
Therapeutic Potential of Targeting the SUMO Pathway in Cancer

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Basic Principles of SUMOylation and Its Role in Physiology
3. Altered Expression and Prognostic Significance of SUMO Pathway in Cancer
Regulation of SUMO Machinery Expression and Activity in Cancer
4. Genetic Changes Targeting SUMO Machinery in Cancer
4.1. Germline Variants
4.2. Somatic Mutations
5. SUMOylation Regulates Key Cancer Genes and Hallmark Properties of Cancer Cells
5.1. Substrates of SUMO Relevant for Cancer
5.2. SUMOylation in Cellular Processes Relevant for Cancer
6. SUMOylation in Hematologic Malignancies and Solid Tumors
6.1. Acute Promyelocytic Leukemia
6.2. Acute Myeloid Leukemias Other Than APL
6.3. Lymphomas
6.4. Multiple Myeloma
6.5. Breast Cancer
6.6. Prostate Cancer
6.7. Kidney Cancer
6.8. Lung Cancer
6.9. Hepatocellular Carcinoma
6.10. Gliomas
7. Therapeutic Targeting of the SUMO Pathway
7.1. SUMO E1 Inhibitors
7.2. SUMO E2 Inhibitors
7.3. SENP Inhibitors
7.4. Current Stage of Clinical Development of SUMO Pathway Inhibitors
7.5. Potential Rational Combinations
8. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabakaran, S.; Lippens, G.; Steen, H.; Gunawardena, J. Post-translational modification: Nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 565–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conibear, A.C. Deciphering protein post-translational modifications using chemical biology tools. Nat. Rev. Chem. 2020, 4, 674–695. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Zhang, X.; Sun, F.; Wang, J. Emerging Role of Protein Post-Translational Modification in the Potential Clinical Application of Cancer. Nano Life 2020, 10, 2040008. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Sundvall, M. Role of ubiquitin and SUMO in intracellular trafficking. Curr. Issues Mol. Biol. 2020, 35, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay-Kumar, S.; Bugg, C.E.; Cook, W.J. Structure of ubiquitin refined at 1.8 Å resolution. J. Mol. Biol. 1987, 194, 531–544. [Google Scholar] [CrossRef]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998, 280, 275–286. [Google Scholar] [CrossRef]
- Guo, D.; Li, M.; Zhang, Y.; Yang, P.; Eckenrode, S.; Hopkins, D.; Zheng, W.; Purohit, S.; Podolsky, R.H.; Muir, A.; et al. A functional variant of SUMO4, a new IκBα modifier, is associated with type diabetes. Nat. Genet. 2004, 36, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Bohren, K.M.; Nadkarni, V.; Song, J.H.; Gabbay, K.H.; Owerbach, D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 2004, 279, 27233–27238. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wansleeben, C.; Zhao, S.; Miao, P.; Paschen, W.; Yang, W. SUMO 2 is essential while SUMO 3 is dispensable for mouse embryonic development. EMBO Rep. 2014, 15, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.C.; Lee, C.C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef] [PubMed]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; D’Souza, R.C.J.; Yang, B.; Verlaan-De Vries, M.; Mann, M.; Vertegaal, A.C.O. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Tammsalu, T.; Matic, I.; Jaffray, E.G.; Ibrahim, A.F.M.; Tatham, M.H.; Hay, R.T. Proteome-wide identification of SUMO2 modification sites. Sci. Signal. 2014, 7, rs2. [Google Scholar] [CrossRef] [Green Version]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.P.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric Chains of SUMO-2 and SUMO-3 are Conjugated to Protein Substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [Green Version]
- Matic, I.; van Hagen, M.; Schimmel, J.; Macek, B.; Ogg, S.C.; Tatham, M.H.; Hay, R.T.; Lamond, A.I.; Mann, M.; Vertegaal, A.C.O. In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol. Cell. Proteom. 2008, 7, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Durrin, L.K.; Wilkinson, T.A.; Krontiris, T.G.; Chen, Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 14373–14378. [Google Scholar] [CrossRef] [Green Version]
- Hecker, C.M.; Rabiller, M.; Haglund, K.; Bayer, P.; Dikic, I. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 2006, 281, 16117–16127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, K.; Piller, T.; Müller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131, jcs211904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Erdene, T.; Nagamalleswari, K.; Yin, L.; Wu, K.; Pan, Z.Q.; Wilkinson, K.D. Identification and characterization of DEN1, a deneddylase of the ULP family. J. Biol. Chem. 2003, 278, 28892–28900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, A.; Müller, S. SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol. 2014, 15, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.S.; Schwienhorst, I.; Dohmen, R.J.; Blobel, G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997, 16, 5509–5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desterro, J.M.P.; Rodriguez, M.S.; Kemp, G.D.; Ronald, T.H. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 1999, 274, 10618–10624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuma, T.; Honda, R.; Ichikawa, G.; Tsumagari, N.; Yasuda, H. In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem. Biophys. Res. Commun. 1999, 254, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jääskeläinen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Pichler, A.; Gast, A.; Seeler, J.S.; Dejean, A.; Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 2002, 108, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Cappadocia, L.; Pichler, A.; Lima, C.D. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat. Struct. Mol. Biol. 2015, 22, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagey, M.H.; Melhuish, T.A.; Wotton, D. The polycomb protein Pc2 is a SUMO E3. Cell 2003, 113, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xu, X.; Chang, C.W.; Liu, Y. TRIM28 functions as the SUMO E3 ligase for PCNA in prevention of transcription induced DNA breaks. Proc. Natl. Acad. Sci. USA 2020, 117, 23588–23596. [Google Scholar] [CrossRef]
- Hietakangas, V.; Anckar, J.; Blomster, H.A.; Fujimoto, M.; Palvimo, J.J.; Nakai, A.; Sistonen, L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 2006, 103, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Stabell, M.; Sæther, T.; Røhr, Å.K.; Gabrielsen, O.S.; Myklebost, O. Methylation-dependent SUMOylation of the architectural transcription factor HMGA2. Biochem. Biophys. Res. Commun. 2021, 552, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Anckar, J.; Sistonen, L. SUMO: Getting it on. Biochem. Soc. Trans. 2007, 35, 1409–1413. [Google Scholar] [CrossRef] [Green Version]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.-P.; Mikkonen, L.; Toppari, J.; Palvimo, J.J.; Thesleff, I.; Jänne, O.A. Sumo-1 Function Is Dispensable in Normal Mouse Development. Mol. Cell. Biol. 2008, 28, 5381–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T.H. SUMO-Specific Protease 1 Is Essential for Stabilization of HIF1α during Hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef] [Green Version]
- Chiu, S.Y.; Asai, N.; Costantini, F.; Hsu, W. SUMO-specific protease 2 is essential for modulating p53-mdm2 in development of trophoblast stem cell niches and lineages. PLoS Biol. 2008, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.; Yang, K.; Wang, Z.; Sun, X.; Zou, Q.; Yu, X.; Cheng, J.; Tong, X.; Yeh, E.T.H.; Yang, J.; et al. DeSUMOylation of MKK7 kinase by the SUMO2/3 protease SENP3 potentiates lipopolysaccharide-induced inflammatory signaling in macrophages. J. Biol. Chem. 2018, 293, 3965–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Mink, S.; Wong, K.A.; Stein, N.; Getman, C.; Dempsey, P.W.; Wu, H.; Shuai, K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 2004, 5, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.; Sustmann, C.; Kieslinger, M.; Gilmozzi, A.; Irmer, D.; Kremmer, E.; Turck, C.; Grosschedl, R. PIASy-Deficient Mice Display Modest Defects in IFN and Wnt Signaling. J. Immunol. 2004, 173, 6189–6199. [Google Scholar] [CrossRef] [Green Version]
- Santti, H.; Mikkonen, L.; Anand, A.; Hirvonen-Santti, S.; Toppari, J.; Panhuysen, M.; Vauti, F.; Perera, W.; Corte, G.; Wurst, W.; et al. Disruption of the murine PIASx gene results in reduced testis weight. J. Mol. Endocrinol. 2005, 34, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Campla, C.K.; Breit, H.; Dong, L.; Gumerson, J.D.; Roger, J.E.; Swaroop, A. Pias3 is necessary for dorso-ventral patterning and visual response of retinal cones but is not required for rod photoreceptor differentiation. Biol. Open 2017, 6, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.J.; Zhu, H.Y.; Yang, C.; Ji, F. SENP2 regulates hepatocellular carcinoma cell growth by modulating the stability of β-catenin. Asian Pac. J. Cancer Prev. 2012, 13, 3583–3587. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.Y.; Mu, X.Y.; Liu, B.; Wang, Y.; Bao, E.D.; Qiu, J.X.; Fan, Y. SUMO-specific protease 2 suppresses cell migration and invasion through inhibiting the expression of MMP13 in bladder cancer cells. Cell. Physiol. Biochem. 2013, 32, 542–548. [Google Scholar] [CrossRef]
