
Playing on the Dark Side: SMYD3 Acts as a Cancer Genome Keeper in Gastrointestinal Malignancies

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
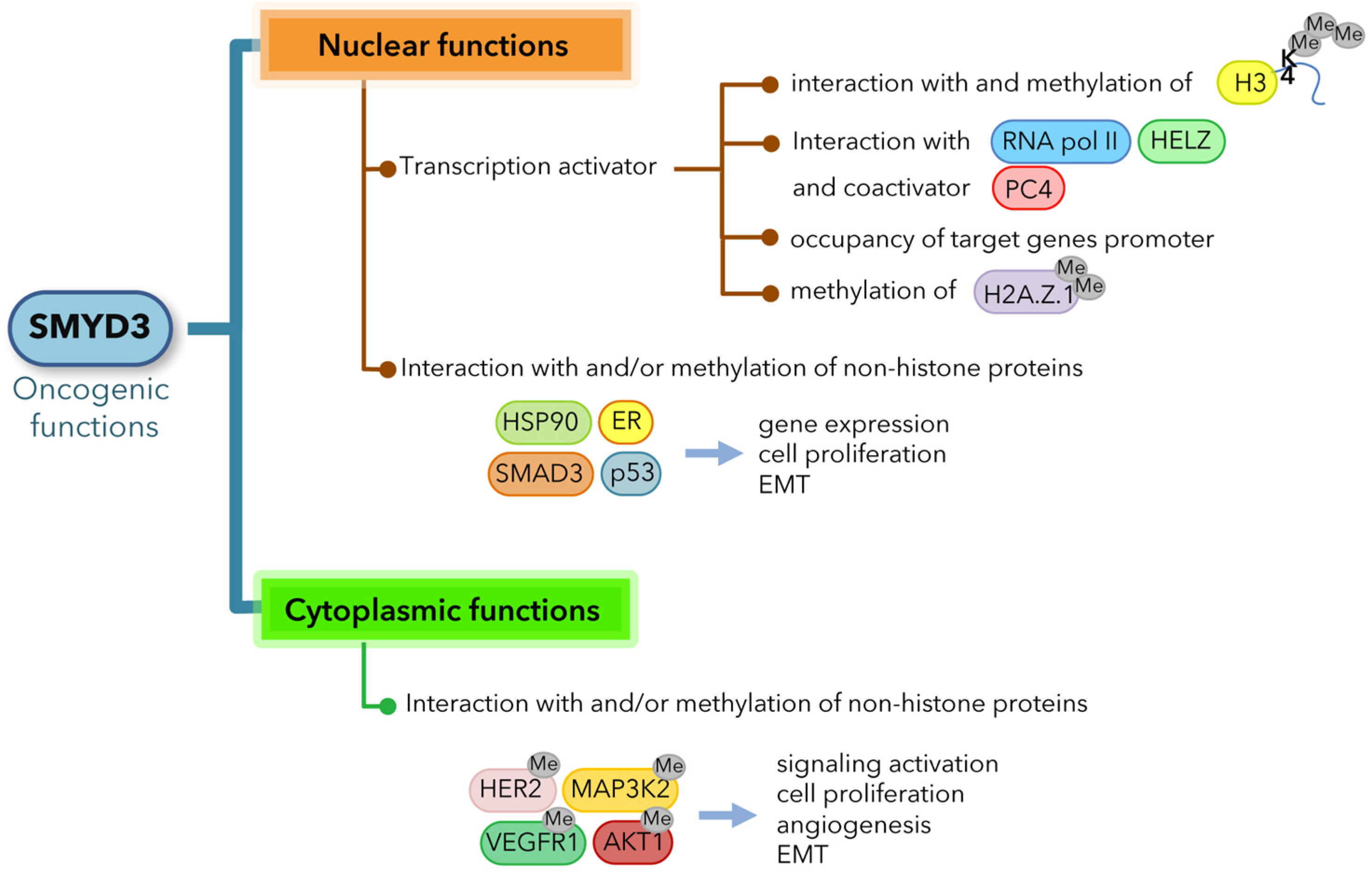
2. SMYD3 Oncogenic Functions
2.1. SMYD3 Exerts Oncogenic Effects through Multiple Mechanisms
2.2. SMYD3 and Cancer Cell Growth: An Emerging Debate
3. SMYD3 Alterations in GI Cancers
4. Oncogenic Role of SMYD3 in GI Cancers
4.1. Role of SMYD3 in Controlling Cell Cycle Progression
4.2. Role of SMYD3 in DNA Damage: From Tumorigenesis to Cancer Progression
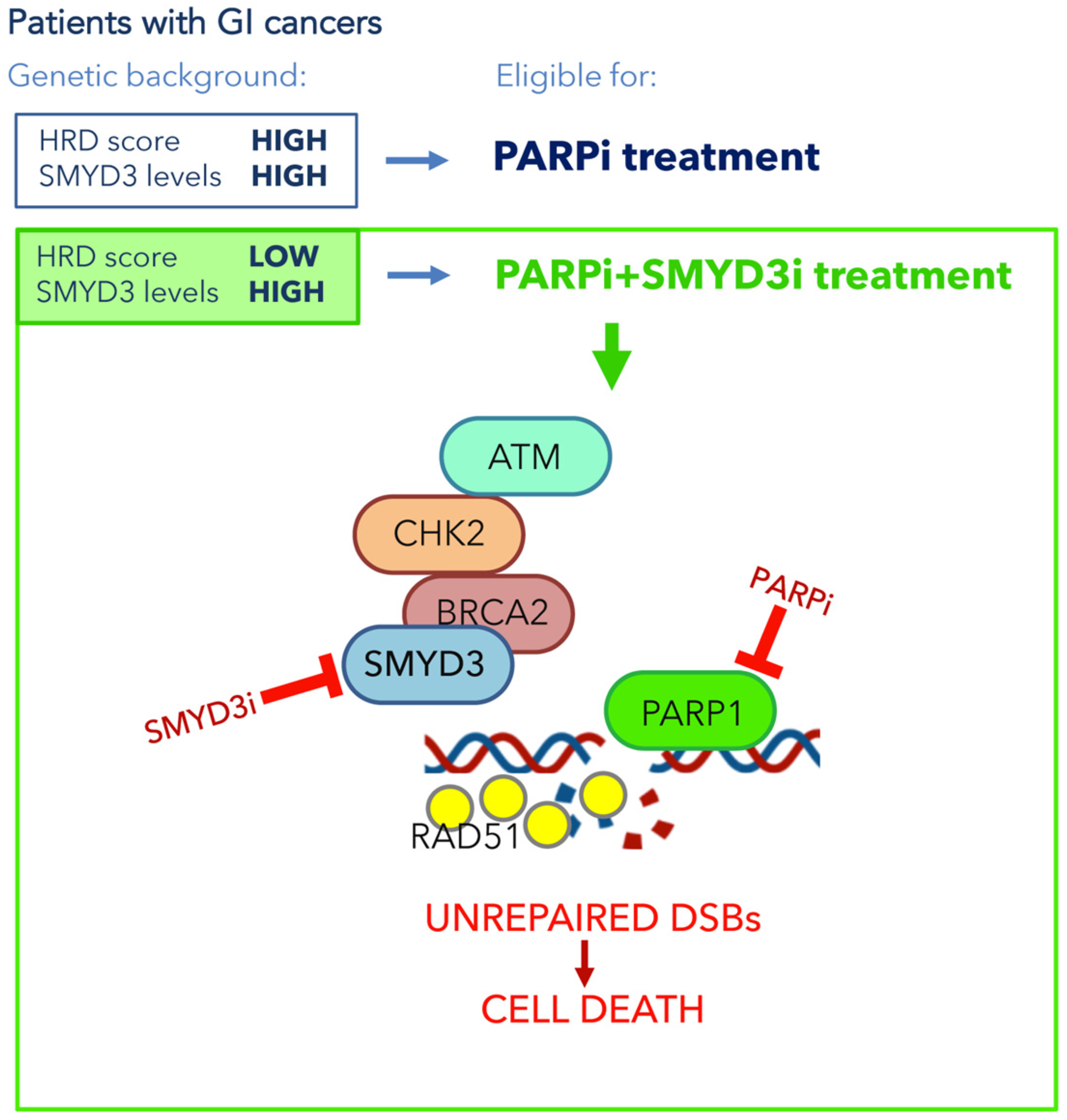
5. Clinical Impact of SMYD3 Inhibition for New Therapeutic Strategies in GI Cancers
5.1. Alterations of DNA Damage Checkpoint Factors in GI Cancer Initiation and Development
5.2. SMYD3 as a Promising Pharmacological Target Involved in DNA Damage Checkpoints in GI Cancers
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bottino, C.; Peserico, A.; Simone, C.; Caretti, G. SMYD3: An Oncogenic Driver Targeting Epigenetic Regulation and Signaling Pathways. Cancers 2020, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Foreman, K.W.; Brown, M.; Park, F.; Emtage, S.; Harriss, J.; Das, C.; Zhu, L.; Crew, A.; Arnold, L.; Shaaban, S.; et al. Structural and Functional Profiling of the Human Histone Methyltransferase SMYD3. PLoS ONE 2011, 6, e22290. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, C.; Yang, Z. Is MYND Domain-Mediated Assembly of SMYD3 Complexes Involved in Calcium Dependent Signaling? Front. Mol. Biosci. 2019, 6, 121. [Google Scholar] [CrossRef]
