
Current Landscape of Non-Small Cell Lung Cancer: Epidemiology, Histological Classification, Targeted Therapies, and Immunotherapy

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Trends in Epidemiology
3. Advancements in Histopathological Classification
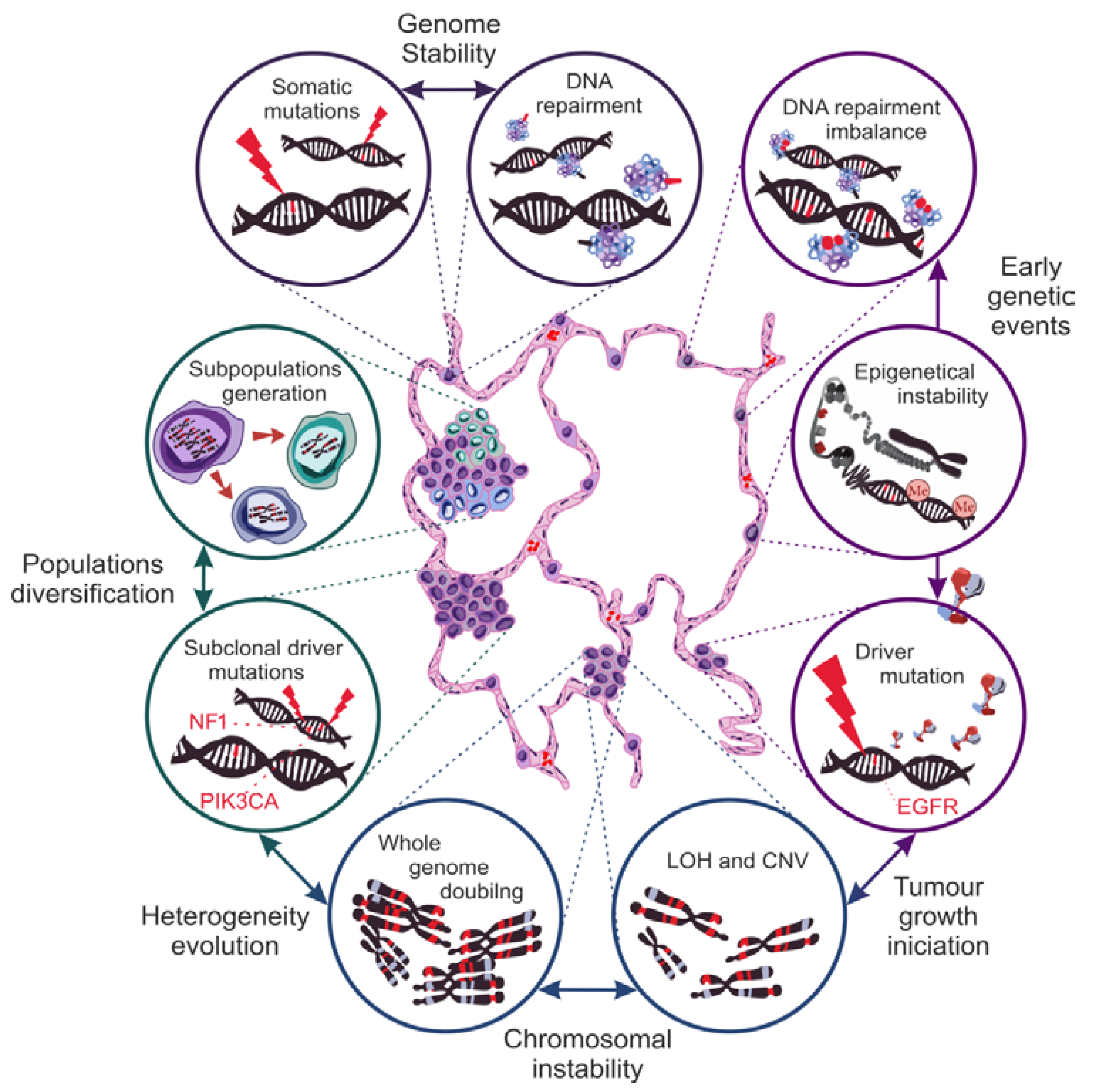
4. Genetic Basis of NSCLC Heterogeneity
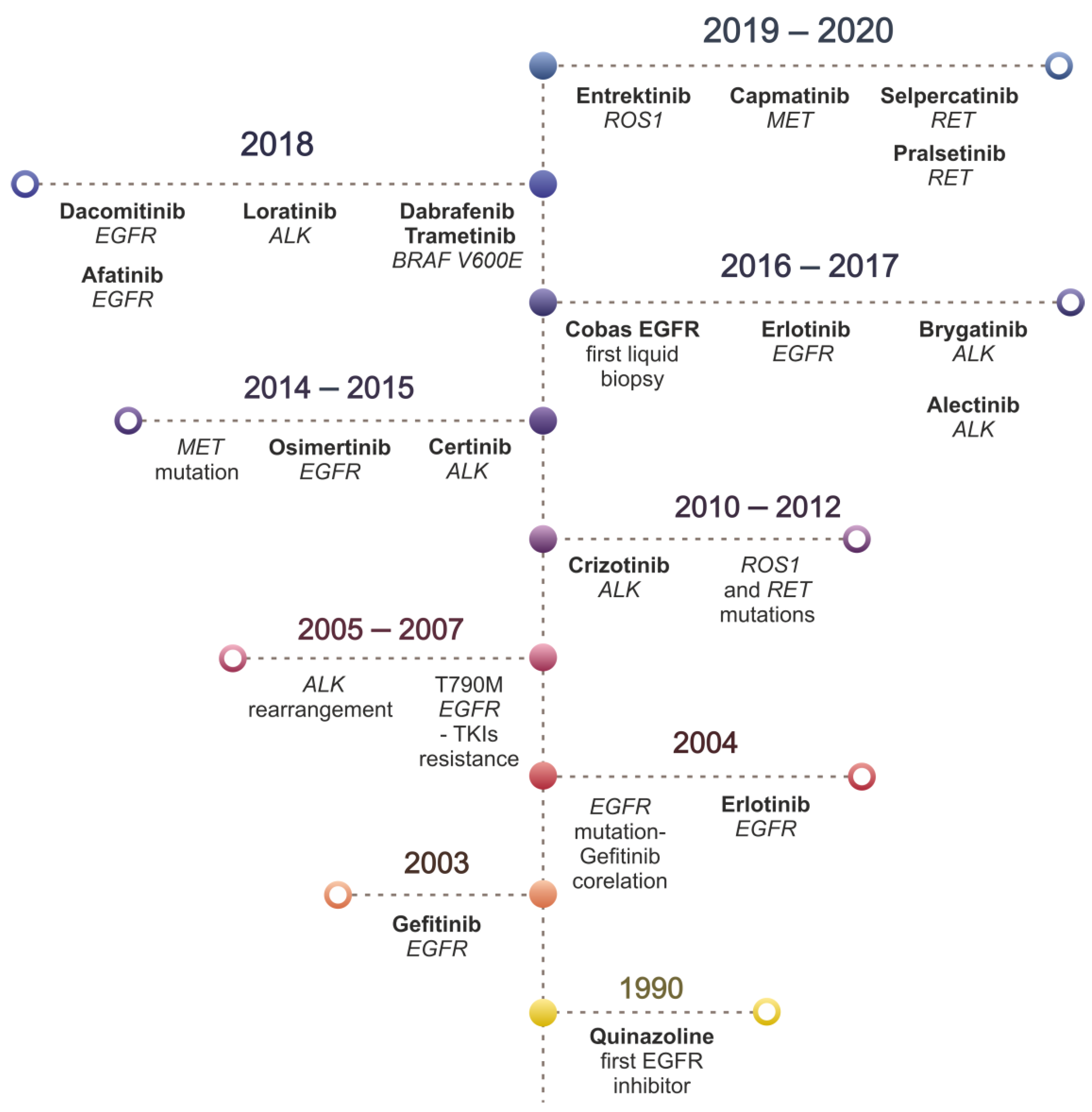
5. Overview of Targeted Therapy for NSCLC
5.1. Protein Genes from the Epidermal Growth Factor Receptor (EGFR) Family
5.2. Anaplastic Lymphoma Kinase (ALK) Receptor Gene
5.3. The C-Ros Oncogene 1 of the Receptor Tyrosine Kinase (ROS1) Gene
5.4. Tyrosine-Protein Kinase MET Gene—MET
5.5. Tyrosine-Protein Kinase RET Gene—RET
5.6. Neurotrophic Tyrosine Kinase Receptor Type 1—NTRK1
5.7. V600E Mutation of the BRAF1 Gene (Rapidly Accelerated Fibrosarcoma Homolog B)
5.8. KRAS Gene Mutation (Kirsten Rat Sarcoma Viral Oncogene Homolog)
5.9. FDA Approved TKIs
6. Overview of Immunotherapy for NSCLC
6.1. Immune Checkpoint Inhibitors
6.2. Combination Treatment Strategies
6.2.1. Immune Checkpoint Inhibitor Combined with Chemotherapy
6.2.2. Combined Immune Checkpoint Inhibitors: ICI PD-1/PD-L1 Combined with Anti-CTLA-4
Combined Immune Checkpoint Inhibitors with EGFR-TKI
6.3. Predictive Biomarkers
7. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung, cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global epidemiology of lung cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, K.; Toyokawa, G.; Shoji, F.; Okamoto, T.; Maehara, Y. The Significance of the PD-L1 Expression in Non–Small-Cell Lung Cancer: Trenchant Double Swords as Predictive and Prognostic Markers. Clin. Lung Cancer 2018, 19, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Liu, S.V.; Subramaniam, D.S.; Giaccone, G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat. Rev. Clin. Oncol. 2015, 12, 511–526. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Arjun Kumar, E.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Kaderbhai, C.G.; Boidot, R.; Beltjens, F.; Chevrier, S.; Arnould, L.; Favier, L.; Lagrange, A.; Coudert, B.; Ghiringhelli, F. Use of dedicated gene panel sequencing using next generation sequencing to improve the personalized care of lung cancer. Oncotarget 2016, 7, 24860–24870. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Thorac. Oncol. 2013, 8, 823–859. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; Govindan, R. Clinical implications of genomic discoveries in lung cancer. N. Engl. J. Med. 2016, 374, 1864–1873. [Google Scholar] [CrossRef] [Green Version]
- Politi, K.; Herbst, R.S. Lung cancer in the era of precision medicine. Clin. Cancer Res. 2015, 21, 2213–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łaczmańska, I.; Dębicka, I.; Gil, J.; Michałowska, D.; Pawlak, I.; Sąsiadek, M.M. Personalised medicine in lung cancer. Genet. Oncol. Nowotw. J. Oncol. 2021, 71, 122–128. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Riely, G.J. New pathologic classification of lung cancer: Relevance for clinical practice and clinical trials. J. Clin. Oncol. 2013, 31, 992–1001. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [Green Version]
- Moldaver, D.; Hurry, M.; Evans, W.K.; Cheema, P.K.; Sangha, R.; Burkes, R.; Melosky, B.; Tran, D.; Boehm, D.; Venkatesh, J.; et al. Development, validation and results from the impact of treatment evolution in non-small cell lung cancer (iTEN) model. Lung Cancer 2020, 139, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Meza, R.; Meernik, C.; Jeon, J.; Cote, M.L. Lung cancer incidence trends by gender, race and histology in the United States, 1973-2010. PLoS ONE 2015, 10, e0121323. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.W.; Ruiz, B.A.; Hsieh, M.-C.; Wu, X.-C.; Ries, L.A.G.; Lewis, D.R. Analysis of stage and clinical/prognostic factors for lung cancer from SEER registries: AJCC staging and collaborative stage data collection system. Cancer 2014, 120, 3781–3792. [Google Scholar] [CrossRef]
- Henschke, C.I.; McCauley, D.I.; Yankelevitz, D.F.; Naidich, D.P.; McGuinness, G.; Miettinen, O.S.; Libby, D.M.; Pasmantier, M.W.; Koizumi, J.; Altorki, N.K.; et al. Early Lung Cancer Action Project: Overall design and findings from baseline screening. Lancet 1999, 354, 99–105. [Google Scholar] [CrossRef]
- The National Lung Screening Trial Research Team Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [CrossRef] [Green Version]
- Wadowska, K.; Bil-Lula, I.; Trembecki, Ł.; Śliwińska-Mossoń, M. Genetic markers in lung cancer diagnosis: A review. Int. J. Mol. Sci. 2020, 21, 4569. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, P.T.; Sanford, T.; Choudhary, A.; Mydlarz, W.W.; Brown, D.; Adai, A.T.; Ochs, M.F.; Ahrendt, S.A.; Mambo, E.; Califano, J.A. Serum microrna biomarkers for detection of non-small cell lung cancer. PLoS ONE 2012, 7, e32307. [Google Scholar] [CrossRef]
- Jiang, M.; Li, X.; Quan, X.; Li, X.; Zhou, B. Clinically Correlated MicroRNAs in the Diagnosis of Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Kadota, T.; Matsuzaki, J.; Yoshida, Y.; Yamamoto, Y.; Nakagawa, K.; Takizawa, S.; Aoki, Y.; Nakamura, E.; Miura, J.; et al. A miRNA-based diagnostic model predicts resectable lung cancer in humans with high accuracy. Commun. Biol. 2020, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann. Oncol. 2017, 28, 1117–1123. [Google Scholar] [CrossRef]
