
Generation of Antibody-Drug Conjugate Resistant Models


Abstract
Simple Summary
Abstract
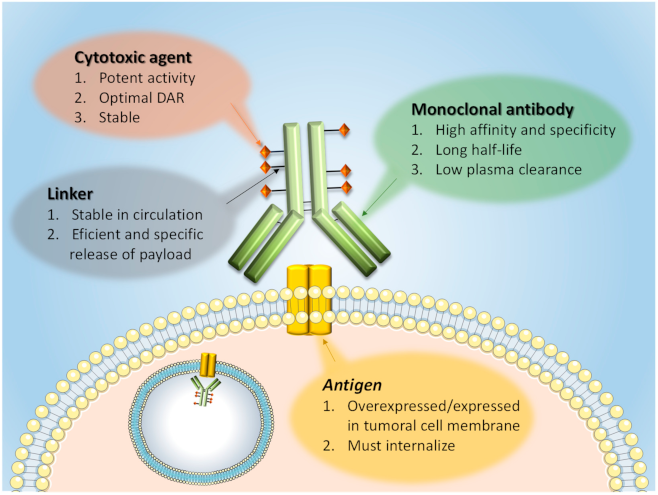
1. Introduction: ADC Structure and Mechanism of Action
2. Approved ADCs
3. Preclinical Models of ADC Resistance
3.1. In Vitro and Ex Vivo Models of Resistance to ADCs
3.1.1. Primary Resistance to ADCs in Established Cell Lines
3.1.2. Secondary Resistance to ADCs in Established Cell Lines
Continuous Treatment
Intermittent Treatment
3.1.3. Resistance to ADCs in Primary Cultures of Human Tumoral Cells
3.2. In Vivo Models
3.2.1. Cell Line Derived Xenografts (CDXs)
Implantation of ADC Resistant Cells
Generation of ADC Resistance In Vivo
3.2.2. Patient-Derived Xenografts (PDXs)
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Kostova, V.; Désos, P.; Starck, J.B.; Kotschy, A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef]
- Nolting, B. Linker technologies for antibody-drug conjugates. Methods Mol. Biol. 2013, 1045, 71–100. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired Resistance to Antibody-Drug Conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Herbst, R.S.; Chen, L. Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol. 2018, 39, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef]
- Ouellette, M.M.; McDaniel, L.D.; Wright, W.E.; Shay, J.W.; Schultz, R.A. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 2000, 9, 403–411. [Google Scholar] [CrossRef]
- Mitra, A.; Mishra, L.; Li, S. Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol. 2013, 31, 347–354. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Sun, Y.L.; Patel, A.; Kumar, P.; Chen, Z.S. Role of ABC transporters in cancer chemotherapy. Chin. J. Cancer 2012, 31, 51–57. [Google Scholar] [CrossRef]
- Le Joncour, V.; Martins, A.; Puhka, M.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H.; Barok, M. A Novel Anti-HER2 Antibody-Drug Conjugate XMT-1522 for HER2-Positive Breast and Gastric Cancers Resistant to Trastuzumab Emtansine. Mol. Cancer Ther. 2019, 18, 1721–1730. [Google Scholar] [CrossRef]
- Li, G.; Guo, J.; Shen, B.Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 1441–1453. [Google Scholar] [CrossRef]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef]
- Takegawa, N.; Nonagase, Y.; Yonesaka, K.; Sakai, K.; Maenishi, O.; Ogitani, Y.; Tamura, T.; Nishio, K.; Nakagawa, K.; Tsurutani, J. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int. J. Cancer 2017, 141, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Jimi, S.; Hara, S.; Takamatsu, Y.; Suzumiya, J.; Tamura, K. Importance of inducible multidrug resistance 1 expression in HL-60 cells resistant to gemtuzumab ozogamicin. Leuk. Lymphoma 2012, 53, 1399–1405. [Google Scholar] [CrossRef]