- Pei, H.; Chen, L.; Liao, Q.M.; Wang, K.J.; Chen, S.G.; Liu, Z.J.; Zhang, Z.C. Sumo-specific protease 2 (Senp2) functions as a tumosuppressor in osteosarcoma via sox9 degradation. Exp. Ther. Med. 2018, 16, 5359–5365. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.L.; Wang, S.F.; Liang, X.T.; Liang, H.X.; Wang, T.T.; Wu, S.Q.; Qiu, Z.J.; Zhan, R.; Xu, Z.S. SENP2 exerts an anti-tumor effect on chronic lymphocytic leukemia cells through the inhibition of the Notch and NF-κB signaling pathways. Int. J. Oncol. 2019, 54, 455–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-κB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef]
- Li, X.; Meng, Y. SUMOylation Regulator-Related Molecules Can Be Used as Prognostic Biomarkers for Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 689. [Google Scholar] [CrossRef]
- Taheri, M.; Oskooei, V.K.; Ghafouri-Fard, S. Protein inhibitor of activated STAT genes are differentially expressed in breast tumor tissues. Pers. Med. 2019, 16, 277–285. [Google Scholar] [CrossRef]
- Tuccilli, C.; Baldini, E.; Sorrenti, S.; Di Gioia, C.; Bosco, D.; Ascoli, V.; Mian, C.; Barollo, S.; Rendina, R.; Coccaro, C.; et al. Papillary thyroid cancer is characterized by altered expression of genes involved in the sumoylation process. J. Biol. Regul. Homeost. Agents 2015, 29, 655–662. [Google Scholar]
- Wang, J.; Ni, J.; Yi, S.; Song, D.; Ding, M. Protein inhibitor of activated STAT xα depresses cyclin D and cyclin D kinase, and contributes to the inhibition of osteosarcoma cell progression. Mol. Med. Rep. 2016, 13, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.S.; Zhang, Z.D.; Pei, L.; Bai, Z.Z.; Yang, Y.; Yang, H.; Tian, Q.H. CBX4 exhibits oncogenic activities in breast cancer via Notch1 signaling. Int. J. Biochem. Cell Biol. 2018, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gou, J.; Li, H.; Yang, X. Bioinformatic analysis of the expression and prognostic value of chromobox family proteins in human breast cancer. Sci. Rep. 2020, 10, 17739. [Google Scholar] [CrossRef] [PubMed]
- Alshareeda, A.T.; Negm, O.H.; Green, A.R.; Nolan, C.; Tighe, P.; Albarakati, N.; Sultana, R.; Madhusudan, S.; Ellis, I.O.; Rakha, E.A. SUMOylation proteins in breast cancer. Breast Cancer Res. Treat. 2014, 144, 519–530. [Google Scholar] [CrossRef]
- Agboola, A.; Musa, A.; Banjo, A.; Ayoade, B.; Deji-Agboola, M.; Nolan, C.; Rakha, E.; Ellis, I.; Green, A. PIASγ expression in relation to clinicopathological, tumour factors and survival in indigenous black breast cancer women. J. Clin. Pathol. 2014, 67, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Dabir, S.; Kluge, A.; Kresak, A.; Yang, M.; Fu, P.; Groner, B.; Wildey, G.; Dowlati, A. Low PIAS3 expression in malignant mesothelioma is associated with increased STAT3 activation and poor patient survival. Clin. Cancer Res. 2014, 20, 5124–5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Xu, Y.; Ruan, N.; Li, J.; Lv, Q.; Zhang, Q.; Chen, Y.; Wang, Q.; Xia, Q.; Li, Q. Genetic alteration and clinical significance of sumoylation regulators in multiple cancer types. J. Cancer 2020, 11, 6823–6833. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Z.; Xia, Z.; Wang, X.; Ma, Y.; Sheng, Z.; Gu, Q.; Shen, G.; Zhou, L.; Zhu, H.; et al. SAE1 promotes human glioma progression through activating AKT SUMOylation-mediated signaling pathways. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, Y.; Long, X.D.; Wang, W.; Jiao, H.K.; Mei, Z.; Yin, Q.Q.; Ma, L.N.; Zhou, A.W.; Wang, L.S.; et al. Cbx4 governs HIF-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 2014, 25, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Jiao, H.K.; Xu, Y.; Li, J.; Wang, W.; Mei, Z.; Long, X.D.; Chen, G.Q. Prognostic significance of Cbx4 expression and its beneficial effect for transarterial chemoembolization in hepatocellular carcinoma. Cell Death Dis. 2015, 6, e1689. [Google Scholar] [CrossRef] [Green Version]
- Chanda, A.; Chan, A.; Deng, L.; Kornaga, E.N.; Enwere, E.K.; Morris, D.G.; Bonni, S. Identification of the SUMO E3 ligase PIAS1 as a potential survival biomarker in breast cancer. PLoS ONE 2017, 12, e0177639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tang, J.; Liao, D.; Wang, G.; Zhang, M.; Sang, Y.; Cao, J.; Wu, Y.; Zhang, R.; Li, S.; et al. Chromobox homolog 4 is correlated with prognosis and tumor cell growth in hepatocellular carcinoma. Ann. Surg. Oncol. 2013, 20, 684–692. [Google Scholar] [CrossRef]
- Qian, J.; Luo, Y.; Gu, X.; Wang, X. Inhibition of SENP6-Induced Radiosensitization of Human Hepatocellular Carcinoma Cells by Blocking Radiation-Induced NF-κB Activation. Cancer Biother. Radiopharm. 2013, 28, 196–200. [Google Scholar] [CrossRef]
- Stefanska, B.; Cheishvili, D.; Suderman, M.; Arakelian, A.; Huang, J.; Hallett, M.; Han, Z.G.; Al-Mahtab, M.; Akbar, S.M.F.; Khan, W.A.; et al. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets. Clin. Cancer Res. 2014, 20, 3118–3132. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Tan, X.; Zhao, A.; Zhu, L.; Yin, B.; Yuan, J.; Qiang, B.; Peng, X. microRNA-214-mediated UBC9 expression in glioma. BMB Rep. 2012, 45, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Tang, Y.; Guo, W.; Du, Y.; Wang, Y.; Li, P.; Zang, W.; Yin, X.; Wang, H.; Chu, H.; et al. Up-regulation of microRNA-138 induce radiosensitization in lung cancer cells. Tumor Biol. 2014, 35, 6557–6565. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tao, W.; Ni, S.; Chen, Q.; Zhao, Z.; Ma, L.; Fu, Y.; Jiao, Z. Tumor-suppressive microRNA-145 induces growth arrest by targeting SENP1 in human prostate cancer cells. Cancer Sci. 2015, 106, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, J.; Wang, Q.; Liu, W.; Zhou, J.; Liu, R.; Zeng, Q.; Peng, X.; Huang, C.; Cao, P.; et al. MicroRNA-195 functions as a tumor suppressor by inhibiting CBX4 in hepatocellular carcinoma. Oncol. Rep. 2015, 33, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Du, J.; Wang, Y.; Shi, H.; Jiang, Q.; Wang, Y.; Zhang, H.; Wei, Y.; Xue, W.; Pu, Z.; et al. MicroRNA-497-5p induces cell cycle arrest of cervical cancer cells in s phase by targeting cbx4. Onco-Targets Ther. 2019, 12, 10535–10545. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, W.; Zhang, R.; Liu, P.; Ye, Y.; Yu, W.; Guo, X.; Yu, J. Cancer exosome-derived miR-9 and miR-181a promote the development of early-stage MDSCs via interfering with SOCS3 and PIAS3 respectively in breast cancer. Oncogene 2020, 39, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Kluge, A.; Dabir, S.; Vlassenbroeck, I.; Eisenberg, R.; Dowlati, A. Protein inhibitor of activated STAT3 expression in lung cancer. Mol. Oncol. 2011, 5, 256–264. [Google Scholar] [CrossRef]
- Brantley, E.C.; Nabors, L.B.; Gillespie, G.Y.; Choi, Y.H.; Palmer, C.A.; Harrison, K.; Roarty, K.; Benveniste, E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin. Cancer Res. 2008, 14, 4694–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Han, Y.; Wang, Y.; Sun, X.; Yan, S.; Yeh, E.T.H.; Chen, Y.; Cang, H.; Li, H.; Shi, G.; et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009, 28, 2748–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Wang, M.; Li, J.; Xiao, M.; Chin, Y.E.; Cheng, J.; Yeh, E.T.H.; Yang, J.; Yi, J. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene 2016, 35, 5826–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, K.; Wagner, K.; Mendler, L.; Hölper, S.; Dehne, N.; Müller, S. SUMO Signaling by Hypoxic Inactivation of SUMO-Specific Isopeptidases. Cell Rep. 2016, 16, 3075–3086. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Li, J.; Zou, Y.; Yi, J.; Zhang, H.; Cao, M.; Yeh, E.T.H.; Cheng, J. PIASy stimulates HIF1α SUMOylation and negatively regulates HIF1α activity in response to hypoxia. Oncogene 2010, 29, 5568–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dünnebier, T.; Bermejo, J.L.; Haas, S.; Fischer, H.P.; Pierl, C.B.; Justenhoven, C.; Brauch, H.; Baisch, C.; Gilbert, M.; Harth, V.; et al. Common variants in the UBC9 gene encoding the SUMO-conjugating enzyme are associated with breast tumor grade. Int. J. Cancer 2009, 125, 596–602. [Google Scholar] [CrossRef]
- Dünnebier, T.; Bermejo, J.L.; Haas, S.; Fischer, H.P.; Pierl, C.B.; Justenhoven, C.; Brauch, H.; Baisch, C.; Gilbert, M.; Harth, V.; et al. Polymorphisms in the UBC9 and PIAS3 genes of the SUMO-conjugating system and breast cancer risk. Breast Cancer Res. Treat. 2010, 121, 185–194. [Google Scholar] [CrossRef]
- Mirecka, A.; Morawiec, Z.; Wozniak, K. Genetic Polymorphism of SUMO-Specific Cysteine Proteases—SENP1 and SENP2 in Breast Cancer. Pathol. Oncol. Res. 2016, 22, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; You, S.; Wang, J.; Fan, S.; Shi, J.; Peng, A.; Yu, T. Association between sumoylation-related gene rs77447679 polymorphism and risk of gastric cancer(GC) in a Chinese population. J. Cancer 2017, 8, 3226–3231. [Google Scholar] [CrossRef] [Green Version]