- Sirinupong, N.; Brunzelle, J.; Doko, E.; Yang, Z. Structural Insights into the Autoinhibition and Posttranslational Activation of Histone Methyltransferase SmyD3. J. Mol. Biol. 2011, 406, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Foreman, K.; Harriss, J.; Das, C.; Zhu, L.; Edwards, M.; Shaaban, S.; Tucker, H. C-terminal domain of SMYD3 serves as a unique HSP90-regulated motif in oncogenesis. Oncotarget 2015, 6, 4005–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 Promotes Cancer Invasion by Epigenetic Upregulation of the Metalloproteinase MMP-9. Cancer Res. 2011, 72, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.-G.; Zhang, C.-L.; Zhao, W.-W.; Liu, Z.-P.; Liu, L.; Mu, A.; Guo, S.; Wang, N.; Zhou, H.; Zhang, T.-C. Histone methyltransferase SMYD3 promotes MRTF-A-mediated transactivation of MYL9 and migration of MCF-7 breast cancer cells. Cancer Lett. 2013, 344, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Germani, A.; Sanese, P.; Barbosa, A.M.; Di Virgilio, V.; Fittipaldi, R.; Fabini, E.; Bertucci, C.; Varchi, G.; Moyer, M.P.; et al. A SMYD3 Small-Molecule Inhibitor Impairing Cancer Cell Growth. J. Cell. Physiol. 2015, 230, 2447–2460. [Google Scholar] [CrossRef] [Green Version]
- Van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.-F.; McDevitt, P.; Sinnamon, R.; Le, B.; Mas, G.; et al. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 2012, 7, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Sarris, M.E.; Moulos, P.; Haroniti, A.; Giakountis, A.; Talianidis, I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Heo, K.; Kim, J.H.; Kim, K.; Choi, J.; An, W. Requirement of Histone Methyltransferase SMYD3 for Estrogen Receptor-mediated Transcription. J. Biol. Chem. 2009, 284, 19867–19877. