
Vorinostat (SAHA) and Breast Cancer: An Overview

 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epigenetic Basis of Cancer
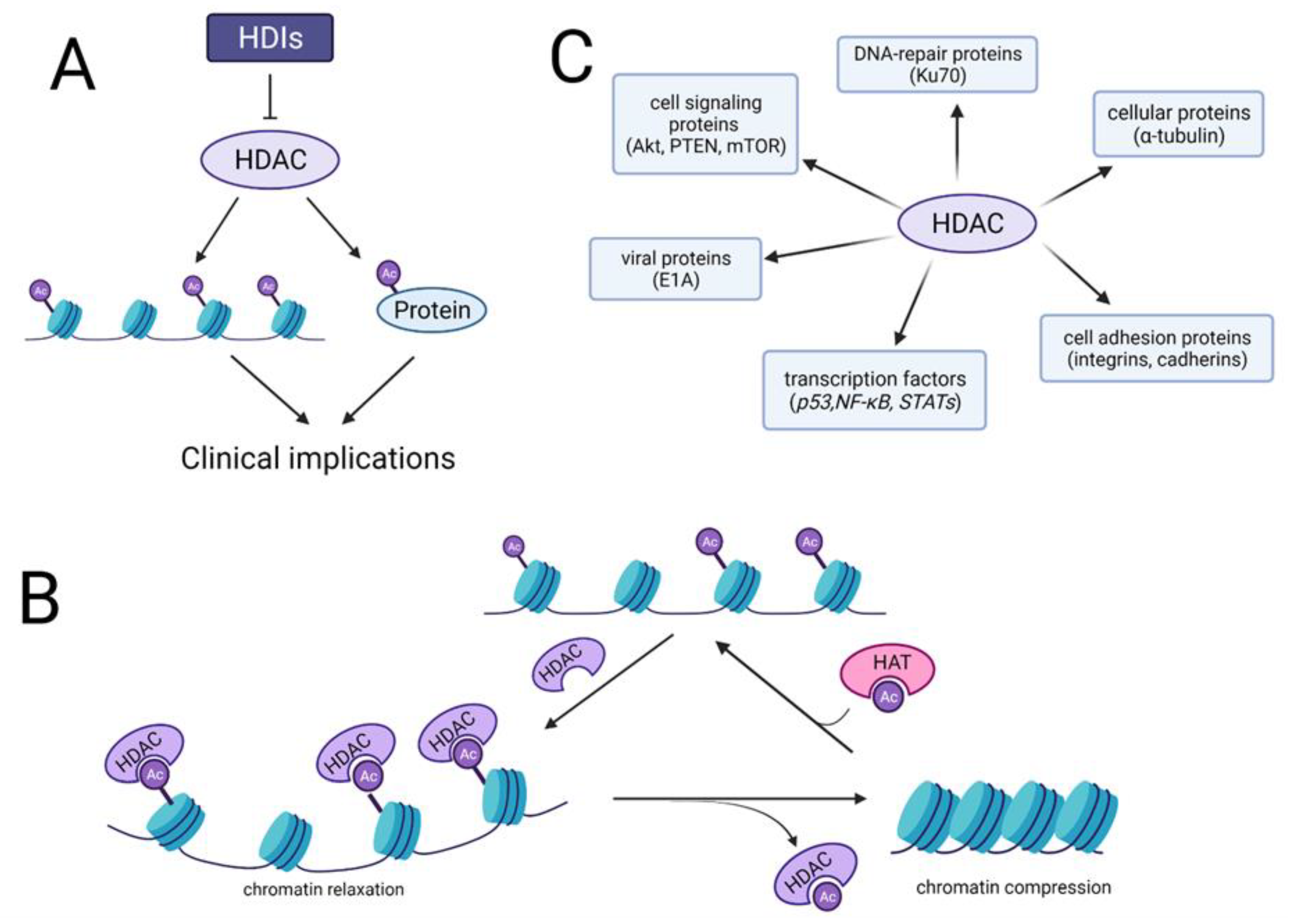
3. Histone Deacetylase Inhibitors (HDIs)
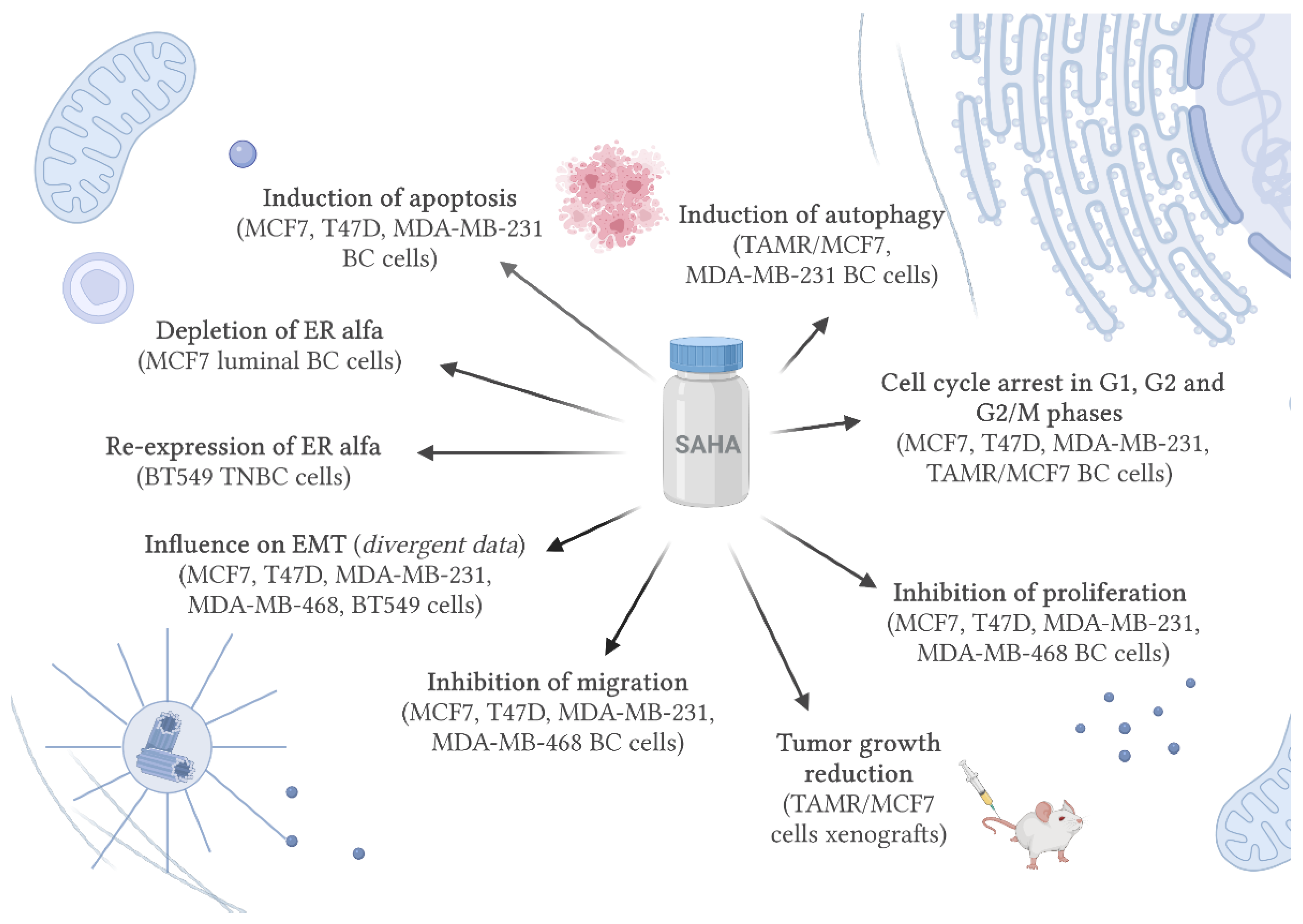
4. SAHA and Breast Cancer
4.1. SAHA Modulates Receptor Status
4.2. SAHA Induces Cell Death and Cell Cycle Arrest
4.3. SAHA Affects Migration and Epithelial-Mesenchymal Transition (EMT)
5. “SAHA et al.” and Breast Cancer
5.1. SAHA and Cisplatin (CDDP)
5.2. SAHA and Taxanes
5.3. SAHA and Trastuzumab
5.4. SAHA and Olaparib
5.5. SAHA and Idasanutlin (RG7388)
5.6. SAHA and Tozasertib (MK-0457)
5.7. SAHA and Epigallocatechin-3-Gallate (EGCG)
5.8. SAHA and Sodium Butyrate (NaB)
5.9. SAHA and Clarithromycin (CAM) + Bortezomib (BZ)
5.10. SAHA and Tumor Necrosis FACTOR Related Apoptosis Inducing Ligand (TRAIL)
5.11. SAHA and Soluble CD137 Receptor
6. SAHA in Clinical Trials
7. Chimeric HDAC-Based Inhibitors
8. SAHA and Drug Carriers
9. Discussion
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Msc, M.L.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2020, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Easton, D.F. Models of genetic susceptibility to breast cancer. Oncogene 2006, 25, 5898–5905. [Google Scholar] [CrossRef] [Green Version]
- Ataollahi, M.; Sharifi, J.; Paknahad, A. Breast cancer and associated factors: A review. J. Med. Life 2015, 8, 6–11. [Google Scholar]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular subtypes and local-regional control of breast cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [Green Version]
- Cheang, M.C.U.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 Index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonacho, T.; Rodrigues, F.; Liberal, J. Immunohistochemistry for diagnosis and prognosis of breast cancer: A review. Biotech. Histochem. 2019, 95, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Szymiczek, A.; Lone, A.; Akbari, M.R. Molecular intrinsic versus clinical subtyping in breast cancer: A comprehensive review. Clin. Genet. 2020, 99, 613–637. [Google Scholar] [CrossRef] [PubMed]
- Yersal, S.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Abubakar, M.; Figueroa, J.; Ali, H.R.; Blows, F.; Lissowska, J.; Caldas, C.; Easton, D.F.; Sherman, M.E.; Garcia-Closas, M.; Dowsett, M.; et al. Combined quantitative measures of ER, PR, HER2, and KI67 provide more prognostic information than categorical combinations in luminal breast cancer. Mod. Pathol. 2019, 32, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Bedard, P.L. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Ortiz, A.; Sanchez-Muñoz, A.; Parrado, M.R.C.; Álvarez, M.; Ribelles, N.; Dominguez, A.R.; Alba, E. Deciphering HER2 Breast cancer disease: Biological and clinical implications. Front. Oncol. 2019, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.; Tan, K.; Rajan, R.S.; Mokhtar, N.; Adzmi, E.M.; Rahman, W.W.A.; Din, T.T.; Balakrishnan, V. Triple negative breast cancer: A review of present and future diagnostic modalities. Medicina 2021, 57, 62. [Google Scholar] [CrossRef] [PubMed]
- Toft, D.J.; Cryns, V.L. Minireview: Basal-like breast cancer: From molecular profiles to targeted therapies. Mol. Endocrinol. 2011, 25, 199–211. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Rosso, K.; Nathanson, S.D. Pathogenesis, prevention, diagnosis and treatment of breast cancer. World J. Clin. Oncol. 2014, 5, 283–298. [Google Scholar] [CrossRef]
- Moo, T.-A.; Sanford, R.; Dang, C.; Morrow, M. Overview of breast cancer therapy. PET Clin. 2018, 13, 339–354. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2019, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhu, J.; Zhou, J.; Wang, X.; Li, D.; Han, W.; Fang, Y.; Pan, H. Epigenetic modifications as regulatory elements of autophagy in cancer. Cancer Lett. 2015, 360, 106–113. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Hałasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kałafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a marker of cancer progression and potential target of anti-cancer therapy. Cells 2019, 8, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, P.; Chen, W.; Li, H.; Li, M.; Li, L. The histone acetylation modifications of breast cancer and their therapeutic implications. Pathol. Oncol. Res. 2018, 24, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Selvi, R.B.; Kundu, T.K. Reversible acetylation of chromatin: Implication in regulation of gene expression, disease and therapeutics. Biotechnol. J. 2009, 4, 375–390. [Google Scholar] [CrossRef]
- Ilisso, C.P.; Cave, D.D.; Mosca, L.; Pagano, M.; Coppola, A.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine regulates apoptosis and autophagy in MCF-7 breast cancer cells through the modulation of specific microRNAs. Cancer Cell Int. 2018, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, L.; Wang, S.; Wang, M.; Zhao, J.; Zhang, Z.; Li, X.; Jia, L.; Han, Y. SIRT3 deacetylates and promotes degradation of P53 in PTEN-defective non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2017, 144, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-J.; Liu, Y.-P.; Dai, H.-Y.; Shiue, Y.-L.; Tsai, C.-J.; Huang, M.-S.; Yeh, Y.-T. Nuclear HDAC6 inhibits invasion by suppressing NF-κB/MMP2 and is inversely correlated with metastasis of non-small cell lung cancer. Oncotarget 2015, 6, 30263–30276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Tao, Y.; Li, M.; Che, T.; Qu, J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 2020, 20, 2923–2940. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, J.; Luo, Y.; Liu, F.; Yuan, Y.; Zhuang, S. A p53/lnc-Ip53 negative feedback loop regulates tumor growth and chemoresistance. Adv. Sci. 2020, 7, 2001364. [Google Scholar] [CrossRef]
- Zhu, X.; Leboeuf, M.; Liu, F.; Grachtchouk, M.; Seykora, J.T.; Morrisey, E.E.; Dlugosz, A.A.; Millar, S.E. HDAC1/2 control proliferation and survival in adult epidermis and pre-basal cell carcinoma via p16 and p53. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef]
- Huang, K.; Liu, Y.; Gu, C.; Liu, D.; Zhao, B. Trichostatin A augments esophageal squamous cell carcinoma cells migration by inducing acetylation of RelA at K310 leading epithelia–mesenchymal transition. Anti-Cancer Drugs 2020, 31, 567–574. [Google Scholar] [CrossRef]
- Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Mani, J.; Vallo, S.; Tsaur, I.; Roos, F.; Chun, F.K.-H.; Blaheta, R.A. HDAC inhibition counteracts metastatic re-activation of prostate cancer cells induced by chronic mTOR suppression. Cells 2018, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Wang, X.; Long, X.; Liu, W.; Xiang, C.; Bao, F.; Wang, D. Sirtuin 7 promotes colorectal carcinoma proliferation and invasion through the inhibition of E-cadherin. Exp. Ther. Med. 2017, 15, 2333–2342. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, F.M.; Vesci, L.; Anastasi, A.M.; Chiapparino, C.; Rosi, A.; Giannini, G.; Taddei, M.; Cini, E.; Faltoni, V.; Petricci, E.; et al. ErbB2 targeted epigenetic modulation: Anti-tumor efficacy of the ADC Trastuzumab-HDACi ST8176AA1. Front. Oncol. 2020, 9, 1534. [Google Scholar] [CrossRef]
- Gong, P.; Wang, Y.; Jing, Y. Apoptosis induction by histone deacetylase inhibitors in cancer cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaiah, M.J.; Kumar, K.R. mTOR-Rictor-EGFR axis in oncogenesis and diagnosis of glioblastoma multiforme. Mol. Biol. Rep. 2021, 48, 4813–4835. [Google Scholar] [CrossRef]
- Fujimoto, T.; Inoue-Mochita, M.; Iraha, S.; Tanihara, H.; Inoue, T. Suberoylanilide hydroxamic acid (SAHA) inhibits transforming growth factor-beta 2-induced increases in aqueous humor outflow resistance. J. Biol. Chem. 2021, 297, 101070. [Google Scholar] [CrossRef]
- Barber, M.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, S.; Wu, N.; Cho, K.-S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, G.M.; Gherardi, S.; Xu, N.; Neiron, Z.; Trahair, T.; Scarlett, C.J.; Chang, D.; Liu, P.Y.; Jankowski, K.; Iraci, N.; et al. Transcriptional upregulation of histone deacetylase 2 promotes Myc-induced oncogenic effects. Oncogene 2010, 29, 5957–5968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; An, J.; Yang, Y.; Wu, D.; Bai, Y.; Cao, W.; Ma, L.; Chen, J.; Yu, Z.; He, Y.; et al. Dual inhibition of AKT-m TOR and AR signaling by targeting HDAC 3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol. Med. 2018, 10, e8478. [Google Scholar] [CrossRef]
- Mao, S.; Ma, J.; Yu, H. Sirtuin-7 knockdown inhibits the growth of endometrial cancer cells by inducing apoptosis via the NF-κB signaling pathway. Oncol. Lett. 2018, 17, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Hai, R.; He, L.; Shu, G.; Yin, G. Characterization of histone deacetylase mechanisms in cancer development. Front. Oncol. 2021, 11, 2839. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Blal, H.A.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Costa-Machado, L.F.; Fernandez-Marcos, P.J. The sirtuin family in cancer. Cell Cycle 2019, 18, 2164–2196. [Google Scholar] [CrossRef]
- Takai, N.; Kawamata, N.; Gui, D.; Said, J.W.; Miyakawa, I.; Koeffler, H.P. Human ovarian carcinoma cells: Histone deacetylase inhibitors exhibit antiproliferative activity and potently induce apoptosis. Cancer 2004, 101, 2760–2770. [Google Scholar] [CrossRef]
- Takai, N.; Desmond, J.C.; Kumagai, T.; Gui, D.; Said, J.W.; Whittaker, S.; Miyakawa, I.; Koeffler, H.P. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin. Cancer Res. 2004, 10, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Zhou, L.-M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Li, H.; Zhu, M.; Hu, P.; Liu, X.; Qing, Y.; Wang, X.; Wang, H.; Wang, Z.; Xu, J.; et al. Involvement of p53 acetylation in growth suppression of cutaneous T-cell lymphomas induced by HDAC inhibition. J. Investig. Dermatol. 2020, 140, 2009–2022. [Google Scholar] [CrossRef]
- McLeod, A.B.; Stice, J.P.; Wardell, S.E.; Alley, H.M.; Chang, C.; McDonnell, D.P. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2017, 78, 266–277. [Google Scholar] [CrossRef]
- Lee, C.M.; Chung, I.Y.; Park, Y.; Yun, K.W.; Jo, H.G.; Park, H.J.; Lee, H.J.; Lee, S.B.; Kim, H.J.; Ko, B.S.; et al. The impact of androgen receptor and histone deacetylase 1 expression on the prognosis of ductal carcinoma in situ. J. Breast Cancer 2020, 23, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Jiang, C.-Y.; Zhang, Y.; Zhao, J.; Han, B.-M.; Xia, S.-J. SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in prostate cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Végan, F.; Humblin, E.; Boidot, R.; Rébé, C.; Derangère, V.; et al. Sirtuin-1 activation controls tumor growth by impeding Th17 differentiation via STAT3 deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockhammer, P.; Okumus, Ö.; Hegedus, L.; Rittler, D.; Ploenes, T.; Herold, T.; Kalbourtzis, S.; Bankfalvi, A.; Sucker, A.; Kimmig, R.; et al. HDAC inhibition induces cell cycle arrest and mesenchymal-epithelial transition in a novel pleural-effusion derived uterine carcinosarcoma cell line. Pathol. Oncol. Res. 2021, 27, 636088. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic acid and breast cancer: State of the art in 2021. Cancers 2021, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Whittaker, S.J.; Demierre, M.-F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.; Cashen, A.F. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Future Oncol. 2015, 11, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of panobinostat for the treatment of multiple myeloma. J. Oncol. 2020, 2020, 7131802. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Sun, W.; Lv, S.; Li, H.; Cui, W.; Wang, L. Enhancing the anticancer efficacy of immunotherapy through combination with histone modification inhibitors. Genes 2018, 9, 633. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, S.; Redon, C.E.; Peer, C.J.; Bryla, C.; Lee, M.-J.; Trepel, J.B.; Tomita, Y.; Rajan, A.; Giaccone, G.; Bonner, W.M.; et al. Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anti-Cancer Drugs 2018, 29, 457–465. [Google Scholar] [CrossRef]
- Gumbarewicz, E.; Tylżanowski, P.; Łuszczki, J.; Kałafut, J.; Czerwonka, A.; Szumiło, J.; Wawruszak, A.; Kupisz, K.; Polberg, K.; Smok-Kalwat, J.; et al. Differential molecular response of larynx cancer cell lines to combined VPA/CDDP treatment. Am. J. Cancer Res. 2021, 11, 2821–2837. [Google Scholar] [PubMed]
- Gumbarewicz, E.; Luszczki, J.; Wawruszak, A.; Dmoszynska-Graniczka, M.; Grabarska, A.; Jarząb, A.; Polberg, K.; Stepulak, A. Isobolographic analysis demonstrates additive effect of cisplatin and HDIs combined treatment augmenting their anti-cancer activity in lung cancer cell lines. Am. J. Cancer Res. 2016, 6, 2831–2845. [Google Scholar] [PubMed]