- Thun, M.J.; Carter, B.D.; Feskanich, D.; Freedman, N.D.; Prentice, R.; Lopez, A.D.; Hartge, P.; Gapstur, S.M. 50-Year trends in smoking-related mortality in the United States. N. Engl. J. Med. 2013, 368, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Lortet-Tieulent, J.; Renteria, E.; Sharp, L.; Weiderpass, E.; Comber, H.; Baas, P.; Bray, F.; Coebergh, J.W.; Soerjomataram, I. Convergence of decreasing male and increasing female incidence rates in major tobacco-related cancers in Europe in 1988–2010. Eur. J. Cancer 2015, 51, 1144–1163. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.A.; Fedewa, S.A.; Henley, S.J.; Pollack, L.A.; Jemal, A. Proportion of Never Smokers among Men and Women with Lung Cancer in 7 US States. JAMA Oncol. 2021, 7, 302. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- Khaddour, K.; Jonna, S.; Deneka, A.; Patel, J.D.; Abazeed, M.E.; Golemis, E.; Borghaei, H.; Boumber, Y. Targeting the epidermal growth factor receptor in egfr-mutated lung cancer: Current and emerging therapies. Cancers 2021, 13, 3164. [Google Scholar] [CrossRef]
- Turner, M.C.; Krewski, D.; Pope, C.A.; Chen, Y.; Gapstur, S.M.; Thun, M.J. Long-term ambient fine particulate matter air pollution and lung cancer in a large cohort of never-smokers. Am. J. Respir. Crit. Care Med. 2011, 184, 1374–1381. [Google Scholar] [CrossRef]
- Raaschou-Nielsen, O.; Andersen, Z.J.; Beelen, R.; Samoli, E.; Stafoggia, M.; Weinmayr, G.; Hoffmann, B.; Fischer, P.; Nieuwenhuijsen, M.J.; Brunekreef, B.; et al. Air pollution and lung cancer incidence in 17 European cohorts: Prospective analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet. Oncol. 2013, 14, 813–822. [Google Scholar] [CrossRef]
- Goss, P.E.; Strasser-Weippl, K.; Lee-Bychkovsky, B.L.; Fan, L.; Li, J.; Chavarri-Guerra, Y.; Liedke, P.E.R.; Pramesh, C.S.; Badovinac-Crnjevic, T.; Sheikine, Y.; et al. Challenges to effective cancer control in China, India, and Russia. Lancet Oncol. 2014, 15, 489–538. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global Cancer Statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart, 4th ed.; WHO: Geneva, Switzerland, 2015; ISBN 9789283224365. [Google Scholar]
- Rekhtman, N.; Travis, W.D. Large No More: The Journey of Pulmonary Large Cell Carcinoma from Common to Rare Entity. J. Thorac. Oncol. 2019, 14, 1125–1127. [Google Scholar] [CrossRef] [PubMed]
- Kashima, J.; Kitadai, R.; Okuma, Y. Molecular and morphological profiling of lung cancer: A foundation for “next-generation” pathologists and oncologists. Cancers 2019, 11, 599. [Google Scholar] [CrossRef] [Green Version]
- Laing, G.M.; Kerr, K.M. The 2015 World Health Organisation Classification of Lung Cancer; Springer: Cham, Switzerlnad, 2018; pp. 57–75. [Google Scholar]
- Shih, A.R.; Uruga, H.; Bozkurtlar, E.; Chung, J.; Hariri, L.P.; Minami, Y.; Wang, H.; Yoshizawa, A.; Muzikansky, A.; Moreira, A.L.; et al. Problems in the reproducibility of classification of small lung adenocarcinoma: An international interobserver study. Histopathology 2019, 75, 649–659. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; Van Schil, P.E.; et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma: The cancer genome atlas research network. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Sekido, Y.; Fong, K.M.; Minna, J.D. Molecular Genetics of Lung Cancer. Annu. Rev. Med. 2003, 54, 73–87. [Google Scholar] [CrossRef]
- Nenclares, P.; Harrington, K.J. The biology of cancer. Medicine 2020, 48, 67–72. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining “chromosomal instability”. Trends Genet. 2008. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Casula, M.; Manca, A.; Palomba, G.; Sini, M.C.; Doneddu, V.; Cossu, A.; Colombino, M. Genetic Instability Markers in Cancer. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2020; Volume 2055, pp. 133–154. [Google Scholar]
- De, S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011, 27, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lu, Y.P.; Tseng, R.C.; Lin, R.K.; Chang, J.W.; Chen, J.T.; Shih, C.M.; Chen, C.Y. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J. Clin. Investig. 2003, 111, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawes, D.A.; SenGupta, S.; Boulos, P.B. The clinical importance and prognostic implications of microsatellite instability in sporadic cancer. Eur. J. Surg. Oncol. 2003, 29, 201–212. [Google Scholar] [CrossRef]
- Shen, C.; Wang, X.; Tian, L.; Che, G. Microsatellite alteration in multiple primary lung cancer. J. Thorac. Dis. 2014, 6, 1499–1505. [Google Scholar]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Karlic, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahovicek, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Takeshima, H.; Ushijima, T. Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. npj Precis. Oncol. 2019, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sikkink, S.K.; Liloglou, T.; Maloney, P.; Gosney, J.R.; Field, J.K. In-depth analysis of molecular alterations within normal and tumour tissue from an entire bronchial tree. Int. J. Oncol. 2003, 22, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, M.; MacLennan, G.T.; Abdul-Karim, F.W.; Eble, J.N.; Jones, T.D.; Olobatuyi, F.; Eisenberg, R.; Cummings, O.W.; Zhang, S.; et al. Evidence for Common Clonal Origin of Multifocal Lung Cancers. JNCI J. Natl. Cancer Inst. 2009, 101, 560–570. [Google Scholar] [CrossRef]
