
Tumor Nonimmune-Microenvironment-Related Gene Expression Signature Predicts Brain Metastasis in Lung Adenocarcinoma Patients after Surgery: A Machine Learning Approach Using Gene Expression Profiling


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Prediction Modeling Using Machine Learning Methods
2.2. Relationship between the 17-Gene Signature and TNM Stage
2.3. Principal Component Analysis and Pathway Enrichment Analysis
2.4. IHC Analysis
3. Materials and Methods
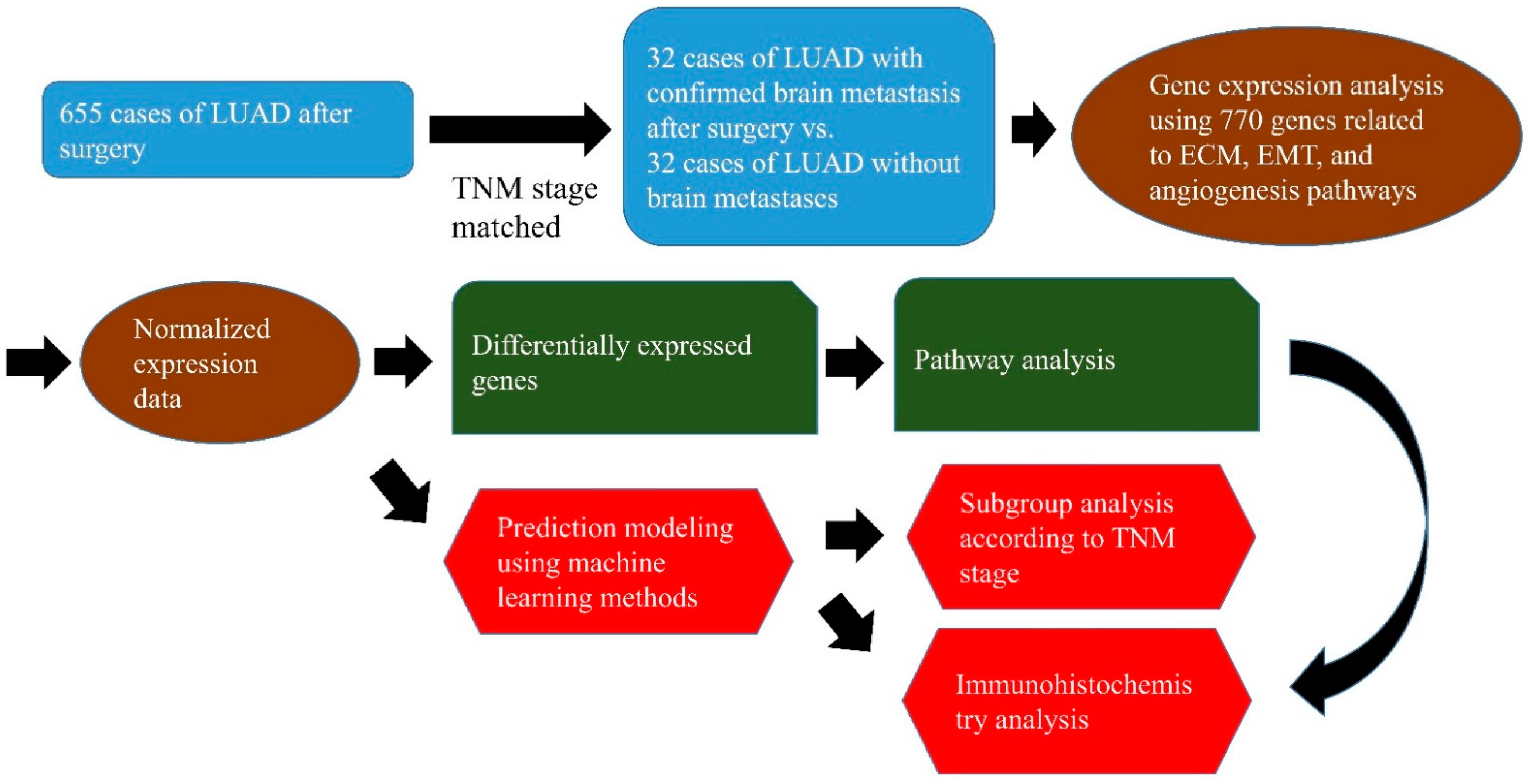
3.1. Clinical Samples
3.2. Gene Expression Analysis Using the NanoString nCounter Assay
3.3. IHC Staining
3.4. Machine Learning Approach and Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, H.; Wang, Z.; Zhang, G.; Zhang, M.; Zhang, X.; Li, H.; Zheng, X.; Ma, Z. Driver genes as predictive indicators of brain metastasis in patients with advanced NSCLC: EGFR, ALK, and RET gene mutations. Cancer Med. 2020, 9, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.; Zhuang, Y.; Zhou, X.; Huang, R.; Liang, X.; Zhan, Q. High probability and frequency of EGFR mutations in non-small cell lung cancer with brain metastases. J. Neuro-Oncol. 2017, 135, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Le Moulec, S.; Mazières, J.; Senellart, H.; Barlesi, F.; Chouaid, C.; Dansin, E.; Bérard, H.; Falchero, L.; Gervais, R.; et al. Bevacizumab in Patients with Nonsquamous Non-Small Cell Lung Cancer and Asymptomatic, Untreated Brain Metastases (BRAIN): A Nonrandomized, Phase II Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1896–1903. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.A.; Welzel, G.; Abo-Madyan, Y.; Wenz, F. Potential toxicities of prophylactic cranial irradiation. Transl. Lung Cancer Res. 2012, 1, 254–262. [Google Scholar] [CrossRef]
- Girigoswami, K.; Saini, D.; Girigoswami, A. Extracellular Matrix Remodeling and Development of Cancer. Stem Cell Rev. Rep. 2020. [Google Scholar] [CrossRef]
- Harati, R.; Hafezi, S.; Mabondzo, A.; Tlili, A. Silencing miR-202-3p increases MMP-1 and promotes a brain invasive phenotype in metastatic breast cancer cells. PLoS ONE 2020, 15, e0239292. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, S.N.; Toraskar, J.; Wilhelm, I.; Hasko, J.; Figenschau, S.L.; Molnar, J.; Seppola, M.; Steigen, S.E.; Steigedal, T.S.; Hadler-Olsen, E.; et al. Nephronectin promotes breast cancer brain metastatic colonization via its integrin-binding domains. Sci. Rep. 2020, 10, 12237. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Pagel, M.A.; Muldoon, L.L.; Fu, R.; Neuwelt, E.A. High αv Integrin Level of Cancer Cells Is Associated with Development of Brain Metastasis in Athymic Rats. Anticancer. Res. 2017, 37, 4029–4040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legras, A.; Pécuchet, N.; Imbeaud, S.; Pallier, K.; Didelot, A.; Roussel, H.; Gibault, L.; Fabre, E.; Le Pimpec-Barthes, F.; Laurent-Puig, P.; et al. Epithelial-to-Mesenchymal Transition and MicroRNAs in Lung Cancer. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeevan, D.S.; Cooper, J.B.; Braun, A.; Murali, R.; Jhanwar-Uniyal, M. Molecular Pathways Mediating Metastases to the Brain via Epithelial-to-Mesenchymal Transition: Genes, Proteins, and Functional Analysis. Anticancer. Res. 2016, 36, 523–532. [Google Scholar] [PubMed]