- Walter, R.B.; Raden, B.W.; Hong, T.C.; Flowers, D.A.; Bernstein, I.D.; Linenberger, M.L. Multidrug resistance protein attenuates gemtuzumab ozogamicin-induced cytotoxicity in acute myeloid leukemia cells. Blood 2003, 102, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Raden, B.W.; Cronk, M.R.; Bernstein, I.D.; Appelbaum, F.R.; Banker, D.E. The peripheral benzodiazepine receptor ligand PK11195 overcomes different resistance mechanisms to sensitize AML cells to gemtuzumab ozogamicin. Blood 2004, 103, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lin, Y.; Song, Z.; Xiao, W.; Chen, L.; Yin, J.; Zhou, Y.; Barta, S.K.; Petrus, M.; Waldmann, T.A.; et al. A20 and RBX1 Regulate Brentuximab Vedotin Sensitivity in Hodgkin Lymphoma Models. Clin. Cancer Res. 2020, 26, 4093–4106. [Google Scholar] [CrossRef]
- Lewis, T.S.; Gordon, K.; Li, F.; Weimann, A.; Bruders, R.; Miyamoto, J.; Chace, D.; Law, C.-L. Abstract 688: Characterization and circumvention of drug resistance mechanisms in SGN-35-resistant HL and ALCL clonal cell lines. Cancer Res. 2014, 74, 688. [Google Scholar] [CrossRef]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.N.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 Overcomes Resistance to Brentuximab Vedotin in Hodgkin Lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef]
- Haag, P.; Viktorsson, K.; Lindberg, M.L.; Kanter, L.; Lewensohn, R.; Stenke, L. Deficient activation of Bak and Bax confers resistance to gemtuzumab ozogamicin-induced apoptotic cell death in AML. Exp. Hematol. 2009, 37, 755–766. [Google Scholar] [CrossRef]
- Amico, D.; Barbui, A.M.; Erba, E.; Rambaldi, A.; Introna, M.; Golay, J. Differential response of human acute myeloid leukemia cells to gemtuzumab ozogamicin in vitro: Role of Chk1 and Chk2 phosphorylation and caspase 3. Blood 2003, 101, 4589–4597. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.B.; Harrington, K.H.; Cordeiro, J.A.; Leung, L.Y.; Putta, S.; Lacayo, N.; Laszlo, G.S.; Gudgeon, C.J.; Hogge, D.E.; Hawtin, R.E.; et al. AKT signaling as a novel factor associated with in vitro resistance of human AML to gemtuzumab ozogamicin. PLoS ONE 2013, 8, e53518. [Google Scholar] [CrossRef]
- Tanner, M.; Kapanen, A.I.; Junttila, T.; Raheem, O.; Grenman, S.; Elo, J.; Elenius, K.; Isola, J. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol. Cancer Ther. 2004, 3, 1585–1592. [Google Scholar]
- Saatci, Ö.; Borgoni, S.; Akbulut, Ö.; Durmuş, S.; Raza, U.; Eyüpoğlu, E.; Alkan, C.; Akyol, A.; Kütük, Ö.; Wiemann, S.; et al. Targeting PLK1 overcomes T-DM1 resistance via CDK1-dependent phosphorylation and inactivation of Bcl-2/xL in HER2-positive breast cancer. Oncogene 2018, 37, 2251–2269. [Google Scholar] [CrossRef]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef]
- Betts, A.; Clark, T.; Jasper, P.; Tolsma, J.; van der Graaf, P.H.; Graziani, E.I.; Rosfjord, E.; Sung, M.; Ma, D.; Barletta, F. Use of translational modeling and simulation for quantitative comparison of PF-06804103, a new generation HER2 ADC, with Trastuzumab-DM1. J. Pharmacokinet. Pharmacodyn. 2020, 47, 513–526. [Google Scholar] [CrossRef]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-low and T-DM1-resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef]
