
Metabolic Plasticity and Combinatorial Radiosensitisation Strategies in Human Papillomavirus-Positive Squamous Cell Carcinoma of the Head and Neck Cell Lines

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Lines and Culture Conditions
2.2. Western Blotting
2.3. Metabolic Profiling Studies
2.4. Clonogenic Survival Assays
3. Results
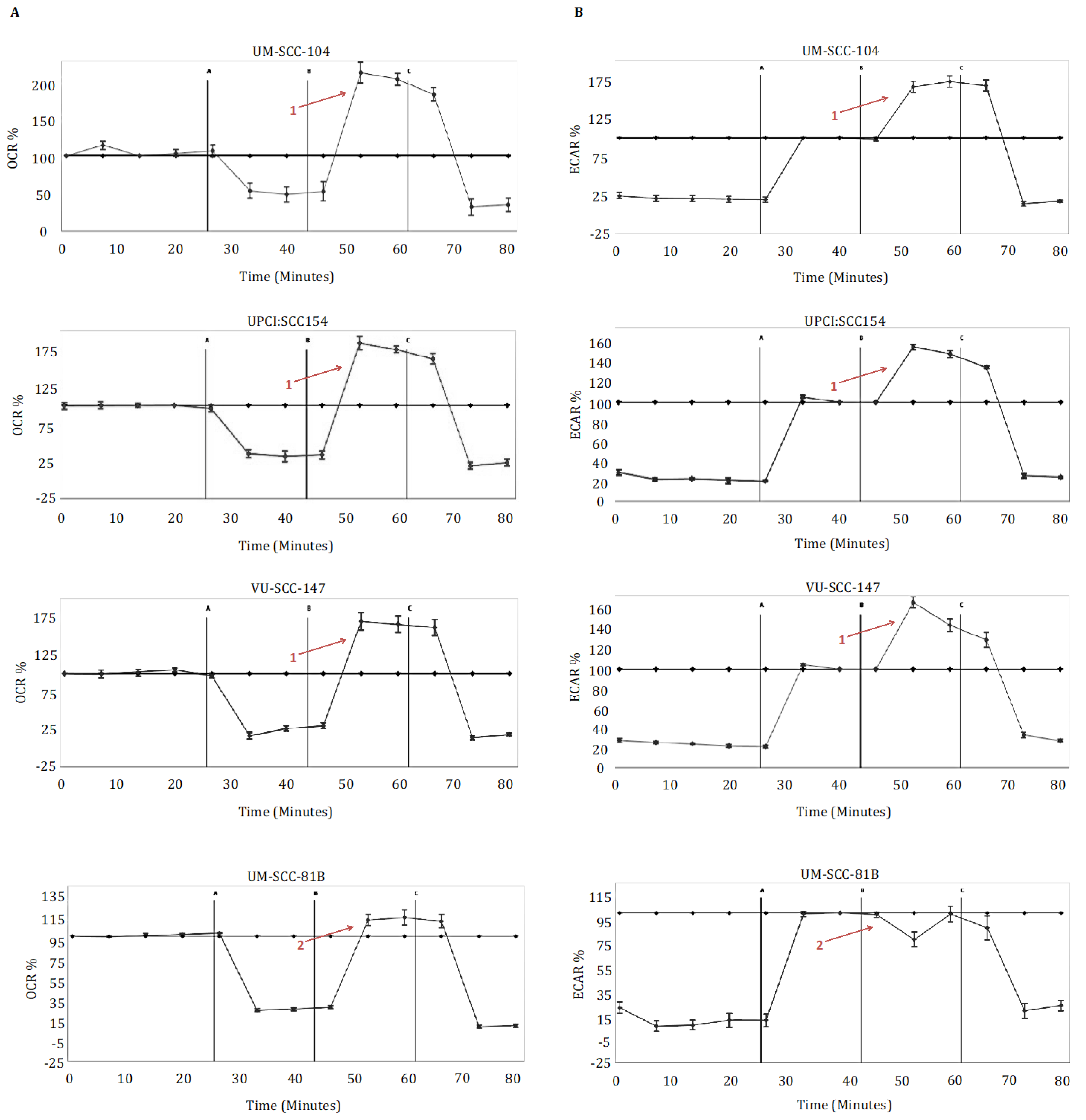
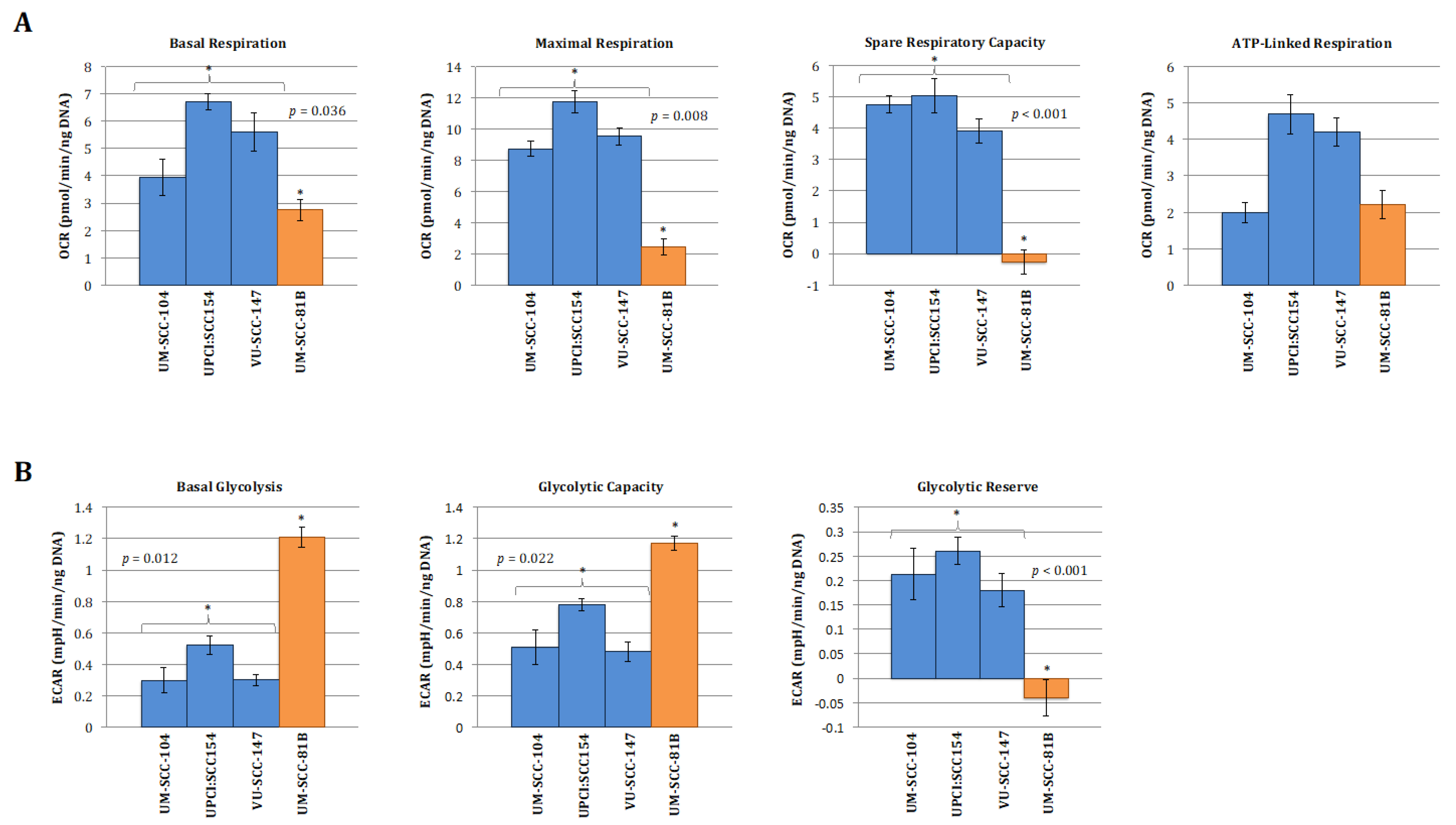
3.1. HPV-Positive SCCHN Cells Maintain Metabolic Diversity
3.2. HPV-Positive SCCHN Metabolic Diversity May Be Related to Residual Wild-Type p53 Function
3.3. HPV-Positive SCCHN Cells Require a Combinatorial Anti-Metabolic Therapeutic Approach
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.S.I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide. Available online: http://globocan.iarc.fr (accessed on 1 February 2021).