- Gazdar, A.; Minna, J. Multifocal lung cancers—Clonality vs field cancerization and does it matter? J. Natl. Cancer Inst. 2009, 101, 541–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquale, R.; Forgione, L.; Roma, C.; Fenizia, F.; Bergantino, F.; Rachiglio, A.; De Luca, A.; Gallo, M.; Maiello, M.; Palumbo, G.; et al. Targeted sequencing analysis of cell-free DNA from metastatic non-small-cell lung cancer patients: Clinical and biological implications. Transl. Lung Cancer Res. 2020, 9, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Nakagomi, T.; Goto, T.; Hirotsu, Y.; Shikata, D.; Yokoyama, Y.; Otake, S.; Amemiya, K.; Oyama, T.; Mochizuki, H.; et al. Identification of Clonality through Genomic Profile Analysis in Multiple Lung Cancers. J. Clin. Med. 2020, 9, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-L.; Kan, M.; Zhang, M.-M.; Yu, S.-S.; Xie, H.-J.; Gu, Z.-H.; Wang, H.-N.; Zhao, S.-X.; Zhou, G.-B.; Song, H.-D.; et al. Multiregion sequencing reveals the intratumor heterogeneity of driver mutations in TP53-driven non-small cell lung cancer. Int. J. Cancer 2017, 140, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013. [CrossRef]
- Huang, J.; Zhang, Y.; Ma, Q.; Zhang, Y.; Wang, M.; Zhou, Y.; Xing, Z.; Jin, M.; Hu, L.; Kong, X. Natural Selection on Exonic SNPs Shapes Allelic Expression Imbalance (AEI) Adaptability in Lung Cancer Progression. Front. Genet. 2020, 11, 665. [Google Scholar] [CrossRef]
- López, S.; Lim, E.L.; Horswell, S.; Haase, K.; Huebner, A.; Dietzen, M.; Mourikis, T.P.; Watkins, T.B.K.; Rowan, A.; Dewhurst, S.M.; et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 2020, 52, 283–293. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246.e13. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Al Bakir, M.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [Green Version]
- Olbromski, M.; Podhorska-Okołów, M.; Dzięgiel, P. Role of SOX Protein Groups F and H in Lung Cancer Progression. Cancers 2020, 12, 3235. [Google Scholar] [CrossRef]
- Risques, R.A.; Kennedy, S.R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism: Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Guillaumet-Adkins, A.; Yañez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and Oxidative Stress in Aging. Oxid. Med. Cell. Longev. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Greaves, M. Cancer causation: The Darwinian downside of past success? Lancet Oncol. 2002, 3, 244–251. [Google Scholar] [CrossRef]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Sharma, K.D.; Takahashi, T.; Iwamoto, R.; Mekada, E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 2002, 13, 2547–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef] [Green Version]
- Arbiser, J.L. Molecular regulation of angiogenesis and tumorigenesis by signal transduction pathways: Evidence of predictable and reproducible patterns of synergy in diverse neoplasms. Semin. Cancer Biol. 2004, 14, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Liu, H.; Cai, M.; Shentu, Y. Inhibition of tumor lymphangiogenesis is an important part that EGFR-TKIs play in the treatment of NSCLC. J. Cancer 2020, 11, 241–250. [Google Scholar] [CrossRef]
- Pirker, R.; Pereira, J.R.; Von Pawel, J.; Krzakowski, M.; Ramlau, R.; Park, K.; De Marinis, F.; Eberhardt, W.E.E.; Paz-Ares, L.; Störkel, S.; et al. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: Analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012, 13, 33–42. [Google Scholar] [CrossRef]
- Bell, D.W.; Gore, I.; Okimoto, R.A.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.V.; Brannigan, B.W.; Mohapatra, G.; Settleman, J.; et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet. 2005, 37, 1315–1316. [Google Scholar] [CrossRef]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or platinum-pemetrexed in EGFR T790M-Positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.H.; Barrett, J.C.; Jänne, P.A. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Papadimitrakopoulou, V.A.; Han, J.; Ahn, M.; Ramalingam, S.S.; Delmonte, A.; Hsia, T.; Laskin, J.; Kim, S.; He, Y.; Tsai, C.; et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: Osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non–small cell lung cancer. Cancer 2020, 126, 373–380. [Google Scholar] [CrossRef]
- Popat, S. Osimertinib as first-line treatment in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 192–193. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR -Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Cho, J.H.; Lim, S.H.; An, H.J.; Kim, K.H.; Park, K.U.; Kang, E.J.; Choi, Y.H.; Ahn, M.S.; Lee, M.H.; Sun, J.M.; et al. Osimertinib for patients with non-small-cell lung cancer harboring uncommon EGFR mutations: A multicenter, open-label, phase II trial (KCSG-Lu15-09). J. Clin. Oncol. Am. Soc. Clin. Oncol. 2020, 38, 488–495. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Arbour, K.C.; Rizvi, H.; Iqbal, A.N.; Gadgeel, S.M.; Girshman, J.; Kris, M.G.; Riely, G.J.; Yu, H.A.; Hellmann, M.D.; et al. Severe immune-related adverse events are common with sequential PD-(L)1 blockade and osimertinib. Ann. Oncol. 2019, 30, 839–844. [Google Scholar] [CrossRef]