- Barok, M.; Le Joncour, V.; Martins, A.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H. ARX788, a novel anti-HER2 antibody-drug conjugate, shows anti-tumor effects in preclinical models of trastuzumab emtansine-resistant HER2-positive breast cancer and gastric cancer. Cancer Lett. 2020, 473, 156–163. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Shen, Y.; Youssef, L.A.; Mohan, N.; Wu, W.J. T-DM1-resistant cells gain high invasive activity via EGFR and integrin cooperated pathways. MAbs 2018, 10, 1003–1017. [Google Scholar] [CrossRef]
- Barok, M.; Tanner, M.; Köninki, K.; Isola, J. Trastuzumab-DM1 is highly effective in preclinical models of HER2-positive gastric cancer. Cancer Lett. 2011, 306, 171–179. [Google Scholar] [CrossRef]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Dugger, T.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.J.; Hutchinson, K.E.; Rexer, B.N.; Estrada, M.V.; Gonzalez Ericsson, P.I.; Sanders, M.E.; Dugger, T.C.; Formisano, L.; Guerrero-Zotano, A.; Red-Brewer, M.; et al. An ERBB1-3 Neutralizing Antibody Mixture with High Activity Against Drug-Resistant HER2+ Breast Cancers with ERBB Ligand Overexpression. J. Natl. Cancer Inst. 2017, 109, 759–765. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- Ríos-Luci, C.; García-Alonso, S.; Díaz-Rodríguez, E.; Nadal-Serrano, M.; Arribas, J.; Ocaña, A.; Pandiella, A. Resistance to the Antibody-Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer Res. 2017, 77, 4639–4651. [Google Scholar] [CrossRef]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R.; et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol. Cancer Ther. 2018, 17, 243–253. [Google Scholar] [CrossRef]
- Cianfriglia, M.; Mallano, A.; Ascione, A.; Dupuis, M.L. Multidrug transporter proteins and cellular factors involved in free and mAb linked calicheamicin-gamma1 (gentuzumab ozogamicin, GO) resistance and in the selection of GO resistant variants of the HL60 AML cell line. Int. J. Oncol. 2010, 36, 1513–1520. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Wang, Q.; Gao, M.; Fu, L.; Li, Y.; Quan, H.; Lou, L. STAT3 activation confers trastuzumab-emtansine (T-DM1) resistance in HER2-positive breast cancer. Cancer Sci. 2018, 109, 3305–3315. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Xu, Y.; Yang, Y.; Chen, X.; Quan, H.; Lou, L. Aberrant intracellular metabolism of T-DM1 confers T-DM1 resistance in human epidermal growth factor receptor 2-positive gastric cancer cells. Cancer Sci. 2017, 108, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Sauveur, J.; Conilh, L.; Beaumel, S.; Chettab, K.; Jordheim, L.P.; Matera, E.L.; Dumontet, C. Characterization of T-DM1-resistant breast cancer cells. Pharmacol. Res. Perspect. 2020, 8, e00617. [Google Scholar] [CrossRef] [PubMed]
- Sauveur, J.; Matera, E.L.; Chettab, K.; Valet, P.; Guitton, J.; Savina, A.; Dumontet, C. Esophageal cancer cells resistant to T-DM1 display alterations in cell adhesion and the prostaglandin pathway. Oncotarget 2018, 9, 21141–21155. [Google Scholar] [CrossRef]
- Nadal-Serrano, M.; Morancho, B.; Escrivá-de-Romaní, S.; Morales, C.B.; Luque, A.; Escorihuela, M.; Espinosa Bravo, M.; Peg, V.; Dijcks, F.A.; Dokter, W.H.A.; et al. The Second Generation Antibody-Drug Conjugate SYD985 Overcomes Resistances to T-DM1. Cancers 2020, 12, 670. [Google Scholar] [CrossRef] [PubMed]