- Lewin, F.; Norell, S.E.; Johansson, H.; Gustavsson, P.; Wennerberg, J.; Biörklund, A.; Rutqvist, L.E. Smoking tobacco, oral snuff, and alcohol in the etiology of squamous cell carcinoma of the head and neck: A population-based case-referent study in Sweden. Cancer 1998, 82, 1367–1375. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Human papillomaviruses. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC (International Agency for Research on Cancer): Lyon, France, 2007; Volume 90, pp. 1–670. [Google Scholar]
- National Cancer Institute. Surveillance Epidemiology and End Results. Available online: http://seer.cancer.gov/faststats/selections.php-Output (accessed on 1 February 2021).
- National Cancer Institute Network. Profile of Head and Neck Cancers. Available online: http://library.ncin.org.uk/docs/100504-OCIUHead_and_Neck_Profi-les.pdf (accessed on 1 February 2021).
- Ramqvist, T.; Dalianis, T. An epidemic of oropharyngeal squamous cell carcinoma (OSCC) due to human papillomavirus (HPV) infection and aspects of treatment and prevention. Anticancer Res. 2011, 31, 1515–1519. [Google Scholar]
- Mehanna, H.; Beech, T.; Nicholson, T.; El-Hariry, I.; McConkey, C.; Paleri, V.; Roberts, S. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer--systematic review and meta-analysis of trends by time and region. Head Neck 2013, 35, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licitra, L.; Perrone, F.; Bossi, P.; Suardi, S.; Mariani, L.; Artusi, R.; Oggionni, M.; Rossini, C.; Cantù, G.; Squadrelli, M.; et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. J. Clin. Oncol. 2006, 24, 5630–5636. [Google Scholar] [CrossRef] [Green Version]
- Mehanna, H.; Olaleye, O.; Licitra, L. Oropharyngeal cancer—Is it time to change management according to human papilloma virus status? Curr. Opin. Otolaryngol. Head Neck Surg. 2012, 20, 120–124. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandulache, V.C.; Skinner, H.D.; Ow, T.J.; Zhang, A.; Xia, X.; Luchak, J.M.; Wong, L.C.; Pickering, C.R.; Zhou, G.; Myers, J.N. Individualizing antimetabolic treatment strategies for head and neck squamous cell carcinoma based on TP53 mutational status. Cancer 2012, 118, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Sandulache, V.C.; Ow, T.J.; Pickering, C.R.; Frederick, M.J.; Zhou, G.; Fokt, I.; Davis-Malesevich, M.; Priebe, W.; Myers, J.N. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer 2011, 117, 2926–2938. [Google Scholar] [CrossRef]
- Wilkie, M.D.; Anaam, E.A.; Lau, A.S.; Rubbi, C.P.; Jones, T.M.; Boyd, M.T.; Vlatković, N. TP53 mutations in head and neck cancer cells determine the Warburg phenotypic switch creating metabolic vulnerabilities and therapeutic opportunities for stratified therapies. Cancer Lett. 2020, 478, 107–121. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.L.; Puricelli, M.D.; Stack, M.S. Virology and molecular pathogenesis of HPV (human papillomavirus)-associated oropharyngeal squamous cell carcinoma. Biochem. J. 2012, 443, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Olthof, N.C.; Huebbers, C.U.; Kolligs, J.; Henfling, M.; Ramaekers, F.C.S.; Cornet, I.; van Lent-Albrechts, J.A.; Stegmann, A.P.A.; Silling, S.; Wieland, U.; et al. Viral load, gene expression and mapping of viral integration sites in HPV16-associated HNSCC cell lines. Int. J. Cancer 2015, 136, E207–E218. [Google Scholar] [CrossRef] [Green Version]