- Murakami, H.; Arnheiter, H. Sumoylation modulates transcriptional activity of MITF in a promoter-specific manner. Pigment Cell Res. 2005, 18, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; De Lichy, M.; Bille, K.; Dessen, P.; D’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- King, R.; Weilbaecher, K.N.; McGill, G.; Cooley, E.; Mihm, M.; Fisher, D.E. Microphthalmia transcription factor: A sensitive and specific melanocyte marker for melanoma diagnosis. Am. J. Pathol. 1999, 155, 731–738. [Google Scholar] [CrossRef]
- Buscà, R.; Berra, E.; Gaggioli, C.; Khaled, M.; Bille, K.; Marchetti, B.; Thyss, R.; Fitsialos, G.; Larribère, L.; Bertolotto, C.; et al. Hypoxia-inducible factor 1α is a new target of microphthalmia- associated transcription factor (MITF) in melanoma cells. J. Cell Biol. 2005, 170, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Levin, N.A.; Bnorka, P.M.; Warnock, M.L.; Gray, J.W.; Christman, M.F. Identification of novel regions of altered DNA copy number in small cell lung tumors. Genes Chromosom. Cancer 1995, 13, 175–185. [Google Scholar] [CrossRef]
- Wang, J.; Qian, J.; Hoeksema, M.D.; Zou, Y.; Espinosa, A.V.; Rahman, S.M.J.; Zhang, B.; Massion, P.P. Integrative genomics analysis identifies candidate drivers at 3q26-29 amplicon in squamous cell carcinoma of the lung. Clin. Cancer Res. 2013, 19, 5580–5590. [Google Scholar] [CrossRef] [Green Version]
- Fields, A.P.; Justilien, V.; Murray, N.R. The chromosome 3q26 OncCassette: A multigenic driver of human cancer. Adv. Biol. Regul. 2016, 60, 47–63. [Google Scholar] [CrossRef] [Green Version]
- La-Touche, S.; Lemetre, C.; Lambros, M.; Stankiewicz, E.; Ng, C.K.Y.; Weigelt, B.; Rajab, R.; Tinwell, B.; Corbishley, C.; Watkin, N.; et al. DNA Copy Number Aberrations, and Human Papillomavirus Status in Penile Carcinoma. Clinico-Pathological Correlations and Potential Driver Genes. PLoS ONE 2016, 11, e0146740. [Google Scholar] [CrossRef]
- Constanzo, J.D.; Tang, K.J.; Rindhe, S.; Melegari, M.; Liu, H.; Tang, X.; Rodriguez-Canales, J.; Wistuba, I.; Scaglioni, P.P. PIAS1-FAK Interaction Promotes the Survival and Progression of Non-Small Cell Lung Cancer. Neoplasia 2016, 18, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltman, I.M.; Vreede, L.A.; Cheng, J.; Looijenga, L.H.J.; Janssen, B.; Schoenmakers, E.F.P.M.; Yeh, E.T.H.; van Kessel, A.G. Fusion of the SUMO/Sentrin-specific protease 1 gene SENP1 and the embryonic polarity-related mesoderm development gene MESDC2 in a patient with an infantile teratoma and a constitutional t(12;15)(q13;q25). Hum. Mol. Genet. 2005, 14, 1955–1963. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, H.; Miura, I.; Suzuki, R.; Suzuki, H.; Hosokawa, Y.; Seto, M. Molecular cytogenetic analysis of the breakpoint region at 6q21-22 in T-cell lymphoma/leukemia cell lines. Genes Chromosom. Cancer 2002, 34, 175–185. [Google Scholar] [CrossRef]
- Xu, H.D.; Shi, S.P.; Chen, X.; Qiu, J.D. Systematic analysis of the genetic variability that impacts sumo conjugation and their involvement in human diseases. Sci. Rep. 2015, 5, 10900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Miao, Y.; Liu, M.; Zeng, Y.; Gao, Z.; Peng, D.; Hu, B.; Li, X.; Zheng, Y.; Xue, Y.; et al. Pan-cancer analysis reveals the functional importance of protein lysine modification in cancer development. Front. Genet. 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [Green Version]
- González-Prieto, R.; Cuijpers, S.A.G.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C.O. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Rabellino, A.; Melegari, M.; Tompkins, V.S.; Chen, W.; Van Ness, B.G.; Teruya-Feldstein, J.; Conacci-Sorrell, M.; Janz, S.; Scaglioni, P.P. PIAS1 Promotes Lymphomagenesis through MYC Upregulation. Cell Rep. 2016, 15, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.X.; Chen, Y.; Su, Y.; Wang, X.; Chauhan, K.M.; Liang, J.; Daniel, C.J.; Sears, R.C.; Dai, M.S. SUMO protease SENP1 deSUMOylates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2018, 115, 10983–10988. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Swatkoski, S.; Holly, A.; Lee, L.C.; Giroux, V.; Lee, C.S.; Hsu, D.; Smith, J.L.; Yuen, G.; Yue, J.; et al. Oncogenesis driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9. Proc. Natl. Acad. Sci. USA 2015, 112, E1724–E1733. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wei, J.; Jiang, C.; Liu, D.; Deng, L.; Zhang, K.; Wang, P. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 2013, 73, 5742–5753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Ollila, S.; Wong, I.P.L.; Vallenius, T.; Palvimo, J.J.; Vaahtomeri, K.; Mäkelä, T.P. SUMOylation of AMPKα1 by PIAS4 specifically regulates mTORC1 signalling. Nat. Commun. 2015, 6, 8979. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Jeong, J.W.; Park, J.A.; Kim, S.H.; Bae, M.K.; Choi, S.J.; Kim, K.W. Sumoylation increases HIF-1α stability and its transcriptional activity. Biochem. Biophys. Res. Commun. 2004, 324, 394–400. [Google Scholar] [CrossRef]
- Carbia-Nagashima, A.; Gerez, J.; Perez-Castro, C.; Paez-Pereda, M.; Silberstein, S.; Stalla, G.K.; Holsboer, F.; Arzt, E. RSUME, a Small RWD-Containing Protein, Enhances SUMO Conjugation and Stabilizes HIF-1α during Hypoxia. Cell 2007, 131, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Mabb, A.M.; Gill, G.B.; Yeh, E.T.H.; Miyamoto, S. NF-κB Induction of the SUMO Protease SENP2: A Negative Feedback Loop to Attenuate Cell Survival Response to Genotoxic Stress. Mol. Cell 2011, 43, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.K.; Choi, K.C.; Yoo, J.Y.; Song, M.; Ko, S.J.; Kim, C.H.; Ahn, J.H.; Chun, K.H.; Yook, J.I.; Yoon, H.G. Reversible SUMOylation of TBL1-TBLR1 Regulates β-Catenin-Mediated Wnt Signaling. Mol. Cell 2011, 43, 203–216. [Google Scholar] [CrossRef]
- Sharma, P.; Kuehn, M.R. SENP1-modulated sumoylation regulates retinoblastoma protein (RB) and Lamin A/C interaction and stabilization. Oncogene 2016, 35, 6429–6438. [Google Scholar] [CrossRef]
- Gostissa, M.; Hengstermann, A.; Fogal, V.; Sandy, P.; Schwarz, S.E.; Scheffner, M.; Del Sal, G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999, 18, 6462–6471. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Midgley, C.A.; Lane, D.P.; Hay, R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999, 18, 6455–6461. [Google Scholar] [CrossRef] [Green Version]
- Xirodimas, D.P.; Chisholm, J.; Desterro, J.M.; Lane, D.P.; Hay, R.T. P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS Lett. 2002, 528, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J. MDM2-ARF complex regulates p53 sumoylation. Oncogene 2003, 22, 5348–5357. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Chiu, S.Y.; Hsu, W. SUMO-specific protease 2 in Mdm2-mediated regulation of p53. Cell Death Differ. 2011, 18, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yan, J.; Zhang, J.; Zhu, S.; Wang, Y.; Shi, T.; Zhu, C.; Chen, C.; Liu, X.; Cheng, J.; et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 2012, 3, 911. [Google Scholar] [CrossRef] [PubMed]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.O.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, Y.; Wang, S.; Hu, N.; Cao, Z.; Wang, W.; Tong, T.; Zhang, X. PIASxα ligase enhances SUMO1 modification of PTEN Protein as a SUMO E3 Ligase. J. Biol. Chem. 2014, 289, 3217–3230. [Google Scholar] [CrossRef] [Green Version]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.S.; Wei, M.; Brandhorst, S.; Longo, V.D. Starvation promotes REV1 SUMOylation and p53-dependent sensitization of melanoma and breast cancer cells. Cancer Res. 2015, 75, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Niskanen, E.A.; Malinen, M.; Sutinen, P.; Toropainen, S.; Paakinaho, V.; Vihervaara, A.; Joutsen, J.; Kaikkonen, M.U.; Sistonen, L.; Palvimo, J.J. Global SUMOylation on active chromatin is an acute heat stress response restricting transcription. Genome Biol. 2015, 16, 153. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Ding, S.; Qiu, C.; Shi, Y.; Song, L.; Wang, Y.; Wang, Y.; Li, J.; Wang, Y.; Sun, Y.; et al. SUMOylation negatively regulates angiogenesis by targeting endothelial NOTCH signaling. Circ. Res. 2017, 121, 636–649. [Google Scholar] [CrossRef]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Garvin, A.J.; Densham, R.M.; Blair-Reid, S.A.; Pratt, K.M.; Stone, H.R.; Weekes, D.; Lawrence, K.J.; Morris, J.R. The deSUMOylase SENP7 promotes chromatin relaxation for homologous recombination DNA repair. EMBO Rep. 2013, 14, 975–983. [Google Scholar] [CrossRef]
- Wagner, K.; Kunz, K.; Piller, T.; Tascher, G.; Hölper, S.; Stehmeier, P.; Keiten-Schmitz, J.; Schick, M.; Keller, U.; Müller, S. The SUMO Isopeptidase SENP6 Functions as a Rheostat of Chromatin Residency in Genome Maintenance and Chromosome Dynamics. Cell Rep. 2019, 29, 480–494.e5. [Google Scholar] [CrossRef] [Green Version]