- Sabbaghi, M.; Gil-Gómez, G.; Guardia, C.; Servitja, S.; Arpí, O.; García-Alonso, S.; Menendez, S.; Arumi-Uria, M.; Serrano, L.; Salido, M.; et al. Defective Cyclin B1 Induction in Trastuzumab-emtansine (T-DM1) Acquired Resistance in HER2-positive Breast Cancer. Clin. Cancer Res. 2017, 23, 7006–7019. [Google Scholar] [CrossRef]
- Linenberger, M.L.; Hong, T.; Flowers, D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Berger, M.S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994. [Google Scholar] [CrossRef]
- Islam, S.S.; Uddin, M.; Noman, A.S.M.; Akter, H.; Dity, N.J.; Basiruzzman, M.; Uddin, F.; Ahsan, J.; Annoor, S.; Alaiya, A.A.; et al. Antibody-drug conjugate T-DM1 treatment for HER2+ breast cancer induces ROR1 and confers resistance through activation of Hippo transcriptional coactivator YAP1. EBioMedicine 2019, 43, 211–224. [Google Scholar] [CrossRef]
- Jung, J.; Seol, H.S.; Chang, S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res. Treat. 2018, 50, 1–10. [Google Scholar] [CrossRef]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Ocaña, A.; García-Alonso, S.; Amir, E.; Pandiella, A. Refining Early Antitumoral Drug Development. Trends Pharmacol. Sci. 2018, 39, 922–925. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Gandullo-Sánchez, L.; Capone, E.; Ocaña, A.; Iacobelli, S.; Sala, G.; Pandiella, A. HER3 targeting with an antibody-drug conjugate bypasses resistance to anti-HER2 therapies. EMBO Mol. Med. 2020, 12, e11498. [Google Scholar] [CrossRef]
- Starling, J.J.; Maciak, R.S.; Hinson, N.A.; Hoskins, J.; Laguzza, B.C.; Gadski, R.A.; Strnad, J.; Rittmann-Grauer, L.; DeHerdt, S.V.; Bumol, T.F.; et al. In vivo selection of human tumor cells resistant to monoclonal antibody-Vinca alkaloid immunoconjugates. Cancer Res. 1990, 50, 7634–7640. [Google Scholar]
- D’Amico, L.; Menzel, U.; Prummer, M.; Müller, P.; Buchi, M.; Kashyap, A.; Haessler, U.; Yermanos, A.; Gébleux, R.; Briendl, M.; et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J. Immunother. Cancer 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M.; et al. A Novel Anti-CD22 Anthracycline-Based Antibody-Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Graziani, E.I.; Sung, M.; Ma, D.; Narayanan, B.; Marquette, K.; Puthenveetil, S.; Tumey, L.N.; Bikker, J.; Casavant, J.; Bennett, E.M.; et al. PF-06804103, A Site-specific Anti-HER2 Antibody-Drug Conjugate for the Treatment of HER2-expressing Breast, Gastric, and Lung Cancers. Mol. Cancer Ther. 2020, 19, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Irie, H.; Kawabata, R.; Fujioka, Y.; Nakagawa, F.; Itadani, H.; Nagase, H.; Ito, K.; Uchida, J.; Ohkubo, S.; Matsuo, K. Acquired resistance to trastuzumab/pertuzumab or to T-DM1 in vivo can be overcome by HER2 kinase inhibition with TAS0728. Cancer Sci. 2020, 111, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Kinneer, K.; Meekin, J.; Tiberghien, A.C.; Tai, Y.T.; Phipps, S.; Kiefer, C.M.; Rebelatto, M.C.; Dimasi, N.; Moriarty, A.; Papadopoulos, K.P.; et al. SLC46A3 as a Potential Predictive Biomarker for Antibody-Drug Conjugates Bearing Noncleavable Linked Maytansinoid and Pyrrolobenzodiazepine Warheads. Clin. Cancer Res. 2018, 24, 6570–6582. [Google Scholar] [CrossRef]