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-M.; Kim, K.; Schmidt, T.; Punj, V.; Tucker, H.; Rice, J.C.; Ulmer, T.S.; An, W. Cooperation between SMYD3 and PC4 drives a distinct transcriptional program in cancer cells. Nucleic Acids Res. 2015, 43, 8868–8883. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-H.; Chen, Y.-J.; Yu, C.-J.; Tzeng, S.-R.; Wu, I.-C.; Kuo, W.-H.; Lin, M.-C.; Chan, N.-L.; Wu, K.-J.; Teng, S.-C. SMYD3-Mediated H2A.Z.1 Methylation Promotes Cell Cycle and Cancer Proliferation. Cancer Res. 2016, 76, 6043–6053. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.; Reynoird, N.; Khatri, P.; Jansen, P.W.T.C.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.J.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Suzuki, T.; Matsuo, Y.; Nakakido, M.; Tsurita, G.; Simone, C.; Watanabe, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. SMYD3-mediated lysine methylation in the PH domain is critical for activation of AKT1. Oncotarget 2016, 7, 75023–75037. [Google Scholar] [CrossRef]
- Kunizaki, M.; Hamamoto, R.; Silva, F.P.; Yamaguchi, K.; Nagayasu, T.; Shibuya, M.; Nakamura, Y.; Furukawa, Y. The Lysine 831 of Vascular Endothelial Growth Factor Receptor 1 Is a Novel Target of Methylation by SMYD3. Cancer Res. 2007, 67, 10759–10765. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, Y.; Suzuki, T.; Matsuo, Y.; Tsurita, G.; Watanabe, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. Protein lysine methyltransferase SMYD3 is involved in tumorigenesis through regulation of HER2 homodimerization. Cancer Med. 2017, 6, 1665–1672. [Google Scholar] [CrossRef]
- Mouse Genome Informatics. Available online: http://www.informatics.jax.org/allele/key/571089 (accessed on 21 March 2021).