- Latif, H.; Liu, S.V. Combining immunotherapy and epidermal growth factor receptor kinase inhibitors: Worth the risk? Ann. Transl. Med. 2019, 7, S76. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Shepherd, F.A.; Kim, D.-W.; Lee, G.-W.; Lee, J.S.; Chang, G.-C.; Lee, S.S.; Wei, Y.-F.; Lee, Y.G.; Laus, G.; et al. Osimertinib Plus Durvalumab versus Osimertinib Monotherapy in EGFR T790M–Positive NSCLC following Previous EGFR TKI Therapy: CAURAL Brief Report. J. Thorac. Oncol. 2019, 14, 933–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: Clinical outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Baglivo, S.; Ludovini, V.; Sidoni, A.; Metro, G.; Ricciuti, B.; Siggillino, A.; Rebonato, A.; Messina, S.; Crinò, L.; Chiari, R. Large Cell Neuroendocrine Carcinoma Transformation and EGFR-T790M Mutation as Coexisting Mechanisms of Acquired Resistance to EGFR-TKIs in Lung Cancer. Mayo Clin. Proc. 2017, 92, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Shiau, C.J.; Babwah, J.P.; Da Cunha Santos, G.; Sykes, J.R.; Boerner, S.L.; Geddie, W.R.; Leighl, N.B.; Wei, C.; Kamel-Reid, S.; Hwang, D.M.; et al. Sample features associated with success rates in population-based EGFR mutation testing. J. Thorac. Oncol. 2014, 9, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, R.N.; Behera, M.; Berry, L.D.; Rossi, M.R.; Kris, M.G.; Johnson, B.E.; Bunn, P.A.; Ramalingam, S.S.; Khuri, F.R. HER2 mutations in lung adenocarcinomas: A report from the Lung Cancer Mutation Consortium. Cancer 2017, 123, 4099–4105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.T.; Shen, R.; Buonocore, D.; Olah, Z.T.; Ni, A.; Ginsberg, M.S.; Ulaner, G.A.; Offin, M.; Feldman, D.; Hembrough, T.; et al. Ado-trastuzumab emtansine for patients with HER2-mutant lung cancers: Results from a phase II basket trial. J. Clin. Oncol. Am. Soc. Clin. Oncol. 2018, 36, 2532–2537. [Google Scholar] [CrossRef]
- Zinner, R.G.; Glisson, B.S.; Fossella, F.V.; Pisters, K.M.W.; Kies, M.S.; Lee, P.M.; Massarelli, E.; Sabloff, B.; Fritsche, H.A.; Ro, J.Y.; et al. Trastuzumab in combination with cisplatin and gemcitabine in patients with Her2-overexpressing, untreated, advanced non-small cell lung cancer: Report of a phase II trial and findings regarding optimal identification of patients with Her2-overexpressing disease. Lung Cancer 2004, 44, 99–110. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, T.; Qin, Z.; Jiang, J.; Wang, Q.; Yang, S.; Rivard, C.; Gao, G.; Ng, T.L.; Tu, M.M.; et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann. Oncol. 2019, 30, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Robichaux, J.P.; Elamin, Y.Y.; Vijayan, R.S.K.; Nilsson, M.B.; Hu, L.; He, J.; Zhang, F.; Pisegna, M.; Poteete, A.; Sun, H.; et al. Pan-Cancer Landscape and Analysis of ERBB2 Mutations Identifies Poziotinib as a Clinically Active Inhibitor and Enhancer of T-DM1 Activity. Cancer Cell 2019, 36, 444–457.e7. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Pikor, L.A.; Ramnarine, V.R.; Lam, S.; Lam, W.L. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung Cancer 2013, 82, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Aronson, S.L.; Engelman, J.A.; Shyr, Y.; Khuri, F.R.; Rudin, C.M.; et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI’s Lung Cancer Mutation Consortium (LCMC). J. Clin. Oncol. 2011, 29, CRA7506. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Wang, D.; Li, D.; Qin, G.; Zhang, W.; Ouyang, J.; Zhang, M.; Xie, L. The Structural Characterization of Tumor Fusion Genes and Proteins. Comput. Math. Methods Med. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Novello, S.; Mazières, J.; Oh, I.-J.; De Castro, J.; Migliorino, M.R.; Helland, A.; Dziadziuszko, R.; Griesinger, F.; Kotb, A.; Zeaiter, A.; et al. Alectinib versus chemotherapy in crizotinib-pretreated anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer: Results from the phase III ALUR study. Ann. Oncol. 2018, 29, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Crinò, L.; Ahn, M.J.; De Marinis, F.; Groen, H.J.M.; Wakelee, H.; Hida, T.; Mok, T.; Spigel, D.; Felip, E.; Nishio, M.; et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: Results from ASCEND-2. J. Clin. Oncol. 2016, 34, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.H.; Han, J.Y.; Lee, J.S.; Hochmair, M.J.; Li, J.Y.C.; Chang, G.C.; Lee, K.H.; et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Lin, Y.T.; Yu, C.J.; Yang, J.C.H.; Shih, J.Y. Anaplastic Lymphoma Kinase (ALK) Kinase Domain Mutation Following ALK Inhibitor(s) Failure in Advanced ALK Positive Non–Small-Cell Lung Cancer: Analysis and Literature Review. Clin. Lung Cancer 2016, 17, e77–e94. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Shaw, A.T.; Solomon, B.J.; Besse, B.; Bauer, T.M.; Lin, C.C.; Soo, R.A.; Riely, G.J.; Ignatius Ou, S.H.; Clancy, J.S.; Li, S.; et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non–small-cell lung cancer. J. Clin. Oncol. 2019, 37, 1370–1379. [Google Scholar] [CrossRef]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.L.; Liu, W.; Dardaei, L.; et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Rimkunas, V.M.; Crosby, K.E.; Li, D.; Hu, Y.; Kelly, M.E.; Gu, T.L.; Mack, J.S.; Silver, M.R.; Zhou, X.; Haack, H. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: Identification of a FIG-ROS1 fusion. Clin. Cancer Res. 2012, 18, 4449–4457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Ou, S.H.I.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Riely, G.J.; Bang, Y.; Kim, D.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.J.; Shapiro, G.I.; Usari, T.; et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126. [Google Scholar] [CrossRef]