- Li, B.T.; Michelini, F.; Misale, S.; Cocco, E.; Baldino, L.; Cai, Y.; Shifman, S.; Tu, H.Y.; Myers, M.L.; Xu, C.; et al. HER2-Mediated Internalization of Cytotoxic Agents in ERBB2 Amplified or Mutant Lung Cancers. Cancer Discov. 2020, 10, 674–687. [Google Scholar] [CrossRef]
- Rizzo, G.; Bertotti, A.; Leto, S.M.; Vetrano, S. Patient-derived tumor models: A more suitable tool for pre-clinical studies in colorectal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 178. [Google Scholar] [CrossRef]
- Pernik, M.N.; Bird, C.E.; Traylor, J.I.; Shi, D.D.; Richardson, T.E.; McBrayer, S.K.; Abdullah, K.G. Patient-Derived Cancer Organoids for Precision Oncology Treatment. J. Pers. Med. 2021, 11, 423. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ADC (Other Names, Commercial Name) | Target (Isotype Immunoglobulin) | Payload (Mechanism of Action) | Linker | Indication (Year of FDA Approval) | Company |
|---|---|---|---|---|---|
| Gemtuzumab ozogamicin (GO, Mylotarg®) | CD33 (IgG4) | Calicheamicin γ1 (CAL, cytotoxic antibiotic, DNA damage) | Cleavable hydrazone linker | Acute myeloid leukemia (AML) (2000–2010, 2017) | Pfizer (New York, U.S.) |
| Brentuximab vedotin (BV, SGN-35, Adcetris®) | CD30 (cAC10 chimeric IgG1) | Monomethyl auristatin E (MMAE, inhibition of microtubule polymerization) | Protease-cleavable linker, maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl linker | Relapsed/refractory Hodgkin lymphoma and anaplastic large cell lymphoma (2011) | Seattle Genetics/Takeda(Washington, U.S./Tokyo, Japan) |
| Ado-trastuzumab emtansine (T-DM1, Kadcyla®) | HER2 (IgG1) | Mertansine (DM1, inhibition of microtubule polymerization) | Non-cleavable thioether linker | HER2+ metastatic breast cancer (2013) Adjuvant treatment of patients with HER2+ early breast cancer who have residual invasive disease after neoadjuvant taxane and trastuzumab based treatment (2019) | Roche (Basel, Switzerland) |
| Inotuzumab ozogamicin (INO, CMC-544, Besponsa®) | CD22 (IgG4) | Calicheamicin derivative (CAL, cytotoxic antibiotic, DNA damage) | Cleavable hydrazone linker | Acute lymphocytic leukemia (ALL) and chronic lymphocytic leukemia (CLL) (2017) | Pfizer (New York, U.S.) |
| Moxetumomab pasudotox (CAT-8015, Lumoxiti®) | CD22 (Fv portion of the mAb) | PE38, a 38 kDa fragment of Pseudomonas exotoxin A (inhibition of protein synthesis) | Payload fused to the antibody using a cleavable amino acid linker | Relapsed or refractory hairy cell leukemia (HCL) (2018) | AstraZeneca (Cambridge, UK) |
| Trastuzumab deruxtecan (T-DXd, DS-8201a, Enhertu®) | HER2 (IgG1) | Deruxtecan (DXd, topoisomerase I targeting) | Enzymatically cleavable maleimide glycynglycynphenylalanyn-glycyn peptide linker | HER2+ metastatic breast cancer (2019) HER2+ locally advanced or metastatic gastric cancer (2021) | AstraZeneca/Daiichi Sankyo (Cambridge, UK/ Tokyo, Japan) |