- Dai, L.; Zhao, J.; Yin, J.; Fu, W.; Chen, G. Cell adhesion molecule 2 (CADM2) promotes brain metastasis by inducing epithelial-mesenchymal transition (EMT) in human non-small cell lung cancer. Ann. Transl. Med. 2020, 8, 465. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Luo, X.; Zeng, H.; Zhan, X.; Yuan, C. SNORA71B promotes breast cancer cells across blood-brain barrier by inducing epithelial-mesenchymal transition. Breast Cancer (Tokyo Jpn.) 2020, 27, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Srivastava, S.K. HER2 mediated de novo production of TGFβ leads to SNAIL driven epithelial-to-mesenchymal transition and metastasis of breast cancer. Mol. Oncol. 2014, 8, 1532–1547. [Google Scholar] [CrossRef]
- Farnsworth, R.H.; Lackmann, M.; Achen, M.G.; Stacker, S.A. Vascular remodeling in cancer. Oncogene 2014, 33, 3496–3505. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Reviews. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef]
- Lorger, M.; Krueger, J.S.; O’Neal, M.; Staflin, K.; Felding-Habermann, B. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. In Proceedings of the National Academy of Sciences of the United States of America, Cambridge, MA, USA, 30 June 2009; Volume 106, pp. 10666–10671. [Google Scholar] [CrossRef] [Green Version]
- Avraham, H.K.; Jiang, S.; Fu, Y.; Nakshatri, H.; Ovadia, H.; Avraham, S. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J. Pathol. 2014, 232, 369–381. [Google Scholar] [CrossRef]
- Lin, C.Y.; Cho, C.F.; Bai, S.T.; Liu, J.P.; Kuo, T.T.; Wang, L.J.; Lin, Y.S.; Lin, C.C.; Lai, L.C.; Lu, T.P.; et al. ADAM9 promotes lung cancer progression through vascular remodeling by VEGFA, ANGPT2, and PLAT. Sci. Rep. 2017, 7, 15108. [Google Scholar] [CrossRef]
- Shintani, Y.; Higashiyama, S.; Ohta, M.; Hirabayashi, H.; Yamamoto, S.; Yoshimasu, T.; Matsuda, H.; Matsuura, N. Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res. 2004, 64, 4190–4196. [Google Scholar] [CrossRef] [Green Version]
- Mamoshina, P.; Volosnikova, M.; Ozerov, I.V.; Putin, E.; Skibina, E.; Cortese, F.; Zhavoronkov, A. Machine Learning on Human Muscle Transcriptomic Data for Biomarker Discovery and Tissue-Specific Drug Target Identification. Front. Genet. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Meng, W.Y.; Li, R.Z.; Wang, Y.W.; Qian, X.; Chan, C.; Yu, Z.F.; Fan, X.X.; Pan, H.D.; Xie, C.; et al. Early lung cancer diagnostic biomarker discovery by machine learning methods. Transl. Oncol. 2021, 14, 100907. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.M. Machine Learning; McGraw-Hill: New York, NY, USA, 1997; p. 414. [Google Scholar]
- Guyon I, E.A. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar]
- Dwivedi, A.K. Artificial neural network model for effective cancer classification using microarray gene expression data. Neural Comput. Appl. 2018, 29, 1545–1554. [Google Scholar] [CrossRef]
- Wilentzik Müller, R.; Gat-Viks, I. Exploring Neural Networks and Related Visualization Techniques in Gene Expression Data. Front. Genet. 2020, 11, 402. [Google Scholar] [CrossRef]