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.-L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired Resistance to Crizotinib from a Mutation in CD74-ROS1. N. Engl. J. Med. 2013, 368, 2395–2401. [Google Scholar] [CrossRef] [Green Version]
- Cargnelutti, M.; Corso, S.; Pergolizzi, M.; Mévellec, L.; Aisner, D.L.; Dziadziuszko, R.; Varella-Garcia, M.; Comoglio, P.M.; Doebele, R.C.; Vialard, J.; et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget 2015, 6, 5182–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.D.; Mahale, S.; Astling, D.P.; Aisner, D.L.; Le, A.T.; Hinz, T.K.; Vaishnavi, A.; Bunn, P.A.; Heasley, L.E.; Tan, A.C.; et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS ONE 2013, 8, e82236. [Google Scholar] [CrossRef] [PubMed]
- McCoach, C.E.; Le, A.T.; Gowan, K.; Jones, K.; Schubert, L.; Doak, A.; Estrada-Bernal, A.; Davies, K.D.; Merrick, D.T.; Bunn, P.A.; et al. Resistance mechanisms to targeted therapies in ROS1þ and ALKþ non–small cell lung cancer. Clin. Cancer Res. 2018, 24, 3334–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, T.; Smith, D.E.; Bunn, P.A.; Aisner, D.L.; Le, A.T.; Hancock, M.; Purcell, W.T.; Bowles, D.W.; Camidge, D.R.; Doebele, R.C. The Incidence of Brain Metastases in Stage IV ROS1-Rearranged Non–Small Cell Lung Cancer and Rate of Central Nervous System Progression on Crizotinib. J. Thorac. Oncol. 2018, 13, 1717–1726. [Google Scholar] [CrossRef] [Green Version]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Cho, B.C.; Lim, S.M.; Kim, H.R.; Lee, J.S.; Lee, K.H.; Lee, Y.G.; Min, Y.J.; Cho, E.K.; Lee, S.S.; Kim, B.S.; et al. Open-label, multicenter, phase II Study of ceritinib in patients with non–small-cell lung cancer harboring ROS1 rearrangement. J. Clin. Oncol. 2017, 35, 2613–2618. [Google Scholar] [CrossRef]
- Drilon, A.; Ou, S.H.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (Tpx-0005) is a next-generation ros1/trk/alk inhibitor that potently inhibits ros1/trk/alk solvent-front mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R.; Gong, B.; Togashi, N.; Miyamoto, M.; Kiga, M.; Iwasaki, S.; Kamai, Y.; Tominaga, Y.; Takeda, Y.; Kagoshima, Y.; et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Awad, M.M.; Leonardi, G.C.; Kravets, S.; Dahlberg, S.E.; Drilon, A.; Noonan, S.A.; Camidge, D.R.; Ou, S.H.I.; Costa, D.B.; Gadgeel, S.M.; et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: A retrospective analysis. Lung Cancer 2019, 133, 96–102. [Google Scholar] [CrossRef]
- Camidge, D.R.; Ou, S.-H.I.; Shapiro, G.; Otterson, G.A.; Villaruz, L.C.; Villalona-Calero, M.A.; Iafrate, A.J.; Varella-Garcia, M.; Dacic, S.; Cardarella, S.; et al. Efficacy and safety of crizotinib in patients with advanced c-MET -amplified non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32, 8001. [Google Scholar] [CrossRef]
- Strickler, J.H.; Weekes, C.D.; Nemunaitis, J.; Ramanathan, R.K.; Heist, R.S.; Morgensztern, D.; Angevin, E.; Bauer, T.M.; Yue, H.; Motwani, M.; et al. First-in-human phase I, dose-escalation and -expansion study of telisotuzumab vedotin, an antibody–drug conjugate targeting c-Met, in patients with advanced solid tumors. J. Clin. Oncol. Am. Soc. Clin. Oncol. 2018, 36, 3298–3306. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in patients with RET-rearranged lung cancers: Results from the global, multicenter RET registry. J. Clin. Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Subbiah, V.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Brose, M.S.; Curigliano, G.; Leboulleux, S.; Zhu, V.W.; Keam, B.; et al. 1913O Results from the registrational phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET mutation-positive medullary thyroid cancer (RET+ MTC). Ann. Oncol. 2020, 31, S1084. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Cui, G.; Liu, D.; Li, W.; Fu, X.; Liang, Y.; Li, Y.; Shi, W.; Chen, X.; Zhao, S. A meta-analysis of the association between BRAF mutation and nonsmall cell lung cancer. Medicine 2017, 96, e6552. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.M.; Kelly, R.J.; et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj, A.; Berenguer, J.; Fernandez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF Mutations Classes I, II, and III in NSCLC Patients Included in the SLLIP Trial: The Need for a New Pre-Clinical Treatment Rationale. Cancers 2019, 11, 1381. [Google Scholar] [CrossRef] [Green Version]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Tsongalis, P.D.; Peterson, J.D.; de Abreu, F.B.; Black, C.C.; Gutmann, E.J.; Liu, X.; Tafe, L.J.; Amos, C.I.; Tsongalis, G.J. The potential utility of re-mining results of somatic mutation testing: KRAS status in lung adenocarcinoma. Cancer Genet. 2016, 209, 195–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.H.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Santos, E.; Martin-Zanca, D.; Reddy, E.P.; Pierotti, M.A.; Della Porta, G.; Barbacid, M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984, 223, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlaender, A.; Drilon, A.; Weiss, G.J.; Banna, G.L.; Addeo, A. KRAS as a druggable target in NSCLC: Rising like a phoenix after decades of development failures. Cancer Treat. Rev. 2020, 85, 101978. [Google Scholar] [CrossRef]