| Polatuzumab vedotin-piiq (PV, DCDS4501A, RG7596, Polivy®) | CD79b (IgG1) | Monomethyl auristatin E (MMAE, inhibition of microtubule polymerization) | Protease-cleavable maleimidocaproyl-valine-citrulline-p-aminobenzoyloxycarbonyl linker | Diffuse large B-cell lymphoma (2019) | Roche (Basel, Switzerland) |
| Enfortumab vedotin (EV, ASG-22ME, AGS-22M6E, Padcev®) | Nectin 4 (IgG1) | Monomethyl auristatin E (MMAE, inhibition of microtubule polymerization) | Cleavable maleimidocaproylvaline-citrulline linker | Locally advanced or metastatic urothelial cancer (2019) | Seattle Genetics/Astellas (Washington, U.S./Tokyo, Japan) |
| Sacituzumab govitecan (SG, IMMU-132, Trodelby®) | TROP-2 (IgG1) | SN-38 (topoisomerase I targeting) | Cleavable ydrolysable Ph sensitive CL2A linker | Metastatic Triple Negative Breast Cancer (2020) | Immunomedics (New Jersey, U.S.) |
| Belantamab mafodotin (GSK2857916, Blenrep®) | BCMA, CD269 (IgG1) | Monomethyl auristatin F (MMAF, inhibition of microtubule polymerization) | Non-cleavable maleimidocaproyl linker | Relapsed and refractory multiple myeloma (2020) | GlaxoSmithKline (Brentford, UK) |
| Loncastuximab tesirine (ADCT-402, Zynlonta®) | CD19 | Pyrrolobenzodiazepine (PBD) dimer toxin or SG3199 (DNA damage) | Valine-alanine cleavable, maleimide type linker | Relapsed or refractory large B-cell lymphoma (2021) | ADC Therapeutics S.A. (Lausanne, Switzerland) |
| Continuous Treatment Strategy | |||||
|---|---|---|---|---|---|
| ADC | Cell Line | Time (Months) | Resistance Mechanism | In Vivo Test | Reference Year of Publication |
| GO | HL-60 | 2 | Reduced CD33 expression | Not reported | [52] 2010 |
| GO | HL-60 | 6 | Increased ABCB1/MDR1/P-gp transporter | Not reported | [28] 2012 |
| BV | DEL Karpas-299 L540cy | Not reported | ABCB1/MDR1/P-gp induction and/or loss of CD30 | Not reported | [32] 2014 |
| BV | Karpas-299 | 3 | Reduced CD30 expression | Not reported | [34] 2015 |
| T-DM1 | BT474 NCI-N87 SKOV-3 MDA-MB-361 | 4–12 | Not described | NCI-N87 resistant cells were also resistant in vivo after several passages in mice treated with T-DM1 | [40] 2016 |
| T-DM1 | BT474 | 3 | Altered lysosomal pH and decreased lysosomal proteolytic activity | Resistance in vivo | [50] 2017 |
| T-DM1 | NCI-N87 | 6 | ABCC2 and ABCG2 upregulation | Resistance in vivo | [26] 2017 |
| T-DM1 | NCI-N87 | 18 | Decreased lysosomal V-ATPase and lysine-MCC-DM1 production | Resistance in vivo | [54] 2017 |
| T-DM1 | KPL-4 | 10 | Decreased HER2 and upregulation of ABCB1/MDR1/P-gp | Lack of resistance | [24] 2018 |
| BT-474M1 | Loss of SLC46A3 and PTEN deficiency | Poor growth | |||
| T-DM1 | BT474 SKBR3 | 9 | PLK1 upregulation | Not reported | [39] 2018 |
| T-DM1 | MDA-MB-361 | 6 | Increased baseline aneuploidy and altered intracellular DM1 trafficking | Not reported | [55] 2020 |
| T-DM1 | OE-19 | 6 | Changes in cell adhesion and the prostaglandin pathway | Not reported | [56] 2018 |
| T-DM1 | BT474 | 12 | STAT3 activation | Resistance in vivo | [53] 2018 |
| T-DM1 | JIMT-1 | 5 | Loss of HER2 and increase of EGFR expression | Not reported | [45] 2018 |