- Chen, Y.C.; Ke, W.C.; Chiu, H.W. Risk classification of cancer survival using ANN with gene expression data from multiple laboratories. Comput. Biol. Med. 2014, 48, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Heckerman, D.; Geiger, D.; Chickering, D.M. Learning Bayesian Networks: The Combination of Knowledge and Statistical Data. Mach. Learn. 1995, 20, 197–243. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.S.; Shahjaman, M.; Rana, M.M.; Mollah, M.N.H. Robustification of Naïve Bayes Classifier and Its Application for Microarray Gene Expression Data Analysis. BioMed Res. Int. 2017, 2017, 3020627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, B.; Gupta, M. Robust approach for estimating probabilities in Naïve–Bayes Classifier for gene expression data. Expert Syst. Appl. 2011, 38, 1293–1298. [Google Scholar] [CrossRef]
- Tin Kam, H. The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Yu, T. A Deep Neural Network Model using Random Forest to Extract Feature Representation for Gene Expression Data Classification. Sci. Rep. 2018, 8, 16477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Li, L.; Wang, L.; Li, X.; Xing, H.; Zhou, J. A random forest classifier predicts recurrence risk in patients with ovarian cancer. Mol. Med. Rep. 2018, 18, 3289–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, M.; Ketany, M.; Manevitz, L.; Showe, L.C.; Showe, M.K. Classification and biomarker identification using gene network modules and support vector machines. BMC Bioinform. 2009, 10, 337. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Yang, B.; Lee, H.; Um, S.W.; Kim, K.; Zo, J.I.; Shim, Y.M.; Jung Kwon, O.; Lee, K.S.; Ahn, M.J.; Kim, H. Incidence of brain metastasis in lung adenocarcinoma at initial diagnosis on the basis of stage and genetic alterations. Lung Cancer 2019, 129, 28–34. [Google Scholar] [CrossRef]
- Tsakonas, G.; Lewensohn, R.; Botling, J.; Ortiz-Villalon, C.; Micke, P.; Friesland, S.; Nord, H.; Lindskog, M.; Sandelin, M.; Hydbring, P.; et al. An immune gene expression signature distinguishes central nervous system metastases from primary tumours in non-small-cell lung cancer. Eur. J. Cancer 2020, 132, 24–34. [Google Scholar] [CrossRef]
- Koh, Y.W.; Han, J.H.; Haam, S.; Lee, H.W. An immune-related gene expression signature predicts brain metastasis in lung adenocarcinoma patients after surgery: Gene expression profile and immunohistochemical analyses. Transl. Lung Cancer Res. 2021, 10, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Hosmer, D.W.; Lemeshow, S.; Sturdivant, R.X. Applied Logistic Regression, 2nd ed.; John Wiley and Sons: New York, NY, USA, 2000; Chapter 5; pp. 160–164. [Google Scholar]
- Jin, X.; Xu, A.; Bie, R.; Guo, P. Machine Learning Techniques and Chi-Square Feature Selection for Cancer Classification Using SAGE Gene Expression Profiles. In International Workshop on Data Mining for Biomedical Applications; Springer-Verlag: Berlin/Heidelberg, Germany, 9 April 2006; pp. 106–115. [Google Scholar]