- Román, M.; Baraibar, I.; López, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 861–878. [Google Scholar] [CrossRef] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence inKRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Riess, J.W.; Frankel, P.; Shackelford, D.; Dunphy, M.; Badawi, R.D.; Nardo, L.; Cherry, S.R.; Lanza, I.; Reid, J.; Gonsalves, W.I.; et al. Phase 1 Trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in Patients With Advanced NSCLC (NCI 10327): Rationale and Study Design. Clin. Lung Cancer 2021, 22, 67–70. [Google Scholar] [CrossRef]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Rybkin, I.I.; Spira, A.I.; Riely, G.J.; Papadopoulos, K.P.; Sabari, J.K.; Johnson, M.L.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S1–S2. [Google Scholar] [CrossRef]
- Kim, E.S.; Kies, M.S.; Fossella, F.V.; Glisson, B.S.; Zaknoen, S.; Statkevich, P.; Munden, R.F.; Summey, C.; Pisters, K.M.W.; Papadimitrakopoulou, V.; et al. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer 2005, 104, 561–569. [Google Scholar] [CrossRef]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Indini, A.; Rijavec, E.; Ghidini, M.; Cortellini, A.; Grossi, F. Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors. Pharmaceutics 2021, 13, 653. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A. Immunotherapy and nsclc: The long and winding road. Cancers 2020, 12, 2512. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; He, Y.; Dziadziuszko, R.; Zhao, S.; Zhang, X.; Deng, J.; Wang, H.; Hirsch, F.R.; Zhou, C.; Yu, H.; et al. T cell immunoglobulin and mucin-domain containing-3 in non-small cell lung cancer. Transl. Lung Cancer Res. 2019, 8, 895–906. [Google Scholar] [CrossRef]
- Spreafico, A.; Janku, F.; Rodon, J.A.; Tolcher, A.W.; Chandana, S.R.; Oliva, M.; Musalli, S.; Knauss, L.; Kragh, M.; Alifrangis, L.; et al. A phase I study of Sym021, an anti-PD-1 antibody (Ab), alone and in combination with Sym022 (anti-LAG-3) or Sym023 (anti-TIM-3). Ann. Oncol. 2019, 30, v488–v489. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Li, X.; Zhang, J. Progress of immune checkpoint LAG-3 in immunotherapy (Review). Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, X.; Li, E.; Zhang, G.; Wang, X.; Tang, T.; Bai, X.; Liang, T. VISTA: An immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 1–13. [Google Scholar] [CrossRef]
- Tagliamento, M.; Agostinetto, E.; Borea, R.; Brandão, M.; Poggio, F.; Addeo, A.; Lambertini, M. VISTA: A Promising Target for Cancer Immunotherapy? ImmunoTargets Ther. 2021, 10, 185–200. [Google Scholar] [CrossRef]
- Socinski, M.A.; Spira, A.I.; Paz-Ares, L.G.; Reck, M.; Lu, S.; Nishio, M.; Li, J.; Zhou, Y.; Rhee, J.W.; Chica Duque, S.; et al. AdvanTIG-302: Anti-TIGIT monoclonal antibody (mAb) ociperlimab (OCI) plus tislelizumab (TIS) versus pembrolizumab (PEM) in programmed death ligand-1 (PD-L1) selected, previously untreated, locally advanced, unresectable or metastatic non-small cell lung c. J. Clin. Oncol. 2021, 39, TPS9128. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Cui, J.; Wei, Y.; Chen, X.; Liu, G.; Gao, X.; Li, W.; Lu, H.; Zhan, P.; Lv, T.; et al. Effectiveness and safety of PD-1/PD-L1 or CTLA4 inhibitors combined with chemotherapy as a first-line treatment for lung cancer: A meta-analysis. J. Thorac. Dis. 2018, 10, 6636–6652. [Google Scholar] [CrossRef]
- Gogishvili, M.; Mobashery, N.; Makharadze, T.; Navarro, M.; Snodgrass, P.; Chen, H.; Lowy, I.; Rietschel, P.; Lee, S. P2.01-26 EMPOWER-Lung 3: Phase 3 Study of Combinations of Cemiplimab and Chemotherapy in First-Line Treatment of Advanced NSCLC. J. Thorac. Oncol. 2019, 14, S649. [Google Scholar] [CrossRef]
- Altorki, N.K.; McGraw, T.E.; Borczuk, A.C.; Saxena, A.; Port, J.L.; Stiles, B.M.; Lee, B.E.; Sanfilippo, N.J.; Scheff, R.J.; Pua, B.B.; et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: A single-centre, randomised phase 2 trial. Lancet Oncol. 2021, 22, 824–835. [Google Scholar] [CrossRef]
- Ahn, M.J.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K. EGFR TKI combination with immunotherapy in non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 465–469. [Google Scholar] [CrossRef]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Hoos, A. Development of immuno-oncology drugs-from CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.-J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Su, W.; Lu, T.; Wang, Y.; Dong, Y.; Qin, Y.; Liu, D.; Sun, L.; Jiao, W. First-Line Immune-Checkpoint Inhibitors in Non-Small Cell Lung Cancer: Current Landscape and Future Progress. Front. Pharmacol. 2020, 11, 578091. [Google Scholar] [CrossRef]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: A multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 2021, 397, 592–604. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Prey, S.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: An open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 848–857. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Park, B.J.; Zakowski, M.F.; Ladanyi, M.; Pao, W.; D’Angelo, S.P.; Kris, M.G.; Shen, R.; Zheng, J.; Azzoli, C.G. Impact on disease-free survival of adjuvant erlotinib or gefitinib in patients with resected lung adenocarcinomas that harbor EGFR mutations. J. Thorac. Oncol. 2011, 6, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.G.; Luft, A.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Fülöp, A.; Rodriguez-Cid, J.; Sugawara, S.; et al. Phase 3 study of carboplatin-paclitaxel/nab-paclitaxel (Chemo) with or without pembrolizumab (Pembro) for patients (Pts) with metastatic squamous (Sq) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36, 105. [Google Scholar] [CrossRef]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Cappuzzo, F.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.-G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. IMpower130: Progression-free survival (PFS) and safety analysis from a randomised phase III study of carboplatin + nab-paclitaxel (CnP) with or without atezolizumab (atezo) as first-line (1L) therapy in advanced non-squamous NSCLC. Ann. Oncol. 2018, 29, viii742–viii743. [Google Scholar] [CrossRef]