- Greaney-Davies, F.S.T.; Risk, J.M.; Robinson, M.; Liloglou, T.; Shaw, R.J.; Schache, A.G. Essential characterisation of human papillomavirus positive head and neck cancer cell lines. Oral. Oncol. 2020, 103, 104613. [Google Scholar] [CrossRef]
- Bradford, C.R.; Zhu, S.; Ogawa, H.; Ogawa, T.; Ubell, M.; Narayan, A.; Johnson, G.; Wolf, G.T.; Fisher, S.G.; Carey, T.E. p53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck 2003, 25, 654–661. [Google Scholar] [CrossRef]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Head and neck squamous cell carcinoma cell lines: Established models and rationale for selection. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.L.; Hauff, S.J.; Owen, J.H.; Graham, M.P.; Czerwinski, M.J.; Je Park, J.; Walline, H.; Papagerakis, S.; Stoerker, J.; McHugh, J.B.; et al. UM-SCC-104: A new human papillomavirus-16-positive cancer stem cell-containing head and neck squamous cell carcinoma cell line. Head Neck 2012, 34, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- White, J.S.; Weissfeld, J.L.; Ragin, C.C.R.; Rossie, K.M.; Martin, C.L.; Shuster, M.; Ishwad, C.S.; Law, J.C.; Myers, E.N.; Johnson, J.T.; et al. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral. Oncol. 2007, 43, 701–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenbergen, R.D.; Hermsen, M.A.; Walboomers, J.M.; Joenje, H.; Arwert, F.; Meijer, C.J.; Snijders, P.J. Integrated human papillomavirus type 16 and loss of heterozygosity at 11q22 and 18q21 in an oral carcinoma and its derivative cell line. Cancer Res. 1995, 55, 5465–5471. [Google Scholar]
- Brady, M.; Vlatkovic, N.; Boyd, M.T. Regulation of p53 and MDM2 activity by MTBP. Mol. Cell. Biol. 2005, 25, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Nair, T.S.; Thomas, T.B.; Yang, L.; Kakaraparthi, B.N.; Morris, A.C.; Clark, A.M.; Campredon, L.P.; Brouwer, A.F.; Eisenberg, M.C.; Meza, R.; et al. Characteristics of head and neck squamous cell carcinoma cell Lines reflect human tumor biology independent of primary etiologies and HPV status. Transl. Oncol. 2020, 13, 100808. [Google Scholar] [CrossRef]
- Bullenkamp, J.; Raulf, N.; Ayaz, B.; Walczak, H.; Kulms, D.; Odell, E.; Thavaraj, S.; Tavassoli, M. Bortezomib sensitises TRAIL-resistant HPV-positive head and neck cancer cells to TRAIL through a caspase-dependent, E6-independent mechanism. Cell Death Dis. 2014, 5, e1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, A.N.; Drury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 1957, 224, 963–969. [Google Scholar] [CrossRef]
- Chen, W.; Gueron, M. The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: Characterization by 31P NMR and metabolic implications. Biochimie 1992, 74, 867–873. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef]
- De Berardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Giessl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, M.D.; Lau, A.S.; Vlatkovic, N.; Jones, T.M.; Boyd, M.T. Metabolic signature of squamous cell carcinoma of the head and neck: Consequences of TP53 mutation and therapeutic perspectives. Oral. Oncol. 2018, 83, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [CrossRef]