- Tanaka, H.Y.; Kitahara, K.; Sasaki, N.; Nakao, N.; Sato, K.; Narita, H.; Shimoda, H.; Matsusaki, M.; Nishihara, H.; Masamune, A.; et al. Pancreatic stellate cells derived from human pancreatic cancer demonstrate aberrant SPARC-dependent ECM remodeling in 3D engineered fibrotic tissue of clinically relevant thickness. Biomaterials 2019, 192, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Siddiqui, A.; Vazakidou, M.E.; Napoli, F.; Böttcher, M.; Menchicchi, B.; Raza, U.; Saatci, Ö.; Krebs, A.M.; Ferrazzi, F.; et al. Polyol Pathway Links Glucose Metabolism to the Aggressiveness of Cancer Cells. Cancer Res. 2018, 78, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Hyldahl, R.D.; Nelson, B.; Xin, L.; Welling, T.; Groscost, L.; Hubal, M.J.; Chipkin, S.; Clarkson, P.M.; Parcell, A.C. Extracellular matrix remodeling and its contribution to protective adaptation following lengthening contractions in human muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 2894–2904. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Luo, X.; Jiang, S.; Wang, H. Bone Morphogenetic Protein 1 Targeting COL1A1 and COL1A2 to Regulate the Epithelial-Mesenchymal Transition Process of Colon Cancer SW620 Cells. J. Nanosci. Nanotechnol. 2020, 20, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Huang, W.; Chen, N.; Shang, Y.; Zhang, H. SMC3 may play an important role in atopic asthma development. Clin. Respir. J. 2016, 10, 469–476. [Google Scholar] [CrossRef]
- Xu, Z.; Gu, C.; Yao, X.; Guo, W.; Wang, H.; Lin, T.; Li, F.; Chen, D.; Wu, J.; Ye, G.; et al. CD73 promotes tumor metastasis by modulating RICS/RhoA signaling and EMT in gastric cancer. Cell Death Dis. 2020, 11, 202. [Google Scholar] [CrossRef]
- Meng, X.; Yang, S.; Zhang, J.; Yu, H. Contribution of alternative splicing to breast cancer metastasis. J. Cancer Metastasis Treat. 2019, 5. [Google Scholar] [CrossRef]
- Anwer, M.; Bolkvadze, T.; Puhakka, N.; Ndode-Ekane, X.E.; Pitkänen, A. Genotype and Injury Effect on the Expression of a Novel Hypothalamic Protein Sushi Repeat-Containing Protein X-Linked 2 (SRPX2). Neuroscience 2019, 415, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Miljkovic-Licina, M.; Hammel, P.; Garrido-Urbani, S.; Bradfield, P.F.; Szepetowski, P.; Imhof, B.A. Sushi repeat protein X-linked 2, a novel mediator of angiogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 4105–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K.; Hirata, K.; Ishida, T.; Takeuchi, S.; Hirase, T.; Rikitake, Y.; Kojima, Y.; Inoue, N.; Kawashima, S.; Yokoyama, M. An anti-proliferative gene BTG1 regulates angiogenesis in vitro. Biochem. Biophys. Res. Commun. 2004, 316, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Massey, K.; Witkiewicz, H.; Schnitzer, J.E. Systems analysis of endothelial cell plasma membrane proteome of rat lung microvasculature. Proteome Sci. 2011, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liu, P.; Gao, H.; Wang, X.; Yan, M. Long non-coding RNA PGM5-AS1 promotes epithelial-mesenchymal transition, invasion and metastasis of osteosarcoma cells by impairing miR-140-5p-mediated FBN1 inhibition. Mol. Oncol. 2020, 14, 2660–2677. [Google Scholar] [CrossRef]