- Zhou, Z.; Jiang, H.; Tu, K.; Yu, W.; Zhang, J.; Hu, Z.; Zhang, H.; Hao, D.; Huang, P.; Wang, J.; et al. ANKHD1 is required for SMYD3 to promote tumor metastasis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, B.-H.; Lin, W.-H.; Huang, Y.-H.; Ni, J.-Y.; Hu, J.; Cui, W.; Zhou, J.; Shen, L.; Xu, L.-F.; et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 2019, 38, 4948–4961. [Google Scholar] [CrossRef]
- Dong, S.-W.; Zhang, H.; Wang, B.-L.; Sun, P.; Wang, Y.-G.; Zhang, P.; Sun, P. Effect of the downregulation of SMYD3 expression by RNAi on RIZ1 expression and proliferation of esophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 1064–1070. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, M.-X.; Zhang, X.-D.; Xu, X.-E.; Wu, Z.-Y.; Liao, L.-D.; Li, L.-Y.; Xie, Y.-M.; Wu, J.-Y.; Zou, H.-Y.; et al. SMYD3 stimulates EZR and LOXL2 transcription to enhance proliferation, migration, and invasion in esophageal squamous cell carcinoma. Hum. Pathol. 2016, 52, 153–163. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, Z.; Chen, C.; Guo, S.; Liao, Z.; Li, Y.; Zhu, Y.; Zou, H.; Wu, J.; Xie, W.; et al. Network analyses elucidate the role of SMYD3 in esophageal squamous cell carcinoma. FEBS Open Bio 2017, 7, 1111–1125. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.-L.; Huang, Q. Overexpression of the SMYD3 Promotes Proliferation, Migration, and Invasion of Pancreatic Cancer. Dig. Dis. Sci. 2019, 65, 489–499. [Google Scholar] [CrossRef]
- Fabini, E.; Manoni, E.; Ferroni, C.; Del Rio, A.; Bartolini, M. Small-molecule inhibitors of lysine methyltransferases SMYD2 and SMYD3: Current trends. Futur. Med. Chem. 2019, 11, 901–921. [Google Scholar] [CrossRef]
- Mitchell, L.H.; Boriack-Sjodin, P.A.; Smith, S.; Thomenius, M.; Rioux, N.; Munchhof, M.; Mills, J.E.; Klaus, C.; Totman, J.; Riera, T.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2015, 7, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Thomenius, M.J.; Totman, J.; Harvey, D.; Mitchell, L.H.; Riera, T.V.; Cosmopoulos, K.; Grassian, A.R.; Klaus, C.; Foley, M.; Admirand, E.A.; et al. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS ONE 2018, 13, e0197372. [Google Scholar] [CrossRef]
- Van Aller, G.S.; Graves, A.P.; Elkins, P.A.; Bonnette, W.G.; McDevitt, P.J.; Zappacosta, F.; Annan, R.S.; Dean, T.W.; Su, D.-S.; Carpenter, C.L.; et al. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24, 774–781. [Google Scholar] [CrossRef]
- Talibov, V.O.; Fabini, E.; FitzGerald, E.A.; Tedesco, D.; Cederfeldt, D.; Talu, M.J.; Rachman, M.M.; Mihalic, F.; Manoni, E.; Naldi, M.; et al. Discovery of an Allosteric Ligand Binding Site in SMYD3 Lysine Methyltransferase. ChemBioChem 2021, 22, 1597–1608. [Google Scholar] [CrossRef]
- Alshiraihi, I.M.; Jarrell, D.K.; Arhouma, Z.; Hassell, K.N.; Montgomery, J.; Padilla, A.; Ibrahim, H.M.; Crans, D.C.; Kato, T.A.; Brown, M.A. In Silico/In Vitro Hit-to-Lead Methodology Yields SMYD3 Inhibitor That Eliminates Unrestrained Proliferation of Breast Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 9549. [Google Scholar] [CrossRef]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef]
- Liu, C.; Fang, X.; Ge, Z.; Jalink, M.; Kyo, S.; Björkholm, M.; Gruber, A.; Sjöberg, J.; Xu, D. The Telomerase Reverse Transcriptase (hTERT) Gene Is a Direct Target of the Histone Methyltransferase SMYD3. Cancer Res. 2007, 67, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Fenizia, C.; Bottino, C.; Corbetta, S.; Fittipaldi, R.; Floris, P.; Gaudenzi, G.; Carra, S.; Cotelli, F.; Vitale, G.; Caretti, G. SMYD3 promotes the epithelial–mesenchymal transition in breast cancer. Nucleic Acids Res. 2018, 47, 1278–1293. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-Z.; Luo, X.-G.; Shen, J.; Zou, J.-N.; Lu, Y.-H.; Xi, T. Knockdown of SMYD3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008, 41, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.-N.; Wang, S.; Yang, J.-S.; Luo, X.-G.; Xie, J.-H.; Xi, T. Knockdown of SMYD3 by RNA interference down-regulates c-Met expression and inhibits cells migration and invasion induced by HGF. Cancer Lett. 2009, 280, 78–85. [Google Scholar] [CrossRef]