- Garvin, A.J.; Walker, A.K.; Densham, R.M.; Chauhan, A.S.; Stone, H.R.; Mackay, H.L.; Jamshad, M.; Starowicz, K.; Daza-Martin, M.; Ronson, G.E.; et al. The deSUMOylase SENP2 coordinates homologous recombination and nonhomologous end joining by independent mechanisms. Genes Dev. 2019, 33, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Despras, E.; Sittewelle, M.; Pouvelle, C.; Delrieu, N.; Cordonnier, A.M.; Kannouche, P.L. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016, 7, 13326. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.-Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef]
- Lee, B.; Muller, M.T. SUMOylation enhances DNA methyltransferase 1 activity. Biochem. J. 2009, 421, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, A.; Viale-Bouroncle, S.; Morsczeck, C.; Muller, S. The SUMO-specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiation. Mol. Cell 2014, 55, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Shankar, S.R.; Wang, Y.; Taneja, R. SUMOylation of G9a regulates its function as an activator of myoblast proliferation. Cell Death Dis. 2019, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tahk, S.; Yee, K.M.; Fan, G.; Shuai, K. The ligase PIAS1 restricts natural regulatory T cell differentiation by epigenetic repression. Science 2010, 330, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Tahk, S.; Yee, K.M.; Yang, R.; Yang, Y.; Mackie, R.; Hsu, C.; Chernishof, V.; O’Brien, N.; Jin, Y.; et al. PIAS1 regulates breast tumorigenesis through selective epigenetic gene silencing. PLoS ONE 2014, 9, e89464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bawa-Khalfe, T.; Cheng, J.; Lin, S.H.; Ittmann, M.M.; Yeh, E.T.H. SENP1 induces prostatic intraepithelial neoplasia through multiple mechanisms. J. Biol. Chem. 2010, 285, 25859–25866. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xia, N.; Li, T.; Xu, Y.; Zou, Y.; Zuo, Y.; Fan, Q.; Bawa-Khalfe, T.; Yeh, E.T.H.; Cheng, J. SUMO-specific protease 1 promotes prostate cancer progression and metastasis. Oncogene 2013, 32, 2493–2498. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Lluva, S.; Tatham, M.H.; Jones, R.C.; Jaffray, E.G.; Edmondson, R.D.; Hay, R.T.; Malliri, A. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat. Cell Biol. 2010, 12, 1078–1085. [Google Scholar] [CrossRef]
- Li, C.; McManus, F.P.; Plutoni, C.; Pascariu, C.M.; Nelson, T.; Alberici Delsin, L.E.; Emery, G.; Thibault, P. Quantitative SUMO proteomics identifies PIAS1 substrates involved in cell migration and motility. Nat. Commun. 2020, 11, 834. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Matsuura, I.; He, D.; Wang, G.; Shuai, K.; Liu, F. Repression of Smad transcriptional activity by PIASy, an inhibitor of activated STAT. Proc. Natl. Acad. Sci. USA 2003, 100, 9791–9796. [Google Scholar] [CrossRef] [Green Version]
- Chandhoke, A.S.; Karve, K.; Dadakhujaev, S.; Netherton, S.; Deng, L.; Bonni, S. The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial-mesenchymal transition in a sumoylation-regulated manner. Cell Death Differ. 2016, 23, 876–888. [Google Scholar] [CrossRef] [Green Version]
- Jang, D.; Kwon, H.; Choi, M.; Lee, J.; Pak, Y. Sumoylation of Flotillin-1 promotes EMT in metastatic prostate cancer by suppressing Snail degradation. Oncogene 2019, 38, 3248–3260. [Google Scholar] [CrossRef] [Green Version]
- Chanda, A.; Ikeuchi, Y.; Karve, K.; Sarkar, A.; Chandhoke, A.S.; Deng, L.; Bonni, A.; Bonni, S. PIAS1 and TIF1γ collaborate to promote SnoN SUMOylation and suppression of epithelial–mesenchymal transition. Cell Death Differ. 2021, 28, 267–282. [Google Scholar] [CrossRef]
- Jiao, J.; Zhang, R.; Li, Z.; Yin, Y.; Fang, X.; Ding, X.; Cai, Y.; Yang, S.; Mu, H.; Zong, D.; et al. Nuclear Smad6 promotes gliomagenesis by negatively regulating PIAS3-mediated STAT3 inhibition. Nat. Commun. 2018, 9, 2504. [Google Scholar] [CrossRef]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.M.; Ayroldi, E.; Migliorati, G.; Thuy, T.T.; Riccardi, C.; Delfino, D.V. SUMO proteins: Guardians of immune system. J. Autoimmun. 2017, 84, 21–28. [Google Scholar] [CrossRef]
- Ran, Y.; Liu, T.-T.; Zhou, Q.; Li, S.; Mao, A.-P.; Li, Y.; Liu, L.-J.; Cheng, J.-K.; Shu, H.-B. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell Biol. 2011, 3, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Deng, H.; Li, X.; Wu, X.; Tang, Q.; Chang, T.-H.; Peng, H.; Rauscher, F.J.; Ozato, K.; Zhu, F. Tripartite Motif-Containing Protein 28 Is a Small Ubiquitin-Related Modifier E3 Ligase and Negative Regulator of IFN Regulatory Factor 7. J. Immunol. 2011, 187, 4754–4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Matsuoka, M.; Xu, S.; Otsuki, N.; Takeda, M.; Kato, A.; Ozato, K. PIASy inhibits virus-induced and interferon-stimulated transcription through distinct mechanisms. J. Biol. Chem. 2011, 286, 8165–8175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-H.; Xu, S.; Tailor, P.; Kanno, T.; Ozato, K. The Small Ubiquitin-like Modifier-Deconjugating Enzyme Sentrin-Specific Peptidase 1 Switches IFN Regulatory Factor 8 from a Repressor to an Activator during Macrophage Activation. J. Immunol. 2012, 189, 3548–3556. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Tailor, P.; Sasaki, T.; Tashiro, M.; Kato, A.; Ozato, K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008, 283, 25660–25670. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Yu, H.; Zheng, X.; Peng, R.; Wang, Q.; Zhou, Y.; Wang, R.; Wang, J.; Qu, B.; Shen, N.; et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog. 2017, 13, e1006156. [Google Scholar] [CrossRef]
- Desterro, J.M.P.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Wu, Z.H.; Miyamoto, S. Sequential Modification of NEMO/IKKγ by SUMO-1 and Ubiquitin Mediates NF-κB Activation by Genotoxic Stress. Cell 2003, 115, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Yang, L.; Sun, Q.; Arguello, M.; Ballard, D.W.; Hiscott, J.; Lin, R. The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat. Immunol. 2007, 8, 592–600. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Wang, Q.; Li, L.; Wang, C. Negative Regulation of TLR Inflammatory Signaling by the SUMO-deconjugating Enzyme SENP6. PLoS Pathog. 2013, 9, e1003480. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Q.; Chen, W.; Wang, C. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013, 24, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Lao, Y.; Teng, X.L.; Li, S.; Zhou, Y.; Wang, F.; Guo, X.; Deng, S.; Chang, Y.; Wu, X.; et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat. Commun. 2018, 9, 3157. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Bian, Q.; Lao, Y.; Yi, J.; Sun, X.; Sun, X.; Yang, J. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Teng, X.L.; Zhang, T.; Yu, X.; Ding, R.; Yi, J.; Deng, L.; Wang, Z.; Zou, Q. SENP3 senses oxidative stress to facilitate STING-dependent dendritic cell antitumor function. Mol. Cell 2021, 81, 940–952.e5. [Google Scholar] [CrossRef] [PubMed]
- Decque, A.; Joffre, O.; Magalhaes, J.G.; Cossec, J.C.; Blecher-Gonen, R.; Lapaquette, P.; Silvin, A.; Manel, N.; Joubert, P.E.; Seeler, J.S.; et al. Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat. Immunol. 2016, 17, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Crowl, J.T.; Stetson, D.B. SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. Proc. Natl. Acad. Sci. USA 2018, 115, 6798–6803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor α gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Longo, L.; Pandolfi, P.P.; Biondi, A.; Rambaldi, A.; Mencarelli, A.; Lo Coco, F.; Diverio, D.; Pegoraro, L.; Avanzi, G.; Tabilio, A.; et al. Rearrangements and aberrant expression of the retinoic acid receptor α gene in acute promyelocytic leukemias. J. Exp. Med. 1990, 172, 1571–1575. [Google Scholar] [CrossRef]
- Kakizuka, A.; Miller, W.H.; Umesono, K.; Warrell, R.P.; Frankel, S.R.; Murty, V.V.V.S.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RARα with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- De Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Daniel, M.; Koken, M.; Romagne, O.; Barbey, S.; Bazarbachi, A.; Stadler, M.; Guillemin, M.; Degos, L.; Chomienne, C.; de Thé, H. PML protein expression in hematopoietic and acute promyelocytic leukemia cells. Blood 1993, 82, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Müller, S.; Ronchetti, S.; Freemont, P.S.; Dejean, A.; Pandolfi, P.P. Role of SUMO-1-modified PML in nuclear body formation. Blood 2000, 95, 2748–2753. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Laukens, K.; Dang, T.H.; van Ostade, X. A Manually Curated Network of the PML Nuclear Body Interactome Reveals an Important Role for PML-NBs in SUMOylation dynamics. Int. J. Biol. Sci. 2010, 6, 51–67. [Google Scholar] [CrossRef]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; David, G.; Wong, C.W.; Dejean, A.; Privalsky, M.L. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor α (RARα) and PLZF-RARα oncoproteins associated with acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 9028–9033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.J.; Evans, R.M. Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol. Cell 2000, 5, 821–830. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, J.; Peres, L.; Riaucoux, F.; Honoré, N.; Kogan, S.; De Thé, H. A sumoylation site in PML/RARA is essential for leukemic transformation. Cancer Cell 2005, 7, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Breitman, T.; Collins, S.; Keene, B. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood 1981, 57, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Lallemand-Breitenbach, V.; Guillemin, M.C.; Janin, A.; Daniel, M.T.; Degos, L.; Kogan, S.C.; Bishop, J.M.; De Thé, H. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J. Exp. Med. 1999, 189, 1043–1052. [Google Scholar] [CrossRef]