- Yang, H.; Sun, B.; Xu, K.; He, Y.; Zhang, T.; Hall, S.R.R.; Tan, S.T.; Schmid, R.A.; Peng, R.-W.; Hu, G.; et al. Pharmaco-transcriptomic correlation analysis reveals novel responsive signatures to HDAC inhibitors and identifies Dasatinib as a synergistic interactor in small-cell lung cancer. EBioMedicine 2021, 69, 103457. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Xu, H.; Chen, Z.; Xiong, F.; Zhang, B.; Chen, K.; Jiang, H.; Luo, C.; Zhang, H. 17-AAG synergizes with Belinostat to exhibit a negative effect on the proliferation and invasion of MDA-MB-231 breast cancer cells. Oncol. Rep. 2020, 43, 1928–1944. [Google Scholar] [CrossRef]
- Franco-Molina, M.A.; Santana-Krímskaya, S.E.; Madrigal-de-León, L.M.; Coronado-Cerda, E.E.; Zárate-Triviño, D.G.; Hernández-Martínez, S.P.; García-Coronado, P.L.; Rodríguez-Padilla, C. Evaluation of the cytotoxic and immunogenic potential of temozolamide, panobinostat, and Lophophora williamsii extract against C6 glioma cells. EXCLI J. 2021, 20, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.; Camilo, V.; Lopes, N.; Moreira-Silva, F.; Correia, M.; Henrique, R.; Jerónimo, C. Hydralazine and panobinostat attenuate malignant properties of prostate cancer cell lines. Pharmaceuticals 2021, 14, 670. [Google Scholar] [CrossRef] [PubMed]
- Broussy, S.; Laaroussi, H.; Vidal, M. Biochemical mechanism and biological effects of the inhibition of silent information regulator 1 (SIRT1) by EX-527 (SEN0014196 or selisistat). J. Enzym. Inhib. Med. Chem. 2020, 35, 1124–1136. [Google Scholar] [CrossRef]
- Lu, B.; Zhang, D.; Wang, X.; Lin, D.; Chen, Y.; Xu, X. Targeting SIRT1 to inhibit the proliferation of multiple myeloma cells. Oncol. Lett. 2021, 21, 306. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, S.; Gordon, L.I. Functional characterization of NAD dependent de-acetylases SIRT1 and SIRT2 in B-Cell Chronic Lymphocytic Leukemia (CLL). Cancer Biol. Ther. 2016, 17, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Marshall, G.M.; Liu, P.Y.; Gherardi, S.; Scarlett, C.J.; Bedalov, A.; Xu, N.; Iraci, N.; Valli, E.; Ling, D.; Thomas, W.; et al. SIRT1 Promotes N-myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-myc protein stability. PLoS Genet. 2011, 7, e1002135. [Google Scholar] [CrossRef] [Green Version]
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Nieva, P.L.; Chantar, M.L.M.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S.; et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Lautz, T.B.; Naiditch, J.A.; Clark, S.; Chu, F.; Madonna, M.B. Efficacy of class I and II vs class III histone deacetylase inhibitors in neuroblastoma. J. Pediatr. Surg. 2012, 47, 1267–1271. [Google Scholar] [CrossRef]
- Hałasa, M.; Łuszczki, J.J.; Dmoszyńska-Graniczka, M.; Baran, M.; Okoń, E.; Stepulak, A.; Wawruszak, A. Antagonistic interaction between histone deacetylase inhibitor: Cambinol and Cisplatin—An isobolographic analysis in breast cancer in vitro models. Int. J. Mol. Sci. 2021, 22, 8573. [Google Scholar] [CrossRef]
- Ceballos, M.P.; Angel, A.; Delprato, C.B.; Livore, V.I.; Ferretti, A.C.; Lucci, A.; Comanzo, C.G.; Alvarez, M.D.L.; Quiroga, A.D.; Mottino, A.D.; et al. Sirtuin 1 and 2 inhibitors enhance the inhibitory effect of sorafenib in hepatocellular carcinoma cells. Eur. J. Pharmacol. 2020, 892, 173736. [Google Scholar] [CrossRef]
- Kim, H.-B.; Lee, S.-H.; Um, J.-H.; Kim, M.-J.; Hyun, S.-K.; Gong, E.-J.; Oh, W.K.; Kang, C.-D.; Kim, S.-H. Sensitization of chemo-resistant human chronic myeloid leukemia stem-like cells to Hsp90 inhibitor by SIRT1 inhibition. Int. J. Biol. Sci. 2015, 11, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Wawruszak, A.; Luszczki, J.; Halasa, M.; Okon, E.; Landor, S.; Sahlgren, C.; Rivero-Muller, A.; Stepulak, A. Sensitization of MCF7 cells with high Notch1 activity by cisplatin and histone deacetylase inhibitors applied together. Int. J. Mol. Sci. 2021, 22, 5184. [Google Scholar] [CrossRef]
- Bubna, A.K. Vorinostat—An overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.; Chatterji, B. HDAC Inhibitors as novel anti-cancer therapeutics. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Suberoylanilide hydroxamic acid induces thioredoxin1-mediated apoptosis in lung cancer cells via up-regulation of miR-129-5p. Mol. Carcinog. 2017, 56, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ida, H.; Ito, K.; Zhang, H.; Ito, Y. Contribution of reactivated RUNX3 to inhibition of gastric cancer cell growth following suberoylanilide hydroxamic acid (vorinostat) treatment. Biochem. Pharmacol. 2007, 73, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, M.B.; Kuschman, H.P.; Bovee, R.; Hickok, J.R.; Thomas, D.D. Vorinostat exhibits anticancer effects in triple-negative breast cancer cells by preventing nitric oxide-driven histone deacetylation. Biol. Chem. 2021, 402, 501–512. [Google Scholar] [CrossRef]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.; Raslan, S.; Tombuloglu, H.; Shamseddin, A.; Cevik, E.; Said, O.A.; Madyan, E.F.; Senel, M.; Bozkurt, A.; Rehman, S.; et al. Vorinostat-loaded titanium oxide nanoparticles (anatase) induce G2/M cell cycle arrest in breast cancer cells via PALB2 upregulation. 3 Biotech 2020, 10, 407. [Google Scholar] [CrossRef]
- Salvo, E.M.; Ramirez, A.O.; Cueto, J.; Law, E.H.; Situ, A.; Cameron, C.; Samjoo, I.A. Risk of recurrence among patients with HR-positive, HER2-negative, early breast cancer receiving adjuvant endocrine therapy: A systematic review and meta-analysis. Breast 2021, 57, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Chainitikun, S.; Fernandez, J.R.E.; Long, J.P.; Iwase, T.; Kida, K.; Wang, X.; Saleem, S.; Lim, B.; Valero, V.; Ueno, N.T. Pathological complete response of adding targeted therapy to neoadjuvant chemotherapy for inflammatory breast cancer: A systematic review. PLoS ONE 2021, 16, e0250057. [Google Scholar] [CrossRef]
- NouriEmamzaden, F.; Word, B.; Cotton, E.; Hawkins, A.; Littlejohn, K.; Moore, R.; Miranda-Carbon, G.; Orish, C.; Lyn-Cook, B. Modulation of estrogen α and progesterone receptors in triple negative breast cancer cell lines: The effects of vorinostat and indole-3-carbinol in vitro. Anticancer Res. 2020, 40, 3669–3683. [Google Scholar] [CrossRef] [PubMed]
