | Plasma | Lymph node metastasis | [240] | |
| Melanoma | miR-17 miR-19a miR-21 miR-126 miR-149 | Plasma | Metastasis | [279] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seibold, T.; Waldenmaier, M.; Seufferlein, T.; Eiseler, T. Small Extracellular Vesicles and Metastasis—Blame the Messenger. Cancers 2021, 13, 4380. https://doi.org/10.3390/cancers13174380
Seibold T, Waldenmaier M, Seufferlein T, Eiseler T. Small Extracellular Vesicles and Metastasis—Blame the Messenger. Cancers. 2021; 13(17):4380. https://doi.org/10.3390/cancers13174380
Chicago/Turabian StyleSeibold, Tanja, Mareike Waldenmaier, Thomas Seufferlein, and Tim Eiseler. 2021. "Small Extracellular Vesicles and Metastasis—Blame the Messenger" Cancers 13, no. 17: 4380. https://doi.org/10.3390/cancers13174380
APA StyleSeibold, T., Waldenmaier, M., Seufferlein, T., & Eiseler, T. (2021). Small Extracellular Vesicles and Metastasis—Blame the Messenger. Cancers, 13(17), 4380. https://doi.org/10.3390/cancers13174380