- Ablain, J.; Leiva, M.; Peres, L.; Fonsart, J.; Anthony, E.; De Thé, H. Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J. Exp. Med. 2013, 210, 647–653. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML-RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Fasci, D.; Anania, V.G.; Lill, J.R.; Salvesen, G.S. SUMO deconjugation is required for arsenic-triggered ubiquitylation of PML. Sci. Signal. 2015, 8, ra56. [Google Scholar] [CrossRef] [Green Version]
- Van Gils, N.; Verhagen, H.J.M.P.; Smit, L. Reprogramming acute myeloid leukemia into sensitivity for retinoic-acid-driven differentiation. Exp. Hematol. 2017, 52, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Baik, H.; Boulanger, M.; Hosseini, M.; Kowalczyk, J.; Zaghdoudi, S.; Salem, T.; Sarry, J.E.; Hicheri, Y.; Cartron, G.; Piechaczyk, M.; et al. Targeting the sumo pathway primes all-trans retinoic acid–induced differentiation of nonpromyelocytic acute myeloid leukemias. Cancer Res. 2018, 78, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Chen, X.; Li, M.; Tan, J.; Zhang, Y.; Yuan, W.; Zhou, J.; Wang, G. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019, 513, 1063–1069. [Google Scholar] [CrossRef]
- Xu, R.; Yu, S.; Zhu, D.; Huang, X.; Xu, Y.; Lao, Y.; Tian, Y.; Zhang, J.; Tang, Z.; Zhang, Z.; et al. hCINAP regulates the DNA-damage response and mediates the resistance of acute myelocytic leukemia cells to therapy. Nat. Commun. 2019, 10, 3812. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Mendez, L.M.; Posey, R.R.; Pandolfi, P.P. The Interplay Between the Genetic and Immune Landscapes of AML: Mechanisms and Implications for Risk Stratification and Therapy. Front. Oncol. 2019, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwamitra, D.; Curry, C.V.; Shi, P.; Alkan, S.; Amin, H.M. SUMOylation Confers Posttranslational Stability on NPM-ALK Oncogenic Protein. Neoplasia 2015, 17, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Sun, H.Y.; Xiao, F.J.; Wang, H.; Yang, Y.; Wang, L.; Gao, C.J.; Guo, Z.K.; Wu, C.T.; Wang, L.S. SENP1 inhibition induces apoptosis and growth arrest of multiple myeloma cells through modulation of NF-κB signaling. Biochem. Biophys. Res. Commun. 2015, 460, 409–415. [Google Scholar] [CrossRef]
- Rizos, H.; Woodruff, S.; Kefford, R.F. p14ARF interacts with the SUMO-conjugating enzyme Ubc9 and promotes the sumoylation of its binding partners. Cell Cycle 2005, 4, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694. [Google Scholar] [CrossRef] [Green Version]
- Kawano, M.; Hirano, T.; Matsuda, T.; Taga, T.; Horii, Y.; Iwato, K.; Asaoku, H.; Tang, B.; Tanabe, O.; Tanaka, H.; et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature 1988, 332, 83–85. [Google Scholar] [CrossRef]
- Klein, B.; Zhang, X.; Jourdan, M.; Content, J.; Houssiau, F.; Aarden, L.; Piechaczyk, M.; Bataille, R. Paracrine rather than autocrine regulation of myeloma-cell growth and differentiation by interleukin-6. Blood 1989, 73, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Bommert, K.; Bargou, R.C.; Stühmer, T. Signalling and survival pathways in multiple myeloma. Eur. J. Cancer 2006, 42, 1574–1580. [Google Scholar] [CrossRef]
- Bawa-Khalfe, T.; Lu, L.S.; Zuo, Y.; Huang, C.; Dere, R.; Lin, F.M.; Yeh, E.T.H. Differential expression of SUMO-specific protease 7 variants regulates epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2012, 109, 17466–17471. [Google Scholar] [CrossRef] [Green Version]
- Cashman, R.; Cohen, H.; Ben-Hamo, R.; Zilberberg, A.; Efroni, S. SENP5 mediates breast cancer invasion via a TGFßRI SUMOylation cascade. Oncotarget 2014, 5, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandhoke, A.S.; Chanda, A.; Karve, K.; Deng, L.; Bonni, S. The PIAS3-Smurf2 sumoylation pathway suppresses breast cancer organoid invasiveness. Oncotarget 2017, 8, 21001–21014. [Google Scholar] [CrossRef]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.-C.; Corbo, L. Sumoylation of the Estrogen Receptor α Hinge Region Regulates Its Transcriptional Activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef]
- Lee, J.Y.; Won, H.Y.; Park, J.H.; Kim, H.Y.; Choi, H.J.; Shin, D.H.; Kang, J.H.; Woo, J.K.; Oh, S.H.; Son, T.; et al. MEL-18 loss mediates estrogen receptor-α downregulation and hormone independence. J. Clin. Investig. 2015, 125, 1801–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traboulsi, T.; El Ezzy, M.; Dumeaux, V.; Audemard, E.; Mader, S. Role of SUMOylation in differential ERα transcriptional repression by tamoxifen and fulvestrant in breast cancer cells. Oncogene 2019, 38, 1019–1037. [Google Scholar] [CrossRef]
- Park, Y.Y.; Kim, K.; Kim, S.B.; Hennessy, B.T.; Kim, S.M.; Park, E.S.; Lim, J.Y.; Li, J.; Lu, Y.; Gonzalez-Angulo, A.M.; et al. Reconstruction of nuclear receptor network reveals that NR2E3 is a novel upstream regulator of ESR1 in breast cancer. EMBO Mol. Med. 2012, 4, 52–67. [Google Scholar] [CrossRef]
- Yang, S.-F.; Hou, M.-F.; Chen, F.-M.; Ou-Yang, F.; Wu, Y.-C.; Chai, C.-Y.; Yeh, Y.-T. Prognostic value of protein inhibitor of activated STAT3 in breast cancer patients receiving hormone therapy. BMC Cancer 2016, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Sundvall, M.; Korhonen, A.; Vaparanta, K.; Anckar, J.; Halkilahti, K.; Salah, Z.; Aqeilan, R.I.; Palvimo, J.J.; Sistonen, L.; Elenius, K. Protein inhibitor of activated STAT3 (PIAS3) protein promotes SUMOylation and nuclear sequestration of the intracellular domain of ErbB4 protein. J. Biol. Chem. 2012, 287, 23216–23226. [Google Scholar] [CrossRef] [Green Version]
- Knittle, A.M.; Helkkula, M.; Johnson, M.S.; Sundvall, M.; Elenius, K. SUMOylation regulates nuclear accumulation and signaling activity of the soluble intracellular domain of the ErbB4 receptor tyrosine kinase. J. Biol. Chem. 2017, 292, 19890–19904. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.Y.; Murphy, M.P.; Golde, T.E.; Carpenter, G. γ-secretase cleavage and nuclear locatization of ErbB-4 receptor tyrosine kinase. Science 2001, 294, 2179–2181. [Google Scholar] [CrossRef]
- Määttä, J.A.; Sundvall, M.; Junttila, T.T.; Peri, L.; Laine, V.J.O.; Isola, J.; Egeblad, M.; Elenius, K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol. Biol. Cell 2006, 17, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Sundvall, M.; Lundin, M.; Lundin, J.; Tanner, M.; Härkönen, P.; Joensuu, H.; Isola, J.; Elenius, K. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005, 65, 1384–1393. [Google Scholar] [CrossRef] [Green Version]
- Dadakhujaev, S.; Salazar-Arcila, C.; Netherton, S.J.; Chandhoke, A.S.; Singla, A.K.; Jirik, F.R.; Bonni, S. A novel role for the SUMO E3 ligase PIAS1 in cancer metastasis. Oncoscience 2014, 1, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; De Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, J.; Zhang, J.; Wang, L.; Cao, J. Depletion of SENP1 suppresses the proliferation and invasion of triple-negative breast cancer cells. Oncol. Rep. 2016, 36, 2071–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotaja, N.; Aittomäki, S.; Silvennoinen, O.; Palvimo, J.J.; Jänne, O.A. ARIP3 (Androgen Receptor-Interacting Protein 3) and Other PIAS (Protein Inhibitor of Activated STAT) Proteins Differ in Their Ability to Modulate Steroid Receptor-Dependent Transcriptional Activation. Mol. Endocrinol. 2000, 14, 1986–2000. [Google Scholar] [CrossRef]
- Gross, M.; Liu, B.; Tan, J.A.; French, F.S.; Carey, M.; Shuai, K. Distinct effects of PIAS proteins on androgen-mediated gene activation in prostate cancer cells. Oncogene 2001, 20, 3880–3887. [Google Scholar] [CrossRef] [Green Version]