- Rydén, L.; Arnlind, M.H.; Vitols, S.; Höistad, M.; Ahlgren, J. Aromatase inhibitors alone or sequentially combined with tamoxifen in postmenopausal early breast cancer compared with tamoxifen or placebo—Meta-analyses on efficacy and adverse events based on randomized clinical trials. Breast 2016, 26, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Spring, L.; Gupta, A.; Reynolds, K.L.; Gadd, M.A.; Ellisen, L.W.; Isakoff, S.J.; Moy, B.; Bardia, A. Neoadjuvant Endocrine Therapy for Estrogen Receptor–Positive Breast Cancer. JAMA Oncol. 2016, 2, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Fowler, A.M.; Alarid, E.T. Amping up estrogen receptors in breast cancer. Breast Cancer Res. 2007, 9, 305. [Google Scholar] [CrossRef]
- Yi, X.; Wei, W.; Wang, S.-Y.; Du, Z.-Y.; Xu, Y.-J.; Yu, X.-D. Histone deacetylase inhibitor SAHA induces ERα degradation in breast cancer MCF-7 cells by CHIP-mediated ubiquitin pathway and inhibits survival signaling. Biochem. Pharmacol. 2008, 75, 1697–1705. [Google Scholar] [CrossRef]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of interactions between cisplatin and two histone deacetylase inhibitors in MCF7, T47D and MDA-MB-231 human breast cancer cell lines—An isobolographic analysis. PLoS ONE 2015, 10, e0143013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Won, A.J.; Lee, J.; Jung, J.H.; Yoon, S.; Lee, B.M.; Kim, H.S. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int. J. Med. Sci. 2012, 9, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Luo, Z.; Yu, P.-J.; Xie, H.; He, Y.-W. Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol. Chem. 2016, 397, 75–83. [Google Scholar] [CrossRef]
- Wawruszak, A.; Gumbarewicz, E.; Okon, E.; Jeleniewicz, W.; Czapinski, J.; Halasa, M.; Okla, K.; Smok-Kalwat, J.; Bocian, A.; Rivero-Muller, A.; et al. Histone deacetylase inhibitors reinforce the phenotypical markers of breast epithelial or mesenchymal cancer cells but inhibit their migratory properties. Cancer Manag. Res. 2019, 11, 8345–8358. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Han, H.; Zou, D.; Zhou, J.; Zhou, W. Suberoylanilide hydroxamic acid-induced specific epigenetic regulation controls Leptin-induced proliferation of breast cancer cell lines. Oncotarget 2016, 8, 3364–3379. [Google Scholar] [CrossRef] [Green Version]
- Vansteenkiste, J.; Van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.L.; Randolph, S.S.; Schöffski, P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Investig. New Drugs 2008, 26, 483–488. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wen, S.; Zhong, Z.; Weng, S.; Jiang, Q.; Mi, H.; Liu, F. The synergistic effects of 5-aminosalicylic acid and vorinostat in the treatment of ulcerative colitis. Front. Pharmacol. 2021, 12, 625543. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Luszczki, J.; Kalafut, J.; Okla, K.; Halasa, M.; Rivero-Muller, A.; Stepulak, A. Additive pharmacological interaction between cisplatin (CDDP) and histone deacetylase inhibitors (HDIs) in MDA-MB-231 triple negative breast cancer (TNBC) cells with altered notch1 activity—An isobolographic analysis. Int. J. Mol. Sci. 2019, 20, 3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garutti, M.; Pelizzari, G.; Bartoletti, M.; Malfatti, M.C.; Gerratana, L.; Tell, G.; Puglisi, F. Platinum salts in patients with breast cancer: A focus on predictive factors. Int. J. Mol. Sci. 2019, 20, 3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caparica, R.; Lambertini, M.; de Azambuja, E. How I treat metastatic triple-negative breast cancer. ESMO Open 2019, 4, e000504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramelo, O.; Silva, C.; Caramelo, F.; Frutuoso, C.; Santos, T.A. The effect of neoadjuvant platinum-based chemotherapy in BRCA mutated triple negative breast cancers -systematic review and meta-analysis. Hered. Cancer Clin. Pract. 2019, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Lynce, F.; Nunes, R. Role of platinums in triple-negative breast cancer. Curr. Oncol. Rep. 2021, 23, 50. [Google Scholar] [CrossRef]
- Shi, Y.-K.; Li, Z.-H.; Han, X.-Q.; Yi, J.-H.; Wang, Z.-H.; Hou, J.-L.; Feng, C.-R.; Fang, Q.-H.; Wang, H.-H.; Zhang, P.-F.; et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances taxol-induced cell death in breast cancer. Cancer Chemother. Pharmacol. 2010, 66, 1131–1140. [Google Scholar] [CrossRef]
- Chang, H.; Jeung, H.-C.; Jung, J.J.; Kim, T.S.; Rha, S.Y.; Chung, H.C. Identification of genes associated with chemosensitivity to SAHA/taxane combination treatment in taxane-resistant breast cancer cells. Breast Cancer Res. Treat. 2010, 125, 55–63. [Google Scholar] [CrossRef]
- Laengle, J.; Kabiljo, J.; Hunter, L.; Homola, J.; Prodinger, S.; Egger, G.; Bergmann, M. Histone deacetylase inhibitors valproic acid and vorinostat enhance trastuzumab-mediated antibody-dependent cell-mediated phagocytosis. J. Immunother. Cancer 2019, 8, e000195. [Google Scholar] [CrossRef] [Green Version]
- Marijon, H.; Lee, D.H.; Ding, L.-W.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Hembruff, S.L.; Rao, R.; Sharma, P.; Balusu, R.; Venkannagari, S.; Smith, J.E.; Peth, K.; Peiper, S.C.; Bhalla, K.N. Co-treatment with vorinostat synergistically enhances activity of Aurora kinase inhibitor against human breast cancer cells. Breast Cancer Res. Treat. 2012, 135, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Steed, K.L.; Jordan, H.R.; Tollefsbol, T.O. SAHA and EGCG promote apoptosis in triple-negative breast cancer cells, possibly through the modulation of cIAP2. Anticancer Res. 2019, 40, 9–26. [Google Scholar] [CrossRef]
- Wang, Z.-T.; Chen, Z.-J.; Jiang, G.-M.; Wu, Y.-M.; Liu, T.; Yi, Y.-M.; Zeng, J.; Du, J.; Wang, H.-S. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell. Signal. 2016, 28, 506–515. [Google Scholar] [CrossRef]