- Naito, Y.; Lee, Y.U.; Yi, T.; Church, S.N.; Solomon, D.; Humphrey, J.D.; Shin’oka, T.; Breuer, C.K. Beyond burst pressure: Initial evaluation of the natural history of the biaxial mechanical properties of tissue-engineered vascular grafts in the venous circulation using a murine model. Tissue Eng. Part A 2014, 20, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhu, H.; Zheng, M.Q.; Dong, Q.R. LncRNA MEG3 Inhibits the Degradation of the Extracellular Matrix of Chondrocytes in Osteoarthritis via Targeting miR-93/TGFBR2 Axis. Cartilage 2019, 1947603519855759. [Google Scholar] [CrossRef]
- Li, M.K.; Liu, L.X.; Zhang, W.Y.; Zhan, H.L.; Chen, R.P.; Feng, J.L.; Wu, L.F. Long non-coding RNA MEG3 suppresses epithelial-to-mesenchymal transition by inhibiting the PSAT1-dependent GSK-3β/Snail signaling pathway in esophageal squamous cell carcinoma. Oncol. Rep. 2020, 44, 2130–2142. [Google Scholar] [CrossRef]
- Magdaleno, F.; Arriazu, E.; Ruiz de Galarreta, M.; Chen, Y.; Ge, X.; Conde de la Rosa, L.; Nieto, N. Cartilage oligomeric matrix protein participates in the pathogenesis of liver fibrosis. J. Hepatol. 2016, 65, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Nakagawa, S.; Yazdani, S.; van Baarlen, J.; Venkatesh, A.; Koh, A.P.; Song, W.M.; Goossens, N.; Watanabe, H.; Beasley, M.B.; et al. Integrin alpha 11 in the regulation of the myofibroblast phenotype: Implications for fibrotic diseases. Exp. Mol. Med. 2017, 49, e396. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, M.; Montero-Montero, L.; Renart, J.; Martín-Villar, E. Podoplanin in Inflammation and Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.J.; Baek, H.S.; Ye, D.J.; Shin, S.; Kim, D.; Chun, Y.J. CYP1B1 Enhances Cell Proliferation and Metastasis through Induction of EMT and Activation of Wnt/β-Catenin Signaling via Sp1 Upregulation. PLoS ONE 2016, 11, e0151598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Juni, R.; Harakalova, M.; Duncker, D.J.; Asselbergs, F.W.; Koolwijk, P.; Hinsbergh, V.V.; Verhaar, M.C.; Mokry, M.; Cheng, C. Indoxyl Sulfate Stimulates Angiogenesis by Regulating Reactive Oxygen Species Production via CYP1B1. Toxins 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martí-Pàmies, I.; Cañes, L.; Alonso, J.; Rodríguez, C.; Martínez-González, J. The nuclear receptor NOR-1/NR4A3 regulates the multifunctional glycoprotein vitronectin in human vascular smooth muscle cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 4588–4599. [Google Scholar] [CrossRef] [Green Version]
- Abbah, S.A.; Thomas, D.; Browne, S.; O’Brien, T.; Pandit, A.; Zeugolis, D.I. Co-transfection of decorin and interleukin-10 modulates pro-fibrotic extracellular matrix gene expression in human tenocyte culture. Sci. Rep. 2016, 6, 20922. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Yang, J.; Yue, J.; Chen, Y.; Zhou, H.; Fan, D.; Zhang, Q.; Buraschi, S.; Iozzo, R.V.; Bi, X. Decorin deficiency promotes epithelial-mesenchymal transition and colon cancer metastasis. Matrix Biol. J. Int. Soc. Matrix Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- McCarty, K.S., Jr.; Szabo, E.; Flowers, J.L.; Cox, E.B.; Leight, G.S.; Miller, L.; Konrath, J.; Soper, J.T.; Budwit, D.A.; Creasman, W.T.; et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res. 1986, 46, 4244s–4248s. [Google Scholar] [PubMed]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data mining toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinform. (Oxf. Engl.) 