- Hoefer, J.; Schfer, G.; Klocker, H.; Erb, H.H.H.; Mills, I.G.; Hengst, L.; Puhr, M.; Culig, Z. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am. J. Pathol. 2012, 180, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Junicho, A.; Matsuda, T.; Yamamoto, T.; Kishi, H.; Korkmaz, K.; Saatcioglu, F.; Fuse, H.; Muraguchi, A. Protein inhibitor of activated STAT3 regulates androgen receptor signaling in prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2000, 278, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Yasuda, H. PIAS1 and PIASxα function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem. 2002, 277, 41311–41317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toropainen, S.; Malinen, M.; Kaikkonen, S.; Rytinki, M.; Jääskeläinen, T.; Sahu, B.; Jänne, O.A.; Palvimo, J.J. SUMO ligase PIAS1 functions as a target gene selective androgen receptor coregulator on prostate cancer cell chromatin. Nucleic Acids Res. 2015, 43, 848–861. [Google Scholar] [CrossRef] [Green Version]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Dietrich, D.; Van Leenders, G.; Uhl, B.; Hoogland, M.; Handle, F.; Schlick, B.; Neuwirt, H.; et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene 2016, 35, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.; Lutz, J.; Nevalainen, M.T. Transcription factor Stat5a/b as a therapeutic target protein for prostate cancer. Int. J. Biochem. Cell Biol. 2010, 42, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ahonen, T.J.; Alanen, K.; Xie, J.; LeBaron, M.J.; Pretlow, T.G.; Ealley, E.L.; Zhang, Y.; Nurmi, M.; Singh, B.; et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004, 64, 4774–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T.H. SUMO-Specific Protease 1 Is Critical for Early Lymphoid Development through Regulation of STAT5 Activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Dagvadorj, A.; Tan, S.H.; Liao, Z.; Xie, J.; Nurmi, M.; Alanen, K.; Rui, H.; Mirtti, T.; Nevalainen, M.T. N-terminal truncation of Stat5a/b circumvents PIAS3-mediated transcriptional inhibition of Stat5 in prostate cancer cells. Int. J. Biochem. Cell Biol. 2010, 42, 2037–2046. [Google Scholar] [CrossRef] [Green Version]
- Poukka, H.; Karvonen, U.; Jänne, O.A.; Palvimo, J.J. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1). Proc. Natl. Acad. Sci. USA 2000, 97, 14145–14150. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Cai, C.; Omwancha, J.; Chen, S.Y.; Baslan, T.; Shemshedini, L. SUMO-3 enhances androgen receptor transcriptional activity through a sumoylation-independent mechanism in prostate cancer cells. J. Biol. Chem. 2006, 281, 4002–4012. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, S.; Jääskeläinen, T.; Karvonen, U.; Rytinki, M.M.; Makkonen, H.; Gioeli, D.; Paschal, B.M.; Palvimo, J.J. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol. Endocrinol. 2009, 23, 292–307. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Bawa, T.; Lee, P.; Gong, L.; Yeh, E.T.H. Role of Desumoylation in the Development of Prostate Cancer. Neoplasia 2006, 8, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Bawa-Khalfe, T.; Cheng, J.; Wang, Z.; Yeh, E.T.H. Induction of the SUMO-specific protease 1 transcription by the androgen receptor in prostate cancer cells. J. Biol. Chem. 2007, 282, 37341–37349. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Wang, D.; Wang, Z.; Yeh, E.T.H. SENP1 Enhances Androgen Receptor-Dependent Transcription through Desumoylation of Histone Deacetylase 1. Mol. Cell. Biol. 2004, 24, 6021–6028. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Tse, A.K.W.; Li, P.; Ma, Q.; Xiang, S.; Nicosia, S.V.; Seto, E.; Zhang, X.; Bai, W. Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 2011, 30, 2207–2218. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Choi, H.J.; Kim, B.; Kim, M.H.; Lee, J.M.; Kim, I.S.; Lee, M.H.; Choi, S.J.; Kim, K.I.; Kim, S.I.; et al. Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nat. Cell Biol. 2006, 8, 631–639. [Google Scholar] [CrossRef]
- Huang, W.; He, T.; Chai, C.; Yang, Y.; Zheng, Y.; Zhou, P.; Qiao, X.; Zhang, B.; Liu, Z.; Wang, J.; et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS ONE 2012, 7, e37693. [Google Scholar] [CrossRef]
- Wu, J.; Lei, H.; Zhang, J.; Chen, X.; Tang, C.; Wang, W.; Xu, H.; Xiao, W.; Gu, W.; Wu, Y.L. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget 2016, 7, 58995–59005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bawa-Khalfe, T.; Yang, F.M.; Ritho, J.; Lin, H.K.; Cheng, J.; Yeh, E.T.H. SENP1 regulates PTEN stability to dictate prostate cancer development. Oncotarget 2017, 8, 17651–17664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangguan, X.; He, J.; Ma, Z.; Zhang, W.; Ji, Y.; Shen, K.; Yue, Z.; Li, W.; Xin, Z.; Zheng, Q.; et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat. Commun. 2021, 12, 1812. [Google Scholar] [CrossRef]
- Wen, D.; Xu, Z.; Xia, L.; Liu, X.; Tu, Y.; Lei, H.; Wang, W.; Wang, T.; Song, L.; Ma, C.; et al. Important role of SUMOylation of spliceosome factors in prostate cancer cells. J. Proteome Res. 2014, 13, 3571–3582. [Google Scholar] [CrossRef]
- Ashikari, D.; Takayama, K.; Tanaka, T.; Suzuki, Y.; Obinata, D.; Fujimura, T.; Urano, T.; Takahashi, S.; Inoue, S. Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene 2017, 36, 6272–6281. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, J. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Van Hagen, M.; Overmeer, R.M.; Abolvardi, S.S.; Vertegaal, A.C.O. RNF4 and VHL regulate the proteasomal degradation of SUMO-conjugated Hypoxia-Inducible Factor-2α. Nucleic Acids Res. 2009, 38, 1922–1931. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Gao, Y.; Kang, X.; Gao, H.; Zhang, J.; Guo, H.; You, M.J.; Xue, W.; Cheng, J.; Huang, Y. SENP1 promotes proliferation of clear cell renal cell carcinoma through activation of glycolysis. Oncotarget 2016, 7, 80435–80449. [Google Scholar] [CrossRef]
- Gerez, J.; Tedesco, L.; Bonfiglio, J.J.; Fuertes, M.; Barontini, M.; Silberstein, S.; Wu, Y.; Renner, U.; Paéz-Pereda, M.; Holsboer, F.; et al. RSUME inhibits VHL and regulates its tumor suppressor function. Oncogene 2015, 34, 4855–4866. [Google Scholar] [CrossRef] [Green Version]
- Tedesco, L.; Elguero, B.; Pacin, D.G.; Senin, S.; Pollak, C.; Garcia Marchiñena, P.A.; Jurado, A.M.; Isola, M.; Labanca, M.J.; Palazzo, M.; et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis. 2019, 10, 266. [Google Scholar] [CrossRef] [Green Version]
- Kadaré, G.; Toutant, M.; Formstecher, E.; Corvol, J.C.; Carnaud, M.; Boutterin, M.C.; Girault, J.A. PIAS1-mediated Sumoylation of Focal Adhesion Kinase Activates Its Autophosphorylation. J. Biol. Chem. 2003, 278, 47434–47440. [Google Scholar] [CrossRef] [Green Version]
- Hung, P.F.; Hong, T.M.; Chang, C.C.; Hung, C.L.; Hsu, Y.L.; Chang, Y.L.; Wu, C.T.; Chang, G.C.; Chan, N.L.; Yu, S.L.; et al. Hypoxia-induced Slug SUMOylation enhances lung cancer metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 5. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific Inhibition of Stat3 Signal Transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Ogata, Y.; Osaki, T.; Naka, T.; Iwahori, K.; Furukawa, M.; Nagatomo, I.; Kijima, T.; Kumagai, T.; Yoshida, M.; Tachibana, I.; et al. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia 2006, 8, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluge, A.; Dabir, S.; Kern, J.; Nethery, D.; Halmos, B.; Ma, P.; Dowlati, A. Cooperative interaction between protein inhibitor of activated signal transducer and activator of transcription-3 with epidermal growth factor receptor blockade in lung cancer. Int. J. Cancer 2009, 125, 1728–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabir, S.; Kluge, A.; McColl, K.; Liu, Y.; Lam, M.; Halmos, B.; Wildey, G.; Dowlati, A. PIAS3 activates the intrinsic apoptotic pathway in non-small cell lung cancer cells independent of p53 status. Int. J. Cancer 2014, 134, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, R.; McColl, K.S.; Kresak, A.; Yang, M.; Chen, Y.; Fu, P.; Wildey, G.; Dowlati, A. PIAS3 expression in squamous cell lung cancer is low and predicts overall survival. Cancer Med. 2015, 4, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.P.; Wong, C.C.L.; Kai, A.K.L.; Ho, D.W.H.; Lau, E.Y.T.; Tsui, Y.M.; Chan, L.K.; Cheung, T.T.; Chok, K.S.H.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef]
- Tao, Y.; Li, R.; Shen, C.; Li, J.; Zhang, Q.; Ma, Z.; Wang, F.; Wang, Z. SENP1 is a crucial promotor for hepatocellular carcinoma through deSUMOylation of UBE2T. Aging 2020, 12, 1563–1576. [Google Scholar] [CrossRef]
- Deng, R.; Zhao, X.; Qu, Y.Y.; Chen, C.; Zhu, C.; Zhang, H.; Yuan, H.; Jin, H.; Liu, X.; Wang, Y.; et al. Shp2 SUMOylation promotes ERK activation and hepatocellular carcinoma development. Oncotarget 2015, 6, 9355–9369. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Wang, L.; Roehn, G.; Pearlstein, R.D.; Ali-Osman, F.; Pan, H.; Goldbrunner, R.; Krantz, M.; Harms, C.; Paschen, W. Small ubiquitin-like modifier 1-3 is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci. 2013, 104, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Olson, J.J.; Hao, C. SUMO1 modification stabilizes CDK6 protein and drives the cell cycle and glioblastoma progression. Nat. Commun. 2014, 5, 4234. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kögel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci. Rep. 2017, 7, 7425. [Google Scholar] [CrossRef]