- Komatsu, S.; Moriya, S.; Che, X.-F.; Yokoyama, T.; Kohno, N.; Miyazawa, K. Combined treatment with SAHA, bortezomib, and clarithromycin for concomitant targeting of aggresome formation and intracellular proteolytic pathways enhances ER stress-mediated cell death in breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 437, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Davis, R.; Singh, K.P.; Kurzrock, R.; Ross, D.D.; Srivastava, R.K. Suberoylanilide hydroxamic acid (Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells orthotopically implanted in BALB/c nude mice. Mol. Cancer Ther. 2009, 8, 1596–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellarosa, D.; Bressan, A.; Bigioni, M.; Parlani, M.; Maggi, C.A.; Binaschi, M. SAHA/Vorinostat induces the expression of the CD137 receptor/ligand system and enhances apoptosis mediated by soluble CD137 receptor in a human breast cancer cell line. Int. J. Oncol. 2012, 41, 1486–1494. [Google Scholar] [CrossRef]
- Nabholtz, J.-M.; Gligorov, J. The role of taxanes in the treatment of breast cancer. Expert Opin. Pharmacother. 2005, 6, 1073–1094. [Google Scholar] [CrossRef]
- Škubník, J.; Pavlíčková, V.; Ruml, T.; Rimpelová, S. Current perspectives on taxanes: Focus on their bioactivity, delivery and combination therapy. Plants 2021, 10, 569. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Mao, J.-W.; Tan, X.-L. Research progress on the source, production, and anti-cancer mechanisms of paclitaxel. Chin. J. Nat. Med. 2020, 18, 890–897. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Trastuzumab Emtansine: A review of its adjuvant use in residual invasive HER2-positive early breast cancer. Drugs 2020, 80, 1723–1730. [Google Scholar] [CrossRef]
- Basho, R.K.; McArthur, H.L. Optimizing (neo)adjuvant treatment of HER2-positive breast cancer. Ther. Adv. Med. Oncol. 2018, 10, 1758835918775697. [Google Scholar] [CrossRef]
- Al-Awadhi, A.; Murray, J.L.; Ibrahim, N.K. Developing anti-HER2 vaccines: Breast cancer experience. Int. J. Cancer 2018, 143, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An overview of PARP inhibitors for the treatment of breast cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- Griguolo, G.; Dieci, M.V.; Miglietta, F.; Guarneri, V.; Conte, P. Olaparib for advanced breast cancer. Future Oncol. 2020, 16, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). OncoTargets Ther. 2019, 12, 2903–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smiley, S.B.; Yun, Y.; Ayyagari, P.; Shannon, H.E.; Pollok, K.E.; Vannier, M.W.; Das, S.K.; Veronesi, M.C. Development of CD133 targeting multi-drug polymer micellar nanoparticles for glioblastoma—In vitro evaluation in glioblastoma stem cells. Pharm. Res. 2021, 38, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, U.; Venkatesan, T.; Radhakrishnan, V.; Samuel, S.; Rasappan, P.; Rathinavelu, A. Cell cycle arrest and cytotoxic effects of SAHA and RG7388 mediated through p21WAF1/CIP1 and p27KIP1 in cancer cells. Medicina 2019, 55, 30. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, U.; Venkatesan, T.; Radhakrishnan, V.; Samuel, S.; Rathinavelu, A. Differential mechanisms of cell death induced by HDAC Inhibitor SAHA and MDM2 inhibitor RG7388 in MCF-7 cells. Cells 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Uniyal, A.; Shantanu, P.A.; Vaidya, S.; Belinskaia, D.A.; Shestakova, N.N.; Kumar, R.; Singh, S.; Tiwari, V. Tozasertib attenuates neuropathic pain by interfering with aurora kinase and KIF11 mediated nociception. ACS Chem. Neurosci. 2021, 12, 1948–1960. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Ji, K.; Sun, Y.-T.; Hou, Y.-B.; Chen, J.-J. Aurora kinase inhibitor tozasertib suppresses mast cell activation in vitro and in vivo. Br. J. Pharmacol. 2020, 177, 2848–2859. [Google Scholar] [CrossRef]
- Giles, F.J.; Cortes, J.; Jones, D.; Bergstrom, D.; Kantarjian, H.; Freedman, S.J. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood 2006, 109, 500–502. [Google Scholar] [CrossRef] [Green Version]
- Bebbington, D.; Binch, H.; Charrier, J.-D.; Everitt, S.; Fraysse, D.; Golec, J.; Kay, D.; Knegtel, R.; Mak, C.; Mazzei, F.; et al. The discovery of the potent aurora inhibitor MK-0457 (VX-680). Bioorganic Med. Chem. Lett. 2009, 19, 3586–3592. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A kinase: A potent oncogene and target for cancer therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Martel, F. The role of EGCG in breast cancer prevention and therapy. Mini-Rev. Med. Chem. 2021, 21, 883–898. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Wang, K.-L.; Chen, H.-Y.; Chiang, Y.-F.; Hsia, S.-M. Protective Effects of Epigallocatechin Gallate (EGCG) on endometrial, breast, and ovarian cancers. Biomolecules 2020, 10, 1481. [Google Scholar] [CrossRef] [PubMed]
- Semaan, J.; El-Hakim, S.; Ibrahim, J.-N.; Safi, R.; Elnar, A.A.; El Boustany, C. Comparative effect of sodium butyrate and sodium propionate on proliferation, cell cycle and apoptosis in human breast cancer cells MCF-7. Breast Cancer 2020, 27, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Salimi, V.; Shahsavari, Z.; Safizadeh, B.; Hosseini, A.; Khademian, N.; Tavakoli-Yaraki, M. Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 2017, 16, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashir, N.Z.; Sharma, P. Clarithromycin as an adjunct to periodontal therapy: A systematic review and meta-analysis. Int. J. Dent. Hyg. 2021. [Google Scholar] [CrossRef]
- Mahashur, A. Management of lower respiratory tract infection in outpatient settings: Focus on clarithromycin. Lung India 2018, 35, 143–149. [Google Scholar] [CrossRef]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical pharmacokinetics and pharmacodynamics of bortezomib. Clin. Pharmacokinet. 2018, 58, 157–168. [Google Scholar] [CrossRef]
- Bladé, J.; Cibeira, M.T.; Rosiñol, L. Bortezomib: A valuable new antineoplastic strategy in multiple myeloma. Acta Oncol. 2005, 44, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Olaparib in Combination with Vorinostat in Patients with Relapsed/Refractory and/or Metastatic Breast. Available online: https://clinicaltrials.gov/ct2/show/NCT03742245?term=vorinostat&cond=Breast+Cancer&draw=2&rank=1 (accessed on 13 August 2021).
- Carboplatin and Nab-Paclitaxel with or without Vorinostat in Treating Women with Newly Diagnosed Operable Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00616967?term=vorinostat&cond=Breast+Cancer&draw=2&rank=6 (accessed on 13 August 2021).
- Pembrolizumab and Tamoxifen with or without Vorinostat for the Treatment of Estrogen Receptor Positive Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04190056?term=vorinostat&cond=Breast+Cancer&draw=3&rank=12 (accessed on 13 August 2021).