- Krupar, R.; Robold, K.; Gaag, D.; Spanier, G.; Kreutz, M.; Renner, K.; Hellerbrand, C.; Hofstaedter, F.; Bosserhoff, A.K. Immunologic and metabolic characteristics of HPV-negative and HPV-positive head and neck squamous cell carcinomas are strikingly different. Virchows Arch. 2014, 465, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.C.; Woo, J.; Moutasim, K.; Mellone, M.; Frampton, S.J.; Mead, A.; Ahmed, W.; Wood, O.; Robinson, H.; Ward, M.; et al. HPV, tumour metabolism and novel target identification in head and neck squamous cell carcinoma. Br. J. Cancer 2019, 120, 356–367. [Google Scholar] [CrossRef]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [Green Version]
- Bernard, X.; Robinson, P.; Nomine, Y.; Masson, M.; Charbonnier, S.; Ramirez-Ramos, J.R.; Deryckere, F.; Trave, G.; Orfanoudakis, G. Proteasomal Degradation of p53 by Human Papillomavirus E6 Oncoprotein Relies on the Structural Integrity of p53 Core Domain. PLoS ONE 2011, 6, e25981. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Pol, S.V.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralser, M.; Wamelink, M.M.; Struys, E.A.; Joppich, C.; Krobitsch, S.; Jakobs, C.; Lehrach, H. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc. Natl. Acad. Sci. USA 2008, 105, 17807–17811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Mehanna, H.; Evans, M.; Beasley, M.; Chatterjee, S.; Dilkes, M.; Homer, J.; O’Hara, J.; Robinson, M.; Shaw, R.; Sloan, P. Oropharyngeal cancer: United Kingdom National Multidisciplinary Guidelines. J. Laryngol. Otol. 2016, 130, S90–S96. [Google Scholar] [CrossRef] [PubMed]
- Dwarakanath, B.S.; Singh, D.; Banerji, A.K.; Sarin, R.; Venkataramana, N.K.; Jalali, R.; Vishwanath, P.N.; Mohanti, B.K.; Tripathi, R.P.; Kalia, V.K.; et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: Present status and future prospects. J. Cancer Res. Ther. 2009, 5 (Suppl. 1), S21–S26. [Google Scholar] [CrossRef]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkie, M.D.; Anaam, E.A.; Lau, A.S.; Rubbi, C.P.; Vlatkovic, N.; Jones, T.M.; Boyd, M.T. Metabolic Plasticity and Combinatorial Radiosensitisation Strategies in Human Papillomavirus-Positive Squamous Cell Carcinoma of the Head and Neck Cell Lines. Cancers 2021, 13, 4836. https://doi.org/10.3390/cancers13194836
Wilkie MD, Anaam EA, Lau AS, Rubbi CP, Vlatkovic N, Jones TM, Boyd MT. Metabolic Plasticity and Combinatorial Radiosensitisation Strategies in Human Papillomavirus-Positive Squamous Cell Carcinoma of the Head and Neck Cell Lines. Cancers. 2021; 13(19):4836. https://doi.org/10.3390/cancers13194836
Chicago/Turabian StyleWilkie, Mark D., Emad A. Anaam, Andrew S. Lau, Carlos P. Rubbi, Nikolina Vlatkovic, Terence M. Jones, and Mark T. Boyd. 2021. "Metabolic Plasticity and Combinatorial Radiosensitisation Strategies in Human Papillomavirus-Positive Squamous Cell Carcinoma of the Head and Neck Cell Lines" Cancers 13, no. 19: 4836. https://doi.org/10.3390/cancers13194836
APA StyleWilkie, M. D., Anaam, E. A., Lau, A. S., Rubbi, C. P., Vlatkovic, N., Jones, T. M., & Boyd, M. T. (2021). Metabolic Plasticity and Combinatorial Radiosensitisation Strategies in Human Papillomavirus-Positive Squamous Cell Carcinoma of the Head and Neck Cell Lines. Cancers, 13(19), 4836. https://doi.org/10.3390/cancers13194836