- Zhang, A.; Tao, W.; Zhai, K.; Fang, X.; Huang, Z.; Yu, J.S.; Sloan, A.E.; Rich, J.N.; Zhou, W.; Bao, S. Protein sumoylation with SUMO1 promoted by Pin1 in glioma stem cells augments glioblastoma malignancy. Neuro-Oncology 2020, 22, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Kroonen, J.S.; Vertegaal, A.C.O. Targeting SUMO Signaling to Wrestle Cancer. Trends Cancer 2021, 7, 496–510. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.I.; Sodeoka, M.; Yoshida, M. Ginkgolic Acid Inhibits Protein SUMOylation by Blocking Formation of the E1-SUMO Intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Uramoto, M.; Saitoh, H.; Kawasaki, H.; Osada, H.; Yoshida, M. Kerriamycin B inhibits protein SUMOylation. J. Antibiot. 2009, 62, 221–224. [Google Scholar] [CrossRef]
- Takemoto, M.; Kawamura, Y.; Hirohama, M.; Yamaguchi, Y.; Handa, H.; Saitoh, H.; Nakao, Y.; Kawada, M.; Khalid, K.; Koshino, H.; et al. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. J. Antibiot. 2014, 67, 335–338. [Google Scholar] [CrossRef]
- Suzawa, M.; Miranda, D.A.; Ramos, K.A.; Ang, K.K.H.; Faivre, E.J.; Wilson, C.G.; Caboni, L.; Arkin, M.R.; Kim, Y.S.; Fletterick, R.J.; et al. A gene-expression screen identifies a non-toxic sumoylation inhibitor that mimics SUMO-less human LRH-1 in liver. eLife 2015, 4. [Google Scholar] [CrossRef]
- Kumar, A.; Ito, A.; Hirohama, M.; Yoshida, M.; Zhang, K.Y.J. Identification of quinazolinyloxy biaryl urea as a new class of SUMO activating enzyme 1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5145–5149. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ito, A.; Hirohama, M.; Yoshida, M.; Zhang, K.Y.J. Identification of sumoylation activating enzyme 1 inhibitors by structure-based virtual screening. J. Chem. Inf. Model. 2013, 53, 809–820. [Google Scholar] [CrossRef]
- Kumar, A.; Ito, A.; Hirohama, M.; Yoshida, M.; Zhang, K.Y.J. Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg. Med. Chem. Lett. 2016, 26, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Riceberg, J.; Soucy, T.; Koenig, E.; Minissale, J.; Gallery, M.; Bernard, H.; Yang, X.; Liao, H.; Rabino, C.; et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 2017, 13, 1164–1171. [Google Scholar] [CrossRef]
- Lv, Z.; Yuan, L.; Atkison, J.H.; Williams, K.M.; Vega, R.; Sessions, E.H.; Divlianska, D.B.; Davies, C.; Chen, Y.; Olsen, S.K. Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat. Commun. 2018, 9, 5145. [Google Scholar] [CrossRef]
- Li, Y.J.; Du, L.; Wang, J.; Vega, R.; Lee, T.D.; Miao, Y.; Aldana-Masangkay, G.; Samuels, E.R.; Li, B.; Ouyang, S.X.; et al. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem. Biol. 2019, 26, 278–288.e6. [Google Scholar] [CrossRef] [PubMed]
- Biederstädt, A.; Hassan, Z.; Schneeweis, C.; Schick, M.; Schneider, L.; Muckenhuber, A.; Hong, Y.; Siegers, G.; Nilsson, L.; Wirth, M.; et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 2020, 69, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Brackett, C.M.; García-Casas, A.; Castillo-Lluva, S.; Blagg, B.S.J. Synthesis and Evaluation of Ginkgolic Acid Derivatives as SUMOylation Inhibitors. ACS Med. Chem. Lett. 2020, 11, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Langston, S.P.; Grossman, S.; England, D.; Afroze, R.; Bence, N.; Bowman, D.; Bump, N.; Chau, R.; Chuang, B.C.; Claiborne, C.; et al. Discovery of TAK-981, a First-in-Class Inhibitor of SUMO-Activating Enzyme for the Treatment of Cancer. J. Med. Chem. 2021, 64, 2501–2520. [Google Scholar] [CrossRef]
- Khattar, M.; Song, K.; Grossman, S.; Xega, K.; He, X.; Idamakanti, N.; Huszar, D. Abstract 3252: TAK-981: A first in class SUMO inhibitor in Phase 1 trials that promotes dendritic cell activation, antigen-presentation, and T cell priming. Cancer Res. 2019, 79, 3252. [Google Scholar] [CrossRef]
- Nakamura, A.; Grossman, S.; Song, K.; Idamakanti, N.; Shapiro, G.; Huszar, D. Abstract 1523: Inhibition of SUMOylation by TAK-981 induces antitumor innate immune responses by modulating macrophage and NK cell function through Type I IFN pathway activation. Cancer Res. 2019, 79, 1523. [Google Scholar] [CrossRef]
- Nakamura, A.; Song, K.; Grossman, S.; Xega, K.; Zhang, Y.; Berger, A.; Berger, A.; Shapiro, G.; Huszar, D. Abstract 552: SUMOylation inhibitor TAK-981 activates NK cells and macrophages via Type I interferon signaling and shows synergistic activity in combination with rituximab and daratumumab in preclinical models. J. Immunother. Cancer 2020, 8, A588. [Google Scholar] [CrossRef]
- Hirohama, M.; Kumar, A.; Fukuda, I.; Matsuoka, S.; Igarashi, Y.; Saitoh, H.; Takagi, M.; Shin-Ya, K.; Honda, K.; Kondoh, Y.; et al. Spectomycin B1 as a novel sumoylation inhibitor that directly binds to SUMO E2. ACS Chem. Biol. 2013, 8, 2635–2642. [Google Scholar] [CrossRef]
- Brandt, M.; Szewczuk, L.M.; Zhang, H.; Hong, X.; McCormick, P.M.; Lewis, T.S.; Graham, T.I.; Hung, S.T.; Harper-Jones, A.D.; Kerrigan, J.J.; et al. Development of a High-Throughput Screen to Detect Inhibitors of TRPS1 Sumoylation. Assay Drug Dev. Technol. 2013, 11, 308–325. [Google Scholar] [CrossRef]
- Kim, Y.S.; Nagy, K.; Keyser, S.; Schneekloth, J.S. An electrophoretic mobility shift assay identifies a mechanistically unique inhibitor of protein sumoylation. Chem. Biol. 2013, 20, 604–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotkowski, K.; Hewitt, W.M.; Sinniah, R.S.; Tropea, J.E.; Needle, D.; Lountos, G.T.; Barchi, J.J.; Waugh, D.S.; Schneekloth, J.S. A Small-Molecule Microarray Approach for the Identification of E2 Enzyme Inhibitors in Ubiquitin-Like Conjugation Pathways. SLAS Discov. 2017, 22, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Wiechmann, S.; Gärtner, A.; Kniss, A.; Stengl, A.; Behrends, C.; Rogov, V.V.; Rodriguez, M.S.; Dötsch, V.; Müller, S.; Ernst, A. Site-specific inhibition of the small ubiquitin-like modifier (SUMO)-conjugating enzyme Ubc9 selectively impairs SUMO chain formation. J. Biol. Chem. 2017, 292, 15340–15351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.H.; Philips, M.R.; Chen, Y.; Lu, L.; Dai, W. K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J. Biol. Chem. 2018, 293, 17574–17581. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Tao, Y.; Gao, J.; Zhou, X.; Tang, S.; Deng, C.; Lai, Z.; Lin, X.; Wang, Q.; Li, T. UBC9 coordinates inflammation affecting development of bladder cancer. Sci. Rep. 2020, 10, 20670. [Google Scholar] [CrossRef]
- Tokarz, P.; Woźniak, K. SENP Proteases as Potential Targets for Cancer Therapy. Cancers 2021, 13, 2059. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Wang, W.; Wang, L.; Wen, D.; Zhao, Y.; Wang, Q.; Meng, Q.; Chen, G.; Wu, Y.; Zhou, H. Design, synthesis, and biological evaluation of benzodiazepine-based SUMO-specific protease 1 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 6389–6392. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, D.; Huang, Z.; Huang, M.; Luo, Y.; Liu, B.; Lu, H.; Wu, Y.; Peng, Y.; Zhang, J. 2-(4-Chlorophenyl)-2-oxoethyl 4-benzamidobenzoate derivatives, a novel class of SENP1 inhibitors: Virtual screening, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 6867–6870. [Google Scholar] [CrossRef] [PubMed]
- Uno, M.; Koma, Y.; Ban, H.S.; Nakamura, H. Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP)1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5169–5173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Zhang, J.; Zhou, H. Identification of SENP1 inhibitors through in silico screening and rational drug design. Eur. J. Med. Chem. 2016, 122, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Lindenmann, U.; Brand, M.; Gall, F.; Frasson, D.; Hunziker, L.; Kroslakova, I.; Sievers, M.; Riedl, R. Discovery of a Class of Potent and Selective Non-competitive Sentrin-Specific Protease 1 Inhibitors. ChemMedChem 2020, 15, 675–679. [Google Scholar] [CrossRef]
- Kumar, A.; Ito, A.; Takemoto, M.; Yoshida, M.; Zhang, K.Y.J. Identification of 1,2,5-oxadiazoles as a new class of SENP2 inhibitors using structure based virtual screening. J. Chem. Inf. Model. 2014, 54, 870–880. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Ye, D.; Smith, J.A.; Lee, Y.J.; Gessler, F.A.; Yasgar, A.; Kouznetsova, J.; Jadhav, A.; Wang, Z.; Pluchino, S.; et al. Quantitative high-throughput screening identifies cytoprotective molecules that enhance SUMO conjugation via the inhibition of SUMO-specific protease (SENP)2. FASEB J. 2018, 32, 1677–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.P.; Liu, K.J.; Kasper, G.; Lin, Q.; Hai, J. Inhibition of SENP3 by URB597 ameliorates neurovascular unit dysfunction in rats with chronic cerebral hypoperfusion. Biomed. Pharmacother. 2017, 91, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Albrow, V.E.; Ponder, E.L.; Fasci, D.; Békés, M.; Deu, E.; Salvesen, G.S.; Bogyo, M. Development of small molecule inhibitors and probes of human SUMO deconjugating proteases. Chem. Biol. 2011, 18, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponder, E.L.; Albrow, V.E.; Leader, B.A.; Békés, M.; Mikolajczyk, J.; Fonović, U.P.; Shen, A.; Drag, M.; Xiao, J.; Deu, E.; et al. Functional characterization of a SUMO deconjugating protease of Plasmodium falciparum using newly identified small molecule inhibitors. Chem. Biol. 2011, 18, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Dobrotă, C.; Fasci, D.; Hădade, N.D.; Roiban, G.-D.; Pop, C.; Meier, V.M.; Dumitru, I.; Matache, M.; Salvesen, G.S.; Funeriu, D.P. Glycine Fluoromethylketones as SENP-Specific Activity Based Probes. ChemBioChem 2012, 13, 80–84. [Google Scholar] [CrossRef]