| T-DM1 | OE-19 NCI-N87 | 7–9 | Slightly decreased expression of HER2, overexpression of ABCC1, ABCC2, and ABCG2, and changes in the disposal of T-DM1 on the secreted extracellular vesicles | RN-87 is resistant in vivo | [23] 2019 |
| T-DM1 | PDX118 | 1.5–3 | Decreased HER2 levels, impairment of lysosomal function, or increased drug efflux | Not reported. Generation of other models of in vivo resistance (Section 3.2.2) | [57] 2020 |
| Pulsed-Selection Strategy | |||||
| ADC | Cell line | Schedule | Resistance mechanism | In vivo test | Reference Year of publication |
| BV | L428 | Treatment with 50 µg/mL of BV until no proliferation was observed. Then, when cells were recovered in free-BV media, BV was added | Increased ABCB1/MDR1/P-gp | Not reported | [34] 2015 |
| BV | KMH2 | Same approach as before | Upregulation ABCB1/MDR1/P-gp | Resistance in vivo | [33] 2020 |
| Anti-HER2 trastuzumab–maytansinoid ADC (TM-ADC), which is structurally similar to T-DM1 | MDA-MB-361 | Multiple cycles for 1.5 to 3 months of 3 days with approximately IC80 of TM-ADC followed by 4–11 days without drug | Increased ABCC1/MRP1 transporter | Resistance in vivo | [25] 2015 |
| JIMT-1 | Decreased HER2 levels | Not reported | |||
| TM-ADC | HCC1954 BT474 | Five cycles at 10 nM of TM-ADC for 3 days, followed by 4–11 days of recovery. Then, the cells were exposed to six cycles extra of 100 nM. Time taken approximately of 4 months | Decreased HER2 | Not reported | [51] 2016 |
| NCI-N87 | Trafficking defect, caveolae-mediated endocytosis of T-DM1 | Resistance in vivo | |||
| T-DM1 | HCC1954 HCC1419 SKBR3 | Three cycles of 3 days on and 3 days off treatment at 1, 2, and 4 µg/mL each.54 days in total. | Defective cyclin B1 induction by T-DM1 | Not reported | [58] 2017 |
| T-DM1 | UACC893 HCC1954 | 3 days on/10 days off for more than 6 months | HCC1954-TDR expressed very low HER2 levels | HCC1954-TDR has also resistance in vivo | [48] 2017 |
| BV | KMH2 L428 | One week at IC90, one week without treatment over 7 weeks | Upregulation of NF-KBsignature genes mediated increasing ABCB1/MDR1/P-gp expression | Not reported | [31] 2020 |
| T-DM1 | HCC1954 | T-DM1 for 4 days, followed by 7 days off treatment. Total time was 8 months | Not described. Attenuated HER2 expression | HCC1954-TDR has also resistance in vivo | [27] 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandullo-Sánchez, L.; Ocaña, A.; Pandiella, A. Generation of Antibody-Drug Conjugate Resistant Models. Cancers 2021, 13, 4631. https://doi.org/10.3390/cancers13184631
Gandullo-Sánchez L, Ocaña A, Pandiella A. Generation of Antibody-Drug Conjugate Resistant Models. Cancers. 2021; 13(18):4631. https://doi.org/10.3390/cancers13184631
Chicago/Turabian StyleGandullo-Sánchez, Lucía, Alberto Ocaña, and Atanasio Pandiella. 2021. "Generation of Antibody-Drug Conjugate Resistant Models" Cancers 13, no. 18: 4631. https://doi.org/10.3390/cancers13184631
APA StyleGandullo-Sánchez, L., Ocaña, A., & Pandiella, A. (2021). Generation of Antibody-Drug Conjugate Resistant Models. Cancers, 13(18), 4631. https://doi.org/10.3390/cancers13184631