- Ren, T.-N.; Wang, J.-S.; He, Y.-M.; Xu, C.-L.; Wang, S.-Z.; Xi, T. Effects of SMYD3 over-expression on cell cycle acceleration and cell proliferation in MDA-MB-231 human breast cancer cells. Med. Oncol. 2010, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, H.; Liu, S.; Lei, Z.; Qin, Z.; Wen, L.; Liu, K.; Wang, X.; Guo, Y.; Liu, Q.; et al. SMYD3 controls a Wnt-responsive epigenetic switch for ASCL2 activation and cancer stem cell maintenance. Cancer Lett. 2018, 430, 11–24. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, Y.; Yang, H.; Li, Y.; Wang, C.; Shi, Y.; Wang, Y. SMYD3 promotes epithelial ovarian cancer metastasis by down-regulating p53 protein stability and promoting p53 ubiquitination. Carcinogenesis 2019, 40, 1492–1503. [Google Scholar] [CrossRef]
- Luo, X.-G.; Ding, Y.; Zhou, Q.-F.; Ye, L.; Wang, S.-Z.; Xi, T. SET and MYND domain-containing protein 3 decreases sensitivity to dexamethasone and stimulates cell adhesion and migration in NIH3T3 cells. J. Biosci. Bioeng. 2007, 103, 444–450. [Google Scholar] [CrossRef]
- Chen, L.-B.; Xu, J.-Y.; Yang, Z.; Wang, G.-B. Silencing SMYD3 in hepatoma demethylates RIZI promoter induces apoptosis and inhibits cell proliferation and migration. World J. Gastroenterol. 2007, 13, 5718–5724. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Q.-T.; Liu, Y.-P.; Dong, Q.-Q.; Hu, H.-J.; Miao, Z.; Li, S.; Liu, Y.; Zhou, H.; Zhang, T.-C.; et al. ATM Signaling Pathway Is Implicated in the SMYD3-mediated Proliferation and Migration of Gastric Cancer Cells. J. Gastric Cancer 2017, 17, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.-G.; Xi, T.; Guo, S.; Liu, Z.-P.; Wang, N.; Jiang, Y.; Zhang, T.-C. Effects of SMYD3 overexpression on transformation, serum dependence, and apoptosis sensitivity in NIH3T3 cells. IUBMB Life 2009, 61, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Breitenbach, M.; Hoffmann, J. Editorial: Cancer Models. Front. Oncol. 2018, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Sanese, P.; Fasano, C.; Buscemi, G.; Bottino, C.; Corbetta, S.; Fabini, E.; Silvestri, V.; Valentini, V.; Disciglio, V.; Forte, G.; et al. Targeting SMYD3 to Sensitize Homologous Recombination-Proficient Tumors to PARP-Mediated Synthetic Lethality. iScience 2020, 23, 101604. [Google Scholar] [CrossRef]
- Liu, N.; Sun, S.; Yang, X. Prognostic significance of stromal SMYD3 expression in colorectal cancer of TNM stage I-III. Int. J. Clin. Exp. Pathol. 2017, 10, 8901–8907. [Google Scholar]
- Liu, Y.; Luo, X.; Deng, J.; Pan, Y.; Zhang, L.; Liang, H. SMYD3 overexpression was a risk factor in the biological behavior and prognosis of gastric carcinoma. Tumor Biol. 2014, 36, 2685–2694. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, J.; Luo, X.; Pan, Y.; Zhang, L.; Zhang, R.; Liang, H. Overexpression of SMYD3 was associated with increased STAT3 activation in gastric cancer. Med. Oncol. 2014, 32, 404. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Luo, X.; Deng, J.; Pan, Y.; Liang, H. Overexpression of SMYD3 and matrix metalloproteinase-9 are associated with poor prognosis of patients with gastric cancer. Tumor Biol. 2015, 36, 4377–4386. