

Morphological Features Extracted by AI Associated with Spatial Transcriptomics in Prostate Cancer

 , , , , , , , , , and
, , , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Prostate Cancer Sections and Spatially Resolved Gene Expression
2.2. Penultimate Layer Activations Are Robust on New Datasets
2.3. Deep Morphological Features Identify Tissue Heterogeneity
2.4. Cancer Sub-Regions Relate to Manual Annotations
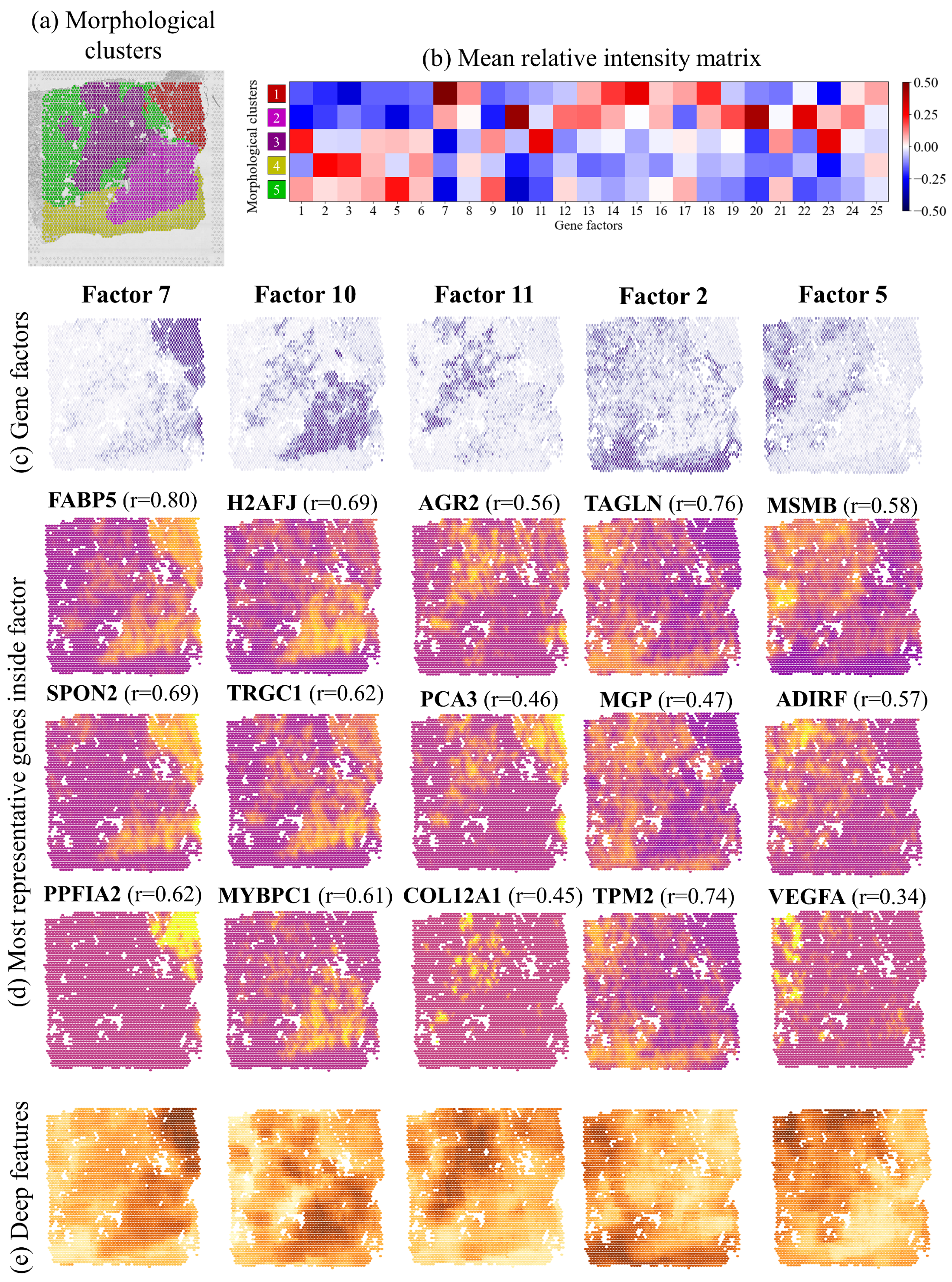
2.5. Morphological Clusters Relate to Gene Expression Factors
2.6. Spatial Expression of Individual Genes Predicted by Morphological