- Trial for Locally Advanced Breast Cancer Using Vorinostat Plus Chemotherapy. Available online: https://clinicaltrials.gov/ct2/show/NCT00574587?term=vorinostat&cond=Breast+Cancer&draw=4&rank=2 (accessed on 13 August 2021).
- HDAC Inhibitor Vorinostat (SAHA) with Capecitabine (Xeloda) Using a New Weekly Dose Regimen for Advanced Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00719875?term=vorinostat&cond=Breast+Cancer&draw=2&rank=4 (accessed on 13 August 2021).
- Search of: Vorinostat | Breast Cancer. Available online: https://clinicaltrials.gov/ct2/results?cond=Breast+Cancer&term=vorinostat&cntry=&state=&city=&dist= (accessed on 13 August 2021).
- Phase I–II Study of Vorinostat, Paclitaxel, and Bevacizumab in Metastatic Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00368875?term=vorinostat&cond=Breast+Cancer&draw=3&rank=14 (accessed on 13 August 2021).
- Vorinostat in Treating Patients with Stage IV Breast Cancer Receiving Hormone Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT01720602?term=vorinostat&cond=Breast+Cancer&draw=5&rank=15 (accessed on 13 August 2021).
- Vorinostat in Treating Patients with Stage IV Breast Cancer Receiving Aromatase Inhibitor Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT01153672?term=vorinostat&cond=Breast+Cancer&draw=5&rank=17 (accessed on 13 August 2021).
- Vorinostat in Treating Women Who Are Undergoing Surgery for Newly Diagnosed Stage I–III Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00262834?term=vorinostat&cond=Breast+Cancer&draw=5&rank=13 (accessed on 13 August 2021).
- Vorinostat and Trastuzumab in Treating Patients with Metastatic or Locally Recurrent Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00258349?term=vorinostat&cond=Breast+Cancer&draw=5&rank=16 (accessed on 13 August 2021).
- Vorinostat in Treating Women with Ductal Carcinoma in Situ of the Breast. Available online: https://clinicaltrials.gov/ct2/show/NCT00788112?term=vorinostat&cond=Breast+Cancer&draw=5&rank=19 (accessed on 13 August 2021).
- Phase II Trial of SAHA & Tamoxifen for Patients with Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00365599?term=vorinostat&cond=Breast+Cancer&draw=7&rank=5 (accessed on 13 August 2021).
- GCC 0845: Vorinostat and Lapatinib in Advanced Solid Tumors and Advanced Breast Cancer to Evaluate Response and Biomarkers. Available online: https://clinicaltrials.gov/ct2/show/NCT01118975?term=vorinostat&cond=Breast+Cancer&draw=8&rank=11 (accessed on 13 August 2021).
- A Study of Vorinostat and Tamoxifen in Newly Diagnosed Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01194427?term=vorinostat&cond=Breast+Cancer&draw=11&rank=9 (accessed on 13 August 2021).
- Suberoylanilide Hydroxamic Acid in Treating Patients with Progressive Stage IV Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00132002?term=vorinostat&cond=Breast+Cancer&draw=15&rank=10 (accessed on 13 August 2021).
- Reversing Therapy Resistance with Epigenetic-Immune Modification. Available online: https://clinicaltrials.gov/ct2/show/NCT02395627?term=vorinostat&cond=Breast+Cancer&draw=16&rank=18 (accessed on 13 August 2021).
- A Clinical Trial of Oral Suberoylanilide Hydroxamic Acid (SAHA) in Patients with Relapsed or Refractory Breast, Colorectal and Non-Small Cell Lung Cancer (0683-011) (TERMINATED). Available online: https://clinicaltrials.gov/ct2/show/NCT00126451?term=vorinostat&cond=Breast+Cancer&draw=16&rank=20 (accessed on 13 August 2021).
- Vorinostat Before Surgery in Treating Patients with Triple-Negative Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01695057?term=vorinostat&cond=Breast+Cancer&draw=17&rank=8 (accessed on 13 August 2021).
- Clinical Trial of SAHA in Patients with Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00416130?term=vorinostat&cond=Breast+Cancer&draw=17&rank=3 (accessed on 13 August 2021).
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.; Doroshow, J.H.; Gandara, D.R.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California cancer consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [Green Version]
- Stearns, V.; Jacobs, L.K.; Fackler, M.; Tsangaris, T.N.; Rudek, M.A.; Higgins, M.; Lange, J.; Cheng, Z.; Slater, S.A.; Jeter, S.C.; et al. Biomarker modulation following short-term vorinostat in women with newly diagnosed primary breast cancer. Clin. Cancer Res. 2013, 19, 4008–4016. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.-H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, B.; Fiskus, W.; Cohen, B.; Pellegrino, C.; Hershman, D.L.; Chuang, E.; Luu, T.; Somlo, G.; Goetz, M.; Swaby, R.; et al. Phase I–II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: Evidence for vorinostat-induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res. Treat. 2011, 132, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Ren, Y.; Mohapatra, A.; Bali, P.; Mandawat, A.; Rao, R.; Herger, B.; Yang, Y.; Atadja, P.; Wu, J.; et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-α levels and transcriptional activity: A result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 2007, 13, 4882–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelucci, A.; Mari, M.; Millimaggi, D.; Giusti, I.; Carta, G.; Bologna, M.; Dolo, V. Suberoylanilide hydroxamic acid partly reverses resistance to paclitaxel in human ovarian cancer cell lines. Gynecol. Oncol. 2010, 119, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Fackler, M.J.; Zhang, Z.; Zhou, X.C.; Goetz, M.P.; Boughey, J.C.; Walsh, B.; Carpenter, J.T.; Storniolo, A.M.; Watkins, S.P.; et al. Tumor and serum DNA methylation in women receiving preoperative chemotherapy with or without vorinostat in TBCRC008. Breast Cancer Res. Treat. 2017, 167, 107–116. [Google Scholar] [CrossRef]
- Fackler, M.J. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res. 2004, 64, 4442–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fackler, M.J.; Bujanda, Z.L.; Umbricht, C.; Teo, W.W.; Cho, S.; Zhang, Z.; Visvanathan, K.; Jeter, S.; Argani, P.; Wang, C.; et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014, 74, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Fackler, M.J.; Umbricht, C.B.; Williams, D.; Argani, P.; Cruz, L.-A.; Merino, V.F.; Teo, W.W.; Zhang, Z.; Huang, P.; Visvananthan, K.; et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011, 71, 6195–6207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Atadja, P.; Davidson, N.E. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol. Ther. 2007, 6, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabnis, G.J.; Goloubeva, O.G.; Kazi, A.A.; Shah, P.; Brodie, A.H. HDAC inhibitor entinostat restores responsiveness of letrozole-resistant MCF-7Ca xenografts to aromatase inhibitors through modulation of Her-2. Mol. Cancer Ther. 2013, 12, 2804–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Peterson, L.M.; Kurland, B.F.; Yan, F.; Jiresova, A.N.; Gadi, V.K.; Specht, J.M.; Gralow, J.R.; Schubert, E.K.; Link, J.M.; Krohn, K.A.; et al. 18F-Fluoroestradiol PET imaging in a phase II trial of vorinostat to restore endocrine sensitivity in ER+/HER2—Metastatic breast cancer. J. Nucl. Med. 2020, 62, 184–190. [Google Scholar] [CrossRef]