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Keely, P.J. The role of focal adhesion kinase in tumor initiation and progression. Cell Adh. Migr. 2009, 3, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Xu, W.W.; Lam, A.K.Y.; Wang, Y.; Hu, H.F.; Guan, X.Y.; Qin, Y.R.; Saremi, N.; Tsao, S.W.; He, Q.Y.; et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget 2017, 8, 38755–38766. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Hui, K.; Wu, S.; Zhang, M.; Que, T.; Gu, Y.; Wang, X.; Wu, K.; Fan, J. MUC15 inhibits cancer metastasis via PI3K/AKT signaling in renal cell carcinoma. Cell Death Dis. 2020, 11, 336. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, Q.F.; Chang, B.Y.; Shen, L.J.; Li, W.; Wu, P.H.; Fan, W.J. TFAP4 Promotes Hepatocellular Carcinoma Invasion and Metastasis via Activating the PI3K/AKT Signaling Pathway. Dis. Markers 2019, 2019, 7129214. [Google Scholar] [CrossRef]
- Oliveira-Barros, E.G.; Branco, L.C.; Da Costa, N.M.; Nicolau-Neto, P.; Palmero, C.; Pontes, B.; Ferreira do Amaral, R.; Alves-Leon, S.V.; Marcondes de Souza, J.; Romão, L.; et al. GLIPR1 and SPARC expression profile reveals a signature associated with prostate Cancer Brain metastasis. Mol. Cell. Endocrinol. 2021, 528, 111230. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Honegger, J.; Schluesener, H.; Schittenhelm, J. Osteonectin expression in surrounding stroma of craniopharyngiomas: Association with recurrence rate and brain infiltration. Int J. Surg. Pathol. 2013, 21, 591–598. [Google Scholar] [CrossRef]
- Bao, J.M.; Dang, Q.; Lin, C.J.; Lo, U.G.; Feldkoren, B.; Dang, A.; Hernandez, E.; Li, F.; Panwar, V.; Lee, C.F.; et al. SPARC is a key mediator of TGF-β-induced renal cancer metastasis. J. Cell. Physiol. 2021, 236, 1926–1938. [Google Scholar] [CrossRef]
- Dong, S.; Zhu, P.; Zhang, S. Expression of collagen type 1 alpha 1 indicates lymph node metastasis and poor outcomes in squamous cell carcinomas of the lung. PeerJ 2020, 8, e10089. [Google Scholar] [CrossRef]
- He, F.; Wang, H.; Li, Y.; Liu, W.; Gao, X.; Chen, D.; Wang, Q.; Shi, G. SRPX2 knockdown inhibits cell proliferation and metastasis and promotes chemosensitivity in esophageal squamous cell carcinoma. Biomed. Pharmacother. 2019, 109, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Lu, L.; Yang, L.; Yin, S.; Wang, Y.; Qi, Z.; Meng, J.; Zang, R.; Yang, G. Fibrillin-1, induced by Aurora-A but inhibited by BRCA2, promotes ovarian cancer metastasis. Oncotarget 2015, 6, 6670–6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Hou, H.; Liu, T.; Su, S.; Xi, X.; Liao, Y.; Xie, R.; Jin, G.; Liu, X.; Zhu, L.; et al. Cartilage Oligomeric Matrix Protein promotes epithelial-mesenchymal transition by interacting with Transgelin in Colorectal Cancer. Theranostics 2020, 10, 8790–8806. [Google Scholar] [CrossRef]