- Xie, W.; Wang, Z.; Zhang, J.; Wang, L.; Zhao, Y.; Zhou, H. Development and evaluation of a highly reliable assay for SUMO-specific protease inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2124–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambaye, N.; Chen, C.H.; Khanna, S.; Li, Y.J.; Chen, Y. Streptonigrin Inhibits SENP1 and Reduces the Protein Level of Hypoxia-Inducible Factor 1α (HIF1α) in Cells. Biochemistry 2018, 57, 1807–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, Y.; Zhang, J.; Ullah, S.; Kang, N.; Zhao, Y.; Zhou, H. Benzothiophene-2-carboxamide derivatives as SENPs inhibitors with selectivity within SENPs family. Eur. J. Med. Chem. 2020, 204, 112553. [Google Scholar] [CrossRef]
- Madu, I.G.; Namanja, A.T.; Su, Y.; Wong, S.; Li, Y.J.; Chen, Y. Identification and characterization of a new chemotype of noncovalent SENP inhibitors. ACS Chem. Biol. 2013, 8, 1435–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Olsen, S.K.; Capili, A.D.; Cisar, J.S.; Lima, C.D.; Tan, D.S. Designed semisynthetic protein inhibitors of Ub/Ubl E1 activating enzymes. J. Am. Chem. Soc. 2010, 132, 1748–1749. [Google Scholar] [CrossRef] [Green Version]
- Puhr, M.; Hoefer, J.; Neuwirt, H.; Eder, I.E.; Kern, J.; Schäfer, G.; Geley, S.; Heidegger, I.; Klocker, H.; Culig, Z. PIAS1 is a crucial factor for prostate cancer cell survival and a valid target in docetaxel resistant cells. Oncotarget 2014, 5, 12043–12056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.C.; Ling, T.Y.; Lu, S.H.; Kuo, H.C.; Ho, H.N.; Yeh, S.D.; Shen, C.N.; Huang, Y.H. Chemotherapeutic sensitivity of testicular germ cell tumors under hypoxic conditions is negatively regulated by SENP1-controlled sumoylation of OCT4. Cancer Res. 2012, 72, 4963–4973. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, R.; Yung, M.M.H.; Sun, J.; Li, Z.; Yang, H.; Zhang, Y.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S.; et al. SENP1-mediated deSUMOylation of JAK2 regulates its kinase activity and platinum drug resistance. Cell Death Dis. 2021, 12, 341. [Google Scholar] [CrossRef] [PubMed]
- Modell, A.E.; Lai, S.; Nguyen, T.M.; Choudhary, A. Bifunctional modalities for repurposing protein function. Cell Chem. Biol. 2021, 28, 1081–1089. [Google Scholar] [CrossRef]

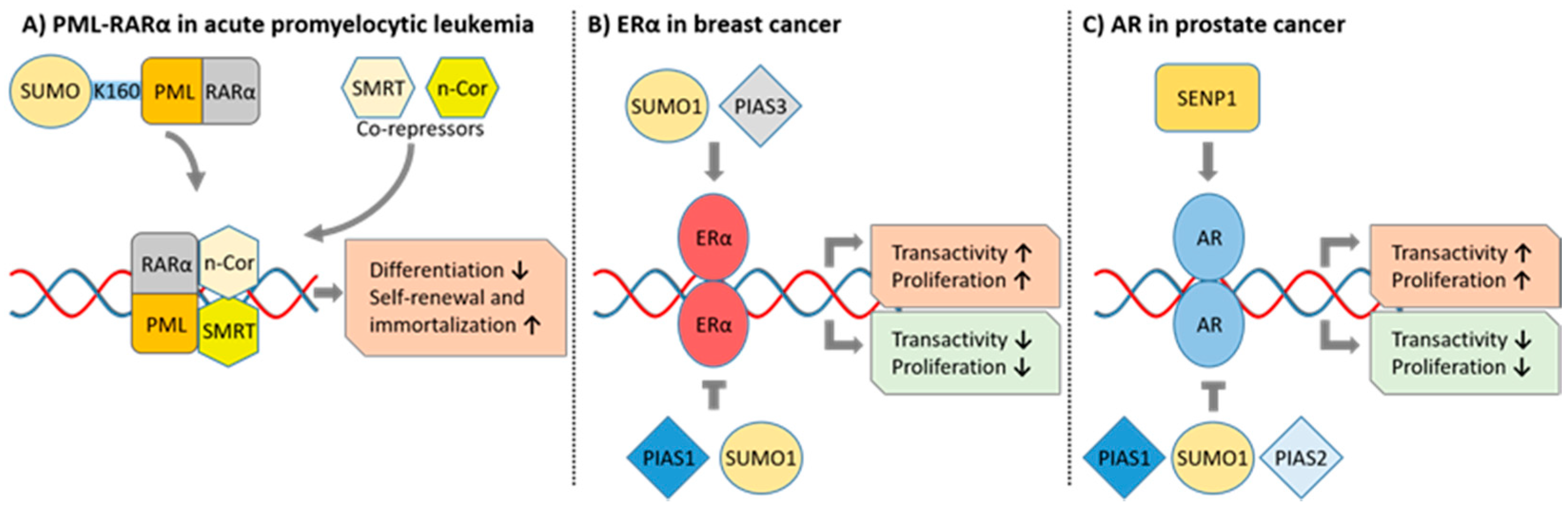

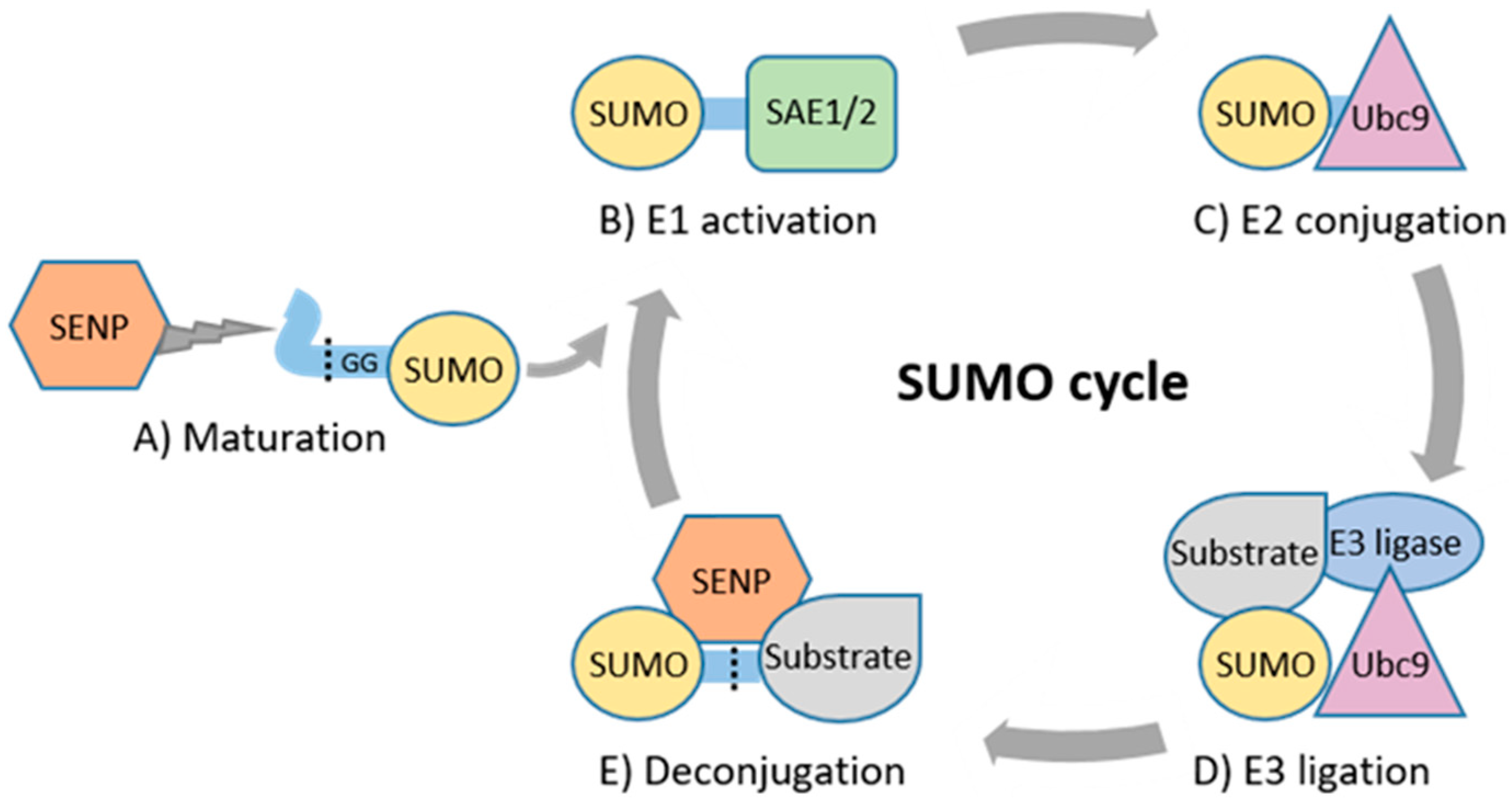{kind=link}
{kind=link}
| High Expression Associates with Poor Prognosis | High Expression Associates with Good Prognosis | ||
|---|---|---|---|
| Cancer Type | Protein(s) | Cancer Type | Protein(s) |
| adrenocortical | PIAS3, PIAS4, SAE1, SAE2, SENP1, SENP3, SUMO1, SUMO2, SUMO4 | bladder | Ubc9 |
| breast | PC2, PIAS3, PIAS4, SAE1, SAE2, SENP5, SENP7L, SUMO1, SUMO2, SUMO3, Ubc9 | breast | PC2, PIAS1, PIAS4 |
| colorectal | SAE2, SENP1, SUMO1 | cervical | PIAS3 |
| gastric | PC2, PIAS2, SAE2, SUMO3, Ubc9 | colorectal | PC2 |
| glioma | SAE1, Ubc9 | gastric | PIAS1, PIAS4 |
| hepatocellular | PC2, PIAS2, PIAS3, PIAS4, SAE1, SAE2, SENP1, SENP3, SENP5, SENP6, SUMO2, Ubc9 | glioma | PIAS3 |
| leukemia | SAE1, SUMO3 | leukemia | PIAS2, SENP5, SENP7 |
| lung | PC2, SAE1, SAE2, SENP1, SUMO2/3, SUMO4, Ubc9 | lung | PIAS3 |
| melanoma (cutaneous) | SAE1 | melanoma (cutaneous) | PIAS1, SENP5, SENP7 |
| melanoma (uveal) | SAE1, SAE2, SUMO3 | melanoma (uveal) | SENP2, Ubc9 |
| mesothelioma | PIAS3, PIAS4, SAE1, SAE2, SENP1 | mesothelioma | PC2, PIAS3, SENP2 |
| multiple myeloma | Ubc9 | ovarian | PIAS2 |
| osteosarcoma | PC2, SENP3 | pancreatic | SENP3 |
| ovarian | SENP3, SENP5 | pheochromocytoma and paraganglioma | Ubc9 |
| pancreatic | SENP2, (SUMO1 and SUMO2/3 together), Ubc9 | renal | PIAS1, PIAS2 |
| prostate | PIAS1, SAE1, SENP1, SENP5, SUMO1, SUMO2 | testicular germ cell | PIAS2 |
| renal | PC2, PIAS3, RSUME, SAE1, SENP1, SENP3, SENP5, SUMO1, SUMO2, Ubc9 | thymoma | PIAS4, SAE1, SAE2, SENP1, Ubc9 |
| sarcoma | PC2, PIAS2, PIAS3, SENP6, SENP7 | ||
| thyroid | PIAS2, SAE1 | ||
| uterine corpus endometrial | PC2, SAE2, SENP2, SENP5, SUMO4 | ||
| Target | Inhibitor | Product Type | Activity | IC50 (μM) | Study |
|---|---|---|---|---|---|
| SAE1/2 | Ginkgolic acid | Natural | In vivo and in vitro | 3.0 | [263] |
| Anacardic acid | Natural | In vivo and in vitro | 2.2 | [263] | |
| Kerriamycin B | Natural | In vitro | 11.7 | [264] | |
| SUMO-AMSN and SUMO-AVSN | Semisynthetic | In vitro | [302] | ||
| Compound 9 | Synthetic | In vitro | 13.4 | [267] | |
| Compound 21 | Synthetic | In vitro | 14.4 | [268] | |
| Pyrazole and thiazole urea containing compounds | Synthetic | In vitro | 13.8–100 | [269] | |
| Davidiin | Natural | In vitro | 0.15 | [265] | |
| Tannic acid | Natural | In vivo and in vitro | 12.8 | [266] | |
| ML-792 | Synthetic | In vitro | 0.003 (SUMO1), 0.011 (SUMO2) | [270] | |
| COH000 | Synthetic | In vivo and in vitro | 0.2 | [271,272] | |
| ML-93 | Synthetic | In vitro | 0.037 | [273] | |
| Ginkgolic acid derivatives | Synthetic | In vitro | 5–50 | [274] | |
| TAK-981 | Synthetic | In vivo and in vitro | nM range | [275] | |
| Ubc9 | GSK145A | Synthetic | In vitro | 12.5 | [280] |
| 2-D08 | Synthetic | In vitro | 6.0 | [281] | |
| Spectomycin B1 | Natural | In vivo and in vitro | 4.4 | [279] | |
| SUBINs | Semisynthetic | In vitro | 0.025 | [283] | |
| Compound 2 | Synthetic | In vitro | 74 | [282] | |
| SENP1 | Compound 38 | Synthetic | In vitro | 9.2 | [287] |
| Triptolide | Natural | In vivo and in vitro | 0.071–0.076 | [236] | |
| Compound J5 | Synthetic | In vitro | 2.385 | [288] | |
| Compound 4 (GN6958) | Synthetic | In vitro | 29.6 | [289] | |
| Compound 13m | Synthetic | In vitro | 3.5 | [290] | |
| Momordin Ic (Mc) | Natural | In vivo and in vitro | 15.37 | [237] | |
| Compounds 6, 7 and 10 | Synthetic | In vitro | 3.7, 0.99, 7.5 | [291] | |
| SENP2 | Compounds 69 and 117 | Synthetic | In vitro | 5.9, 3.7 | [292] |
| Ebselen | Synthetic | In vivo and In vitro | 2.0 | [293] | |
| SENP3 | URB597 | Synthetic | [294] | ||
| SENP1/2/3/5/6/7 | JCP666 | Natural | In vitro | 13.8 (SENP1), 7.0 (SENP2) | [295,296] |
| VEA260 | Synthetic | In vitro | 7.1 (SENP1), 3.7 (SENP2) | [295,296] | |
| VEA499 | Synthetic | In vitro | 3.6 (SENP1), 0.25 (SENP2) | [295] | |
| Compound 1 | Synthetic | In vitro | 5–10 (SENP1), 5–10 (SENP2) | [297] | |
| Compound 3 | Synthetic | In vitro | 3.55 (SENP1), 2.98 (SENP2) | [298] | |
| Streptonigrin | Natural | In vitro | 0.518 (SENP1), 6.919 (SENP2) | [299] | |
| SI2 | Synthetic | In vitro | 1.29 (SENP1), unknown (SENP2), unknown (SENP3) | [240] | |
| Benzothiophene-2-carboxamide derivatives | Synthetic | In vitro | 0.76–35.8 (SENP1), 0.56–75.6 (SENP2), 2.4–100 (SENP5) | [300] | |
| SPI-01 | Synthetic | In vitro | 5.9 (SENP1), 2.9 (SENP2), 3.5 (SENP7) | [301] | |
| VEA561 | Synthetic | In vitro | 5.7 (SENP2), 4.2 (SENP6), 4.3 (SENP7) | [295] |
| Intervention | Condition | Status | Estimated Enrollment | Phase | NCT# |
|---|---|---|---|---|---|
| TAK-981 and Pembrolizumab | Advanced or metastatic solid tumors | Recruiting | 242 | I/II | NCT04381650 |
| TAK-981 and Mezagitamab and Daratumumab/hyaluronidase-fihj | Relapsed and/or refractory multiple myeloma | Recruiting | 81 | I/II | NCT04776018 |
| TAK-981 and Rituximab | Non-Hodgkin lymphoma | Recruiting | 130 | I/II | NCT04074330 |
| TAK-981 | Advanced or metastatic solid tumors, Relapsed/refractory Non-Hodgkin lymphomas | Recruiting | 202 | I/II | NCT03648372 |
| TAK-981 and Cetuximab and Avelumab | Head and neck cancer | Recruiting | 12 | 0/I | NCT04065555 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. https://doi.org/10.3390/cancers13174402
Kukkula A, Ojala VK, Mendez LM, Sistonen L, Elenius K, Sundvall M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers. 2021; 13(17):4402. https://doi.org/10.3390/cancers13174402
Chicago/Turabian StyleKukkula, Antti, Veera K. Ojala, Lourdes M. Mendez, Lea Sistonen, Klaus Elenius, and Maria Sundvall. 2021. "Therapeutic Potential of Targeting the SUMO Pathway in Cancer" Cancers 13, no. 17: 4402. https://doi.org/10.3390/cancers13174402
APA StyleKukkula, A., Ojala, V. K., Mendez, L. M., Sistonen, L., Elenius, K., & Sundvall, M. (2021). Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers, 13(17), 4402. https://doi.org/10.3390/cancers13174402