- Koleva-Kolarova, R.G.; Greuter, M.; Van Kruchten, M.; Vermeulen, K.M.; Feenstra, T.; Buskens, E.; Glaudemans, A.; de Vries, E.; De Vries, E.G.E.; Hospers, G.; et al. The value of PET/CT with FES or FDG tracers in metastatic breast cancer: A computer simulation study in ER-positive patients. Br. J. Cancer 2015, 112, 1617–1625. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.M.; Mankoff, D.A.; Lawton, T.; Yagle, K.; Schubert, E.K.; Stekhova, S.; Gown, A.; Link, J.M.; Tewson, T.; Krohn, K.A. Quantitative imaging of estrogen receptor expression in breast cancer with PET and 18F-fluoroestradiol. J. Nucl. Med. 2008, 49, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.T.; Claret, F.X.; Vu, T.T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A Functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Fessler, S.P.; Wotkowicz, M.T.; Mahanta, S.K.; Bamdad, C. MUC1* is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast Cancer Res. Treat. 2009, 118, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Zhao, F.; Wang, M.; Swaby, R.F.; Sparano, J.A.; Meropol, N.J.; Bhalla, K.N.; Pellegrino, C.M.; Alpaugh, R.K.; Falkson, C.I.; et al. A Phase I/II study of suberoylanilide hydroxamic acid (SAHA) in combination with trastuzumab (Herceptin) in patients with advanced metastatic and/or local chest wall recurrent HER2-amplified breast cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1104). Breast Cancer Res. Treat. 2017, 165, 375–382. [Google Scholar] [CrossRef]
- Deming, D.A.; Ninan, J.; Bailey, H.H.; Kolesar, J.M.; Eickhoff, J.; Reid, J.M.; Ames, M.M.; McGovern, R.M.; Alberti, D.; Marnocha, R.; et al. A Phase I study of intermittently dosed vorinostat in combination with bortezomib in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 323–329. [Google Scholar] [CrossRef]
- Hesham, H.M.; Lasheen, D.; Abouzid, K.A. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef] [PubMed]
- Biersack, B.; Polat, S.; Höpfner, M. Anticancer properties of chimeric HDAC and kinase inhibitors. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 Inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Lai, C.-J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Pursell, N.W.; Samson, M.E.S.; Atoyan, R.; Ma, A.W.; Selmi, A.; Xu, W.; Cai, X.; Voi, M.; Savagner, P.; et al. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol. Cancer Ther. 2013, 12, 925–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Lorusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.A.; Chambers, G.; Ma, A.; et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Sun, J.; Xu, J.; Luo, Z.; Diao, D.; Zhang, Z.; Oberly, P.J.; Minnigh, M.B.; Xie, W.; Poloyac, S.M.; et al. Sensitizing triple negative breast cancer to tamoxifen chemotherapy via a redox-responsive vorinostat-containing polymeric prodrug nanocarrier. Theranostics 2020, 10, 2463–2478. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, K.; Zhu, Q.; Shen, Q.; Zhang, Q.; Yu, J.; Chen, Y.; Lu, W. Synthesis and biological evaluation of paclitaxel and vorinostat co-prodrugs for overcoming drug resistance in cancer therapy in vitro. Bioorganic Med. Chem. 2019, 27, 1405–1413. [Google Scholar] [CrossRef]
- Bhushan, A.; Gonsalves, A.; Menon, J. Current state of breast cancer diagnosis, treatment, and theranostics. Pharmaceutics 2021, 13, 723. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.-J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized phase III trial of endocrine therapy plus entinostat or placebo in hormone receptor–positive advanced breast cancer. A trial of the ECOG-ACRIN cancer research group. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Zhang, F.; Shao, C.; Chen, Z.; Li, Y.; Jing, X.; Huang, Q. Low dose of trichostatin a improves radiation resistance by activating Akt/Nrf2-dependent antioxidation pathway in cancer cells. Radiat. Res. 2021, 195, 366–377. [Google Scholar] [CrossRef]
- Shirbhate, E.; Patel, P.; Patel, V.K.; Veerasamy, R.; Sharma, P.C.; Rajak, H. The combination of histone deacetylase inhibitors and radiotherapy: A promising novel approach for cancer treatment. Future Oncol. 2020, 16, 2457–2469. [Google Scholar] [CrossRef]
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A. Clinical experience with the novel histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid) in patients with relapsed lymphoma. Br. J. Cancer 2006, 95, S7–S12. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.Y.; Yap, C.W.; Wang, Z.; Ho, P.C.; Chan, S.Y.; Ng, K.Y.; Ge, Z.; Lin, H.S. Solubilization of vorinostat by cyclodextrins. J. Clin. Pharm. Ther. 2010, 35, 521–526. [Google Scholar] [CrossRef]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone deacetylase inhibitors and phenotypical transformation of cancer cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.-W.; Yeh, Y.-L.; Wang, Y.-C.; Huang, W.-J.; Chen, Y.-A.; Chiou, Y.-S.; Ho, S.-Y.; Lin, P.; Wang, Y.-J. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS ONE 2013, 8, e76340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, P.; Orlando, L.; Cinieri, S. Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert Opin. Investig. Drugs 2017, 26, 1199–1206. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Subtype of BC | Immunohistochemistry Status | Grade | Prevalence | Outcome | Treatment |
|---|---|---|---|---|---|
| Luminal A | ER+, PR+, HER2−, Ki67− | 1/2 | 23.7% | good | endocrine therapy alone or with chemotherapy |
| Luminal B | ER+, PR+, HER2−, Ki67+ | 2/3 | 38.8% | middle | endocrine therapy and chemotherapy without/with anti-HER2 therapy |
| ER+, PR+, HER2+, Ki67+ | 14% | poor | |||
| HER2-overexpressed | ER−, PR−, HER2+ | 2/3 | 11.2% | poor | chemotherapy and anti-HER2 therapy |
| TNBC | ER−, PR−, HER2−, basal marker+ | 3 | 12.3% | poor | chemotherapy |
| Normal-like | ER+, PR+, HER2−, Ki67− | 1/2/3 | 7.8% | middle | endocrine therapy alone or with chemotherapy |
| Cellular Process | Subtype of BC | Cell Line | Mechanism of Action | References |
|---|---|---|---|---|
| Apoptosis | Luminal | MCF7, T47D | -increase in the number of apoptotic cells | [108] |
| TNBC | MDA-MB-231 | |||
| Autophagy | Luminal | TAMR/MCF7 | -increase in expression of autophagic cell death markers (LC-II and beclin-1) | [109] |
| TNBC | MDA-MB-231 | -induction of autophagy | [99] | |
| Cell cycle | Luminal | MCF7, T47D | -accumulation and reduction of cells in G1 and G2 phases, respectively | [108] |
| TNBC | MDA-MB-231 | |||
| Luminal | TAMR/MCF7 | -cell cycle arrest in G2/M phases | [109] | |
| Proliferation | Lumianal | T47D, MCF7 | -inhibition of proliferation | [110] |
| TNBC | MDA-MB-231, MDA-MB-468 | |||
| Tumor growth | Luminal | TAMR/MCF7 cells xenografts | -tumor growth reduction | [109] |
| Migration | Luminal | MCF7 | -inhibition of migration stimulated by leptin | [111] |
| TNBC | MDA-MB-231, MDA-MB-468 | |||
| Luminal | T47D, MCF7 | -inhibition of migration | [110] | |
| TNBC | MDA-MB-231, MDA-MB-468 | |||
| EMT | TNBC | MDA-MB-231, BT549 | -increase in E-cadherin expression, -decrease in N-cadherin, vimentin and fibronectin expression, -inhibition of EMT through downregulation of FOXA1 expression at both mRNA and protein levels | [112] |
| Luminal | T47D, MCF7 | -increase in E-cadherin expression | [110] | |
| TNBC | MDA-MB-468 | -increase in N-cadherin expression | ||
| ER receptor status | Luminal | MCF7 | -depletion of ERα, -inhibition of ERα mRNA, -ubiquitin-proteasome pathway degradation of ERα (inhibition of cell proliferation, induction of apoptosis) | [107] |
| TNBC | BT549 | -re-expression of ERα (inhibition of cell growth and sensitization to tamoxifen) | [103] |
| Drug-Drug Combination | BC-Subtype | In Vitro/In Vivo Model | Mechanism of Action | Type of Pharmacological Interaction | References |
|---|---|---|---|---|---|
| SAHA and cisplatin (CDDP) | Luminal | MCF7, T47D cells | -cell cycyle progression, -induction of apoptosis, -inhibition of proliferation | -additive (MCF7), -additive with a tendency towards synergism (T47D) | [108] |
| TNBC | MDA-MB-231 cells | -cell cycyle progression, -induction of apoptosis, -inhibition of proliferation, -decrease in Notch1 expression | additive | [108] | |
| Luminal | MCF7 cells with increased and decreased Notch1 activity | -inhibition of proliferation | additive | [92] | |
| TNBC | MDA-MB-231 cells with increased and decreased Notch1 activity | -inhibition of proliferation | -additive (MDA-MB-231 with increased Notch1 activity), -additive with a tendency towards antagonism (MDA-MB-231 with decreased Notch1 activity) | [115] | |