- Primac, I.; Maquoi, E.; Blacher, S.; Heljasvaara, R.; Van Deun, J.; Smeland, H.Y.; Canale, A.; Louis, T.; Stuhr, L.; Sounni, N.E.; et al. Stromal integrin α11 regulates PDGFR-β signaling and promotes breast cancer progression. J. Clin. Investig. 2019, 129, 4609–4628. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, W.; Bi, J.; Wang, J.; Ni, L.; Shi, Q.; Meng, Q. Association of high PDPN expression with pulmonary metastasis of osteosarcoma and patient prognosis. Oncol. Lett. 2019, 18, 6323–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Qu, J.; Ohuchida, K.; Feng, H.; Chong, S.J.F.; Yan, Z.; Piao, Y.; Liu, P.; Sheng, N.; Eguchi, D.; et al. LAMA4 upregulation is associated with high liver metastasis potential and poor survival outcome of Pancreatic Cancer. Theranostics 2020, 10, 10274–10289. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, D.; Liu, Z.; Li, Y.; Zhang, C.; Gong, Y.; Jiang, Y.; Xing, B. THBS1 facilitates colorectal liver metastasis through enhancing epithelial-mesenchymal transition. Clin. Transl. Oncol. 2020, 22, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zhang, D.; Ren, M.; Lu, G.; Zhang, X.; He, S.; Li, Y. THBS4 promotes HCC progression by regulating ITGB1 via FAK/PI3K/AKT pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 10668–10681. [Google Scholar] [CrossRef]
- Liu, W.; Wei, H.; Gao, Z.; Chen, G.; Liu, Y.; Gao, X.; Bai, G.; He, S.; Liu, T.; Xu, W.; et al. COL5A1 may contribute the metastasis of lung adenocarcinoma. Gene 2018, 665, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, B.; Li, D.; Wu, Y.; Wu, X.; Jiao, S.; Xu, C.; Yu, S.; Wang, S.; Yang, J.; et al. Upregulation of IBSP Expression Predicts Poor Prognosis in Patients With Esophageal Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 1117. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shao, T.; Bao, G.; Gao, Z.; Zhang, Y.; Ding, H.; Zhang, W.; Liu, F.; Guo, C. Identification of potential core genes in metastatic renal cell carcinoma using bioinformatics analysis. Am. J. Transl. Res. 2019, 11, 6812–6825. [Google Scholar]
- Bai, X.; Gao, C.; Zhang, L.; Yang, S. Integrin α7 high expression correlates with deteriorative tumor features and worse overall survival, and its knockdown inhibits cell proliferation and invasion but increases apoptosis in breast cancer. J. Clin. Lab. Anal. 2019, 33, e22979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons-Weiler, J.; Patel, S.; Bhattacharya, S. A classification-based machine learning approach for the analysis of genome-wide expression data. Genome Res. 2003, 13, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalman, M.R.; Deeter, A.; Nimishakavi, G.; Duan, Z.H. Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinform. 2012, 13 (Suppl. 2), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.A.; Wishart, D.S. Applications of machine learning in cancer prediction and prognosis. Cancer Inform. 2007, 2, 59–77. [Google Scholar] [CrossRef]
- Xiong, Y.; Ye, M.; Wu, C. Cancer Classification with a Cost-Sensitive Naive Bayes Stacking Ensemble. Comput. Math. Methods Med. 2021, 2021, 5556992. [Google Scholar] [CrossRef]
- Xie, N.N.; Wang, F.F.; Zhou, J.; Liu, C.; Qu, F. Establishment and Analysis of a Combined Diagnostic Model of Polycystic Ovary Syndrome with Random Forest and Artificial Neural Network. Biomed. Res. Int. 2020, 2020, 2613091. [Google Scholar] [CrossRef]