- Socinski, M.A.; Rittmeyer, A.; Shapovalov, D.; Orlandi, F.; McCleod, M.; Soo, R.A.; Palmero, R.; Kozuki, T.; Migliorino, M.R.; Koynov, K.D.; et al. IMpower131: Progression-free survival (PFS) and overall survival (OS) analysis of a randomised phase III study of atezolizumab + carboplatin + paclitaxel or nab-paclitaxel vs carboplatin + nab-paclitaxel in 1L advanced squamous NSCLC. Ann. Oncol. 2018, 29, viii750–viii751. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Cobo, M.; Bordoni, R.; Longeras, P.D.; Szalai, Z.; Ursol, G.; Novello, S.; Orlandi, F.; Ball, S.; Goldschmidt, J., Jr.; et al. IMpower132: PFS and Safety Results with 1L Atezolizumab + Carboplatin/Cisplatin + Pemetrexed in Stage IV Non-Squamous NSCLC. J. Thorac. Oncol. 2018, 13, S332–S333. [Google Scholar] [CrossRef] [Green Version]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Reinmuth, N.; Orlov, S.; Fischer, J.R.; Sugawara, S.; Mandziuk, S.; Marquez-Medina, D.; Novello, S.; Takeda, Y.; Soo, R.; et al. ARCTIC: Durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann. Oncol. 2020, 31, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Chul Cho, B.; Reinmuth, N.; Lee, K.H.; Ahn, M.-J.; Luft, A.; van den Heuvel, M.; Cobo, M.; Smolin, A.; Vicente, D.; et al. Durvalumab with or without tremelimumab vs platinum-based chemotherapy as first-line treatment for metastatic non-small cell lung cancer: MYSTIC. Ann. Oncol. 2018, 29, x40–x41. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.J.; Van Den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab with or Without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Qiao, M.; Jiang, T.; Liu, X.; Mao, S.; Zhou, F.; Li, X.; Zhao, C.; Chen, X.; Su, C.; Ren, S.; et al. Immune Checkpoint Inhibitors in EGFR-Mutated NSCLC: Dusk or Dawn? J. Thorac. Oncol. 2021, 16, 1267–1288. [Google Scholar] [CrossRef]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.M.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Atagi, S. EGFR TKIs and Immune Checkpoint Inhibitors: Is This an Optimal Combination? J. Thorac. Oncol. 2018, 13, 1245–1247. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 1–8. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Govindan, R.; Anders, R.A.; Antonia, S.J.; Sagorsky, S.; Davies, M.J.; Dubinett, S.M.; Ferris, A.; Gandhi, L.; Garon, E.B.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front. Oncol. 2021.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 11, 683419. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- News Release, F. FDA Approves First Cancer Treatment for Any Solid Tumor with a Specific Genetic Feature. In Mol. Cell. Pharmacol.; 2017. Available online: https://www.fda.gov (accessed on 20 May 2021).
- Ismael, N.E.H.S.; El Sheikh, S.A.; Talaat, S.M.; Salem, E.M. Mismatch repair proteins and microsatellite instability in colorectal carcinoma (MLH1, MSH2, MSH6 and PMS2): Histopathological and immunohistochemical study. Maced. J. Med. Sci. 2017, 5, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2020, 25, e147–e159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden–High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification 2004 | Classification 2015 | ||
|---|---|---|---|
| Adenocarcinoma | Adenocarcinoma, mixed subtype Acinar adenocarcinoma | Adenocarcinoma | Lepidic adenocarcinoma Acinar adenocarcinoma |
| Papillary adenocarcinoma | Papillary adenocarcinoma | ||
| Bronchioloalveolar carcinoma | Micropapillary adenocarcinoma | ||
| Solid adenocarcinoma with mucin production | Solid adenocarcinoma | ||
| Fetal adenocarcinoma | Invasive mucinous adenocarcinoma | ||
| Mucinous cystadenocarcinoma | Colloid adenocarcinoma | ||
| Mucinous (“colloid”) carcinoma | Fetal adenocarcinoma | ||
| Signet ring adenocarcinoma | Enteric adenocarcinoma | ||
| Clear cell adenocarcinoma | Minimally invasive adenocarcinoma | ||
| Preinvasive lesions: adenocarcinoma in situ | |||
| Squamous cell carcinoma | Papillary | Squamous cell carcinoma | Keratinizing |
| Clear cell | Nonkeratinizing | ||
| Small cell | Basaloid | ||
| Basaloid | Preinvasive lesions: Squamous cell carcinoma in situ | ||
| Small cell carcinoma | Combined small cell carcinoma | Neuroendocrine tumors | Small cell carcinoma |
| Large cell carcinoma | Large cell neuroendocrine carcinoma | Large cell neuroendocrine carcinoma | |
| Combined large cell neuroendocrine carcinoma | Carcinoid tumors | ||
| Basaloid carcinoma | Preinvasive lesion | ||
| Lymphoepithelioma-like carcinoma | Large cell carcinoma | ||
| Clear cell carcinoma | Adenosquamous carcinoma | ||
| Large cell carcinoma with rhabdoid phenotype | Sarcomatoid carcinomas | ||