| SAHA and taxol | Luminal | MCF7, MCF7/ADR, T47D cells | -induction of apoptosis, -cell-cycle arrest in G2/M phases, -cell growth inhibition | synergistic | [120] |
| TNBC | MDA-MB-231, MDA-MB-453, BT474 cells | ||||
| HER2-overexpressed | SKBR3 cells | ||||
| TNBC | BALB/c mice bearing a BC xenografts | -tumor growth inhibition | |||
| SAHA and paclitaxel (PAX) | Luminal | MCF7, T47D, YCC-B1, YCC-B3, YCC-B5 cells | -upregulation of MAPK13, ATP2C1, ANKDR57, MT1G, RGCR, C12orf49, EXOC6 RAB4A, TM9SF3, IFNGR1 gene expression, -downregulation of DMD, HCG9, KIFC3, SYNGR3, NDRG4, NT5E, EOMES, SMC4, LANCL1, SCHIP1, 2EST gene expression | synergistic | [121] |
| HER2-overexpressed, TNBC | SKBR3, MDA-MB-231, YCC-B2 cells | antagonistic | |||
| SAHA and trastuzumab | HER2-overexpressed | SKBR3 cells | -enhancement of trastuzumab-dependent cytotoxicity and phagocytosis | synergistic | [122] |
| SAHA and olaparib | TNBC | MDA-MB-157, MDA-MB-231, MDA-MB-436 cells | -inhibition of proliferation, -DNA-demage induced cell cycle arrest, -induction of apoptosis | synergistic | [123] |
| SAHA and tozasertib (MK-0457) | TNBC | MDA-MB-231, MDA-MB-468, MDA-MB-474 cells | -induction of apoptosis, -cell cycle arrest in G2/M phases, -induction of multipolar mitotic spindles, -induction of DNA endoreduplication | synergistic | [124] |
| TNBC | mice bearing MDA-MB-231 xenografts | -tumor growth inhibition | |||
| SAHA and epigallocatechin-3-gallate (EGCG) | TNBC | MDA-MB-157, MDA-MB-231 | -inhibition of migration, -induction of apoptosis, -increase in caspase-7 and cIAP2 gene expression | synergistic | [125] |
| SAHA and sodium butyrate (NaB) | TNBC | MDA-MB-231, BT-549 | -inhibition of cell proliferation, -cell cycle arrest in G0/G1 phases, -promotion of apoptosis | synergistic | [126] |
| SAHA and clarithromycin (CAM) + bortezomib (BZ) | TNBC | MDA-MB-231 | -enhancement of ER-stress-mediated cell death, -induction of apoptosis, -increase in CHOP and GADD153 gene expression | synergistic | [127] |
| SAHA and tumor necrosis factor related apoptosis inducing ligand (TRAIL) | TNBC | BALB/c nude mice implanted with MDA-MB-468 TRAIL-resistant cells | -inhibition of tumor growth, metastasis and angiogenesis | synergistic | [128] |
| SAHA and soluble CD137 receptor | TNBC | MDA-MB-231 | -increase in cytotoxicity | synergistic | [129] |
| Clinical Trial | Clinical Trial Number | Drug and Therapy | Type of BC/Condition | Phase | Status | References |
|---|---|---|---|---|---|---|
| Olaparib in combination with vorinostat in patients with relapsed/refractory and/or metastatic breast cancer | NCT03742245 | olaparib + vorinostat | breast cancer, metastatic breast cancer | I | recruiting | [155] |
| Carboplatin and nab-paclitaxel with or without vorinostat in treating women with newly diagnosed operable BC | NCT00616967 | carboplatin + paclitaxel (albumin-stabilized nanoparticle formulation) + vorinostat or placebo | BC | II | active, not recruiting | [156] |
| Pembrolizumab and tamoxifen with or without vorinostat for the treatment of estrogen receptor positive breast cancer | NCT04190056 | Pembrolizumab + tamoxifen + vorinostat | anatomic stage IV breast cancer AJCC v8, prognostic stage IV breast cancer AJCC v8 | II | active, not recruiting | [157] |
| Trial for locally advanced breast cancer using vorinostat plus chemotherapy | NCT00574587 | vorinostat + paclitaxel + trastuzumab + doxorubicin + cyclophosphamide and surgery | breast cancer | I, II | completed | [158] |
| HDAC inhibitor vorinostat (SAHA) with capecitabine (Xeloda) using a new weekly dose regimen for advanced breast cancer | NCT00719875 | vorinostat | advanced breast cancer | I | completed | [159] |
| Ixabepilone and vorinostat in treating patients with metastatic breast cancer | NCT01084057 | vorinostat + ixabepilone | male breast cancer, recurrent breast cancer, stage IV breast cancer | I | completed | [160] |
| Phase I–II study of vorinostat, paclitaxel, and bevacizumab in metastatic breast cancer | NCT00368875 | vorinostat + paclitaxel + bevacizumab | male breast cancer, stage IIIB breast cancer, stage IIIC breast cancer, stage IV breast cancer | I, II | completed | [161] |
| Vorinostat in treating patients with stage IV breast cancer receiving hormone therapy | NCT01720602 | vorinostat + anastrozole + letrozole + exemestane and radiation | male breast cancer, recurrent breast cancer, stage IV breast cancer | N/A | completed | [162] |
| Vorinostat in treating patients with stage IV breast cancer receiving aromatase inhibitor therapy | NCT01153672 | vorinostat and radiation + anastrozole + letrozole + exemestane | male breast cancer, recurrent breast cancer, stage IV breast cancer | N/A | completed | [163] |
| Vorinostat in treating women who are undergoing surgery for newly diagnosed stage I–III breast cancer | NCT00262834 | vorinostat and conventional surgery | breast cancer, stage I breast cancer, stage II breast cancer, stage III breast cancer | II | completed | [164] |
| Vorinostat and trastuzumab in treating patients with metastatic or locally recurrent breast cancer | NCT00258349 | vorinostat + trastuzumab | breast cancer, male breast, cancer recurrent breast cancer, stage IIIB breast cancer, stage IIIC breast cancer, stage IV breast cancer | I, II | completed | [165] |
| Vorinostat in treating women with ductal carcinoma in situ of the breast | NCT00788112 | vorinostat and neoadjuvant therapy and therapeutic conventional surgery | breast cancer | I | completed | [166] |
| Phase II trial of SAHA & tamoxifen for patients with breast cancer | NCT00365599 | vorinostat + tamoxifen | breast cancer | II | completed | [167] |
| GCC 0845:vorinostat and lapatinib in advanced solid tumors and advanced breast cancer to evaluate response and biomarkers | NCT01118975 | vorinostat + lapatinib | breast cancer, neoplasm, metastasis | I, II | terminated | [168] |
| A study of vorinostat and tamoxifen in newly diagnosed breast cancer | NCT01194427 | vorinostat + tamoxifen | stage I breast cancer, stage II breast cancer, stage III breast cancer, invasive breast cancer | II | terminated | [169] |
| Suberoylanilide hydroxamic acid in treating patients with progressive stage IV breast cancer | NCT00132002 | vorinostat | male breast cancer, recurrent breast cancer, stage IV breast cancer | II | terminated | [170] |
| Reversing therapy resistance with epigenetic-immune modification | NCT02395627 | tamoxifen + vorinostat + pembrolizumab | breast neoplasms | II | terminated | [171] |
| A clinical trial of oral suberoylanilide hydroxamic acid (SAHA) in patients with relapsed or refractory breast, colorectal and non-small cell lung cancer (0683-011) | NCT00126451 | MK0683 + vorinostat | breast cancer | II | terminated | [172] |
| Vorinostat before surgery in treating patients with triple-negative breast cancer | NCT01695057 | vorinostat and therapeutic conventional surgery | stage II breast cancer, stage IIIA breast cancer, triple-negative breast cancer | N/A | withdrawn | [173] |
| Clinical trial of SAHA in patients with breast cancer | NCT00416130 | vorinostat | breast cancer | I, II | unknown | [174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. https://doi.org/10.3390/cancers13184700
Wawruszak A, Borkiewicz L, Okon E, Kukula-Koch W, Afshan S, Halasa M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers. 2021; 13(18):4700. https://doi.org/10.3390/cancers13184700
Chicago/Turabian StyleWawruszak, Anna, Lidia Borkiewicz, Estera Okon, Wirginia Kukula-Koch, Syeda Afshan, and Marta Halasa. 2021. "Vorinostat (SAHA) and Breast Cancer: An Overview" Cancers 13, no. 18: 4700. https://doi.org/10.3390/cancers13184700
APA StyleWawruszak, A., Borkiewicz, L., Okon, E., Kukula-Koch, W., Afshan, S., & Halasa, M. (2021). Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers, 13(18), 4700. https://doi.org/10.3390/cancers13184700