- Chen, J.W.; Dhahbi, J. Lung adenocarcinoma and lung squamous cell carcinoma cancer classification, biomarker identification, and gene expression analysis using overlapping feature selection methods. Sci. Rep. 2021, 11, 13323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Y.; Gong, Z.Y.; Ye, H.D.; Zhao, X.K.; Li, J.Y.; Zhang, X.M.; Li, S.; Zhu, W.; Wang, M.; et al. Distinguishing Rectal Cancer from Colon Cancer Based on the Support Vector Machine Method and RNA-sequencing Data. Curr. Med. Sci. 2021, 41, 368–374. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Features | χ² Value | Fold Change | Function | Reference |
|---|---|---|---|---|---|
| 1 | SPARC | 9.375 | 1.67 | ECM remodeling | [40] |
| 2 | SORD | 9.375 | −1.54 | EMT | [41] |
| 3 | COL1A1 | 8.166 | 1.87 | ECM remodeling, EMT | [42,43] |
| 4 | SMC3 | 8.166 | −1.27 | ECM remodeling | [44] |
| 5 | ARHGAP32 | 8.166 | −1.61 | EMT | [45] |
| 6 | SNRPF | 8.166 | −1.61 | EMT | [46] |
| 7 | SRPX2 | 8.166 | 1.6 | ECM remodeling, angiogenesis | [47,48] |
| 8 | BTG1 | 8.166 | −1.38 | Angiogenesis | [49] |
| 9 | ERMP1 | 7.041 | −1.38 | Angiogenesis | [50] |
| 10 | FBN1 | 7.041 | 1.85 | ECM remodeling, EMT | [51,52] |
| 11 | MEG3 | 7.041 | 2.56 | ECM remodeling, EMT | [53,54] |
| 12 | COMP | 7.041 | 2.63 | ECM remodeling | [55] |
| 13 | ITGA11 | 7.041 | 2.03 | ECM remodeling | [56] |
| 14 | PDPN | 7.041 | 1.71 | ECM remodeling | [57] |
| 15 | CYP1B1 | 7.041 | 1.71 | EMT, angiogenesis | [58,59] |
| 16 | NR4A3 | 7.041 | 1.8 | ECM remodeling | [60] |
| 17 | DCN | 7.041 | 1.73 | ECM remodeling, EMT | [61,62] |
| ECM–Receptor Interaction |
|---|
| LAMA4, HSPG2, THBS1, THBS4, COL1A1, COMP, COL1A2, COL5A1, IBSP, COL6A2, COL5A2, ITGA11, ITGA7 |
| Focal adhesion |
| LAMA4, VEGFC, THBS1, EGFR, THBS4, MYLK, COL1A1, COMP, COL1A2, COL5A1, IBSP, COL6A2, COL5A2, ITGA11, ITGA7 |
| PI3K–Akt signaling pathway |
| LAMA4, VEGFC, THBS1, EGFR, THBS4, COL1A1, COMP, NR4A1, COL1A2, COL5A1, IBSP, COL6A2, COL5A2, ITGA11, ITGA7 |
| Protein digestion and absorption |
| COL1A1, COL18A1, COL1A2, COL5A1, COL6A2, COL5A2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haam, S.; Han, J.-H.; Lee, H.W.; Koh, Y.W. Tumor Nonimmune-Microenvironment-Related Gene Expression Signature Predicts Brain Metastasis in Lung Adenocarcinoma Patients after Surgery: A Machine Learning Approach Using Gene Expression Profiling. Cancers 2021, 13, 4468. https://doi.org/10.3390/cancers13174468
Haam S, Han J-H, Lee HW, Koh YW. Tumor Nonimmune-Microenvironment-Related Gene Expression Signature Predicts Brain Metastasis in Lung Adenocarcinoma Patients after Surgery: A Machine Learning Approach Using Gene Expression Profiling. Cancers. 2021; 13(17):4468. https://doi.org/10.3390/cancers13174468
Chicago/Turabian StyleHaam, Seokjin, Jae-Ho Han, Hyun Woo Lee, and Young Wha Koh. 2021. "Tumor Nonimmune-Microenvironment-Related Gene Expression Signature Predicts Brain Metastasis in Lung Adenocarcinoma Patients after Surgery: A Machine Learning Approach Using Gene Expression Profiling" Cancers 13, no. 17: 4468. https://doi.org/10.3390/cancers13174468