| Adenosquamous carcinoma | Other and Unclassified carcinomas | ||
| Sarcomatoid carcinoma | Salivary gland-type tumors | ||
| Carcinoid tumor | Papillomas | ||
| Salivary gland tumors | Adenomas | ||
| Target | Inhibitor | Line of Treatment | Indication | Current-FDA Approval Year | Clinical Trial-Based Approval |
|---|---|---|---|---|---|
| EGFR | Gefitinib | first-line | metastatic NSCLC with exon 19 deletions or exon 21 (L858R) substitution mutations | 2015 | IFUM (NCT01203917) |
| Erlotinib | first- or second-line | metastatic NSCLC with exon 19 deletions or exon 21 (L858R) substitution mutations | 2016 | IUNO trial (NCT01328951) | |
| Afatinib | first- or second-line treatment | metastatic NSCLC with non-resistant EGFR mutations; metastatic, squamous NSCLC progressing after platinum-based chemotherapy | 2018 | LUX-Lung 2 (NCT00525148), LUX-Lung 3 (NCT00949650), and LUX-Lung 6 (NCT01121393) | |
| Osimertinib | first-line or second- treatment | metastatic NSCLC with detected exon 19 deletions or exon 21 L858R mutations or T790M mutation-positive with disease progression on EGFR TKI therapy | 2018 | FLAURA, (NCT02296125) | |
| Dacomitinib | first-line | metastatic NSCLC with detected exon 19 deletions or exon 21 (L858R) substitution mutations | 2018 | ARCHER 1050 (NCT01774721) | |
| ALK | Crizotinib | first-line | locally advanced or metastatic NSCLC | 2011 | PROFILE 1005 (NCT00932451) |
| Ceritinib | first- or second-line | metastatic NSCLC | 2017 | ASCEND-4 (NCT01828099) | |
| Alectinib | first-line | 2017 | ALEX (NCT02075840) | ||
| Brigatinib | second-line | 2017 | ALTA (NCT02094573) | ||
| Lorlatinib | second- or third line | metastatic NSCLC after progression on other ALK TKI therapy | 2018 | Study B7461001 (NCT01970865) | |
| ROS1 | Crizotinib | first-line | metastatic NSCLC | 2016 | PROFILE 1001 (NCT00585195) |
| Entrectinib | first-line | 2019 | STARTRK-1 (NCT02097810) STARTRK-2 (NCT02568267) | ||
| NTRK | Larotrectinib | first-line | solid tumors with detected NTRK gene fusion without a known acquired resistance mutation, independent of tumor origin | 2018 | LOXO-TRK-14001 (NCT02122913), SCOUT (NCT02637687), NAVIGATE (NCT02576431) |
| Entrectinib | first-line | 2019 | STARTRK-1 (NCT02097810) STARTRK-2 (NCT02568267) | ||
| RET | Pralsetinib | first-line | metastatic NSCLC | 2020 | ARROW (NCT03037385) |
| Selpercatinib | first-line | metastatic NSCLC | 2020 | LIBRETTO-001 (NCT03157128) | |
| MET | Capmatinib | first-line | metastatic NSCLC with specific mutations (exon 14 skipping) | 2020 | GEOMETRY (NCT02414139) |
| Tepotinib | first-line | 2021 | VISION (NCT02864992) |
| Checkpoint Inhibitor | Target | Line of Treatment | Indications | Clinical Trial-Based Approval | FDA Approval Year |
|---|---|---|---|---|---|
| Nivolumab | PD-1 | second-line | metastatic squamous NSCLC after chemotherapy; | CheckMate 017 (NCT01642004) | 2015 |
| second-line | extension to non-squamous NSCLC; | CheckMate 057 (NCT01673867) | |||
| Pembrolizumab | PD-1 | first-line | metastatic NSCLC; with no EGFR or ALK mutation; TPS ≥ 50%; | KEYNOTE−024 (NCT02142738) | 2016 |
| second-line | progression after chemotherapy or TKI in metastatic NSCLC; with TPS ≥ 1%; | KEYNOTE-010 (NCT01905657) | |||
| first-line | unresectable stage III or metastatic NSCLC; no possible definitive chemoradiation; with no EGFR or ALK mutation; TPS ≥ 1%; | KEYNOTE-042 (NCT02220894) | 2019 | ||
| Atezolizumab | PDL-1 | second-line | metastatic NSCLC with progression on/after chemotherapy or TKIs; | OAK (NCT02008227) POLAR (NCT01903993) | 2016 |
| first-line | combined with chemotherapy; metastatic non-squamous NSCLC; with no EGFR or ALK mutation; | IMpower150 (NCT02366143) | 2018 | ||
| Durvalumab | PDL-1 | second-line | unresectable Stage III NSCLC; with no progression after chemoradiation therapy; | PACIFIC (NCT02125461) | 2018 |
| Ipilimumab | CTLA-4 | first-line | only in the combination with nivolumab; metastatic NSCLC; with no EGFR or ALK mutation; TPS ≥ 1%; | CheckMate 227 (NCT02477826) | 2020 |
| Cemiplimab | PD-1 | first-line | advanced NSCLC; TPS ≥ 50% | EMPOWER-Lung 1 (NCT03088540) | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodak, O.; Peris-Díaz, M.D.; Olbromski, M.; Podhorska-Okołów, M.; Dzięgiel, P. Current Landscape of Non-Small Cell Lung Cancer: Epidemiology, Histological Classification, Targeted Therapies, and Immunotherapy. Cancers 2021, 13, 4705. https://doi.org/10.3390/cancers13184705
Rodak O, Peris-Díaz MD, Olbromski M, Podhorska-Okołów M, Dzięgiel P. Current Landscape of Non-Small Cell Lung Cancer: Epidemiology, Histological Classification, Targeted Therapies, and Immunotherapy. Cancers. 2021; 13(18):4705. https://doi.org/10.3390/cancers13184705
Chicago/Turabian StyleRodak, Olga, Manuel David Peris-Díaz, Mateusz Olbromski, Marzenna Podhorska-Okołów, and Piotr Dzięgiel. 2021. "Current Landscape of Non-Small Cell Lung Cancer: Epidemiology, Histological Classification, Targeted Therapies, and Immunotherapy" Cancers 13, no. 18: 4705. https://doi.org/10.3390/cancers13184705
APA StyleRodak, O., Peris-Díaz, M. D., Olbromski, M., Podhorska-Okołów, M., & Dzięgiel, P. (2021). Current Landscape of Non-Small Cell Lung Cancer: Epidemiology, Histological Classification, Targeted Therapies, and Immunotherapy. Cancers, 13(18), 4705. https://doi.org/10.3390/cancers13184705