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Kong, F.; Xin, W.; Li, X.; Liang, H.; Jia, Y. Elevated Levels of SET and MYND Domain-Containing Protein 3 Are Correlated with Overexpression of Transforming Growth Factor-β1 in Gastric Cancer. J. Am. Coll. Surg. 2015, 221, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.-Q.; Wang, Q.-T.; Wang, L.; Jiang, Y.-X.; Liu, M.-L.; Hu, H.-J.; Liu, Y.; Zhou, H.; He, H.-P.; Zhang, T.-C.; et al. SMYD3-associated pathway is involved in the anti-tumor effects of sulforaphane on gastric carcinoma cells. Food Sci. Biotechnol. 2018, 27, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lyu, T.; Che, X.; Jia, N.; Li, Q.; Feng, W. Overexpression of SMYD3 in Ovarian Cancer is Associated with Ovarian Cancer Proliferation and Apoptosis via Methylating H3K4 and H4K20. J. Cancer 2019, 10, 4072–4084. [Google Scholar] [CrossRef]
- Jiang, G.L.; Huang, S. Adenovirus expressing RIZ1 in tumor suppressor gene therapy of microsatellite-unstable colorectal cancers. Cancer Res. 2001, 61, 1796–1798. [Google Scholar]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Mikhail, S.; Albanese, C.; Pishvaian, M.J. Cyclin-Dependent Kinase Inhibitors and the Treatment of Gastrointestinal Cancers. Am. J. Pathol. 2015, 185, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Al-Aynati, M.M.; Radulovich, N.; Ho, J.; Tsao, M.-S. Overexpression of G1-S Cyclins and Cyclin-Dependent Kinases during Multistage Human Pancreatic Duct Cell Carcinogenesis. Clin. Cancer Res. 2004, 10, 6598–6605. [Google Scholar] [CrossRef] [Green Version]
- Gansauge, S.; Gansauge, F.; Ramadani, M.; Stobbe, H.; Rau, B.; Harada, N.; Beger, H.G. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res. 1997, 57, 1634–1637. [Google Scholar]
- Matsumoto, M.; Furihata, M.; Ishikawa, T.; Ohtsuki, Y.; Ogoshi, S. Comparison of deregulated expression of cyclin D1 and cyclin E with that of cyclin-dependent kinase 4 (CDK4) and CDK2 in human oesophageal squamous cell carcinoma. Br. J. Cancer 1999, 80, 256–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Matsuura, N.; Sakon, M.; Miyoshi, E.; Noda, K.; Takeda, T.; Umeshita, K.; Nagano, H.; Nakamori, S.; Dono, K.; et al. Expression and prognostic roles of the G1-S modulators in hepatocellular carcinoma: p27 independently predicts the recurrence. Hepatology 1999, 30, 90–99. [Google Scholar] [CrossRef]
- Takano, Y.; Kato, Y.; van Diest, P.J.; Masuda, M.; Mitomi, H.; Okayasu, I. Cyclin D2 Overexpression and Lack of p27 Correlate Positively and Cyclin E Inversely with a Poor Prognosis in Gastric Cancer Cases. Am. J. Pathol. 2000, 156, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Yamaji, T.; Loda, M.; Fuchs, C.S. Loss of nuclear p27 (CDKN1B/KIP1) in colorectal cancer is correlated with microsatellite instability and CIMP. Mod. Pathol. 2006, 20, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Huen, M.S.-Y.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res. 2007, 18, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA repair in cancer: Current state and novel approaches. Cell. Mol. Life Sci. 2019, 77, 677–703. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Tsai, C.-H.; Wang, P.-Y.; Teng, S.-C. SMYD3 Promotes Homologous Recombination via Regulation of H3K4-mediated Gene Expression. Sci. Rep. 2017, 7, 384. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinaro, E.; Andrikou, K.; Casadei-Gardini, A.; Rovesti, G. BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives. Cancers 2020, 12, 3346. [Google Scholar] [CrossRef]
- Parikh, A.R.; He, Y.; Hong, T.S.; Corcoran, R.B.; Clark, J.W.; Ryan, D.P.; Zou, L.; Ting, D.; Catenacci, D.V.; Chao, J.; et al. Analysis of DNA Damage Response Gene Alterations and Tumor Mutational Burden Across 17,486 Tubular Gastrointestinal Carcinomas: Implications for Therapy. Oncologist 2019, 24, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Lorans, M.; Dow, E.; Macrae, F.A.; Winship, I.M.; Buchanan, D.D. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin. Color. Cancer 2018, 17, e293–e305. [Google Scholar] [CrossRef] [Green Version]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Siu, H.C.; Leung, S.Y.; Stratton, M.R. A mutational signature in gastric cancer suggests therapeutic strategies. Nat. Commun. 2015, 6, 8683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [Green Version]
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randon, G.; Fucà, G.; Rossini, D.; Raimondi, A.; Pagani, F.; Perrone, F.; Tamborini, E.; Busico, A.; Peverelli, G.; Morano, F.; et al. Prognostic impact of ATM mutations in patients with metastatic colorectal cancer. Sci. Rep. 2019, 9, 2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgason, H.; Rafnar, T.; Olafsdottir, H.S.; Jonasson, J.G.; Sigurdsson, A.; Stacey, S.N.; Jonasdottir, A.; Tryggvadottir, L.; Alexiusdottir, K.K.; Haraldsson, A.; et al. Loss-of-function variants in ATM confer risk of gastric cancer. Nat. Genet. 2015, 47, 906–910. [Google Scholar] [CrossRef]
- Mirza-Aghazadeh-Attari, M.; Darband, S.G.; Kaviani, M.; Mihanfar, A.; Attari, J.A.; Yousefi, B.; Majidinia, M. DNA damage response and repair in colorectal cancer: Defects, regulation and therapeutic implications. DNA Repair 2018, 69, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Lüttges, J.; Kalthoff, H.; Stürzbecher, H.-W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Galamb, O.; Sipos, F.; Dinya, E.; Spisak, S.; Tulassay, Z.; Molnar, B. mRNA expression, functional profiling and multivariate classification of colon biopsy specimen by cDNA overall glass microarray. World J. Gastroenterol. 2006, 12, 6998–7006. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Ogawa, T.; Baer, R.; Hemmi, H.; Honda, K.; Yamauchi, A.; Inamoto, T.; Ko, K.; Yazumi, S.; Motoda, H.; et al. Abnormal Expression of BRCA1 and BRCA1-Interactive DNA-Repair Proteins in Breast Carcinomas. Int. J. Cancer 2000, 88, 28–36. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, H.; Arbman, G.; Sun, X.-F. The Different Roles of hRAD50 in Microsatellite Stable and Unstable Colorectal Cancers. Dis. Markers 2008, 24, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Madoz-Gúrpide, J.; Cañamero, M.; Sanchez, L.; Solano, J.; Alfonso, P.; Casal, J.I. A Proteomics Analysis of Cell Signaling Alterations in Colorectal Cancer. Mol. Cell. Proteom. 2007, 6, 2150–2164. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- O’Neil, N.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.; de Bono, J. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Arena, S.; Siena, S.; Bardelli, A.; Sartore-Bianchi, A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 2020, 31, 1135–1147. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanese, P.; Fasano, C.; Simone, C. Playing on the Dark Side: SMYD3 Acts as a Cancer Genome Keeper in Gastrointestinal Malignancies. Cancers 2021, 13, 4427. https://doi.org/10.3390/cancers13174427
Sanese P, Fasano C, Simone C. Playing on the Dark Side: SMYD3 Acts as a Cancer Genome Keeper in Gastrointestinal Malignancies. Cancers. 2021; 13(17):4427. https://doi.org/10.3390/cancers13174427
Chicago/Turabian StyleSanese, Paola, Candida Fasano, and Cristiano Simone. 2021. "Playing on the Dark Side: SMYD3 Acts as a Cancer Genome Keeper in Gastrointestinal Malignancies" Cancers 13, no. 17: 4427. https://doi.org/10.3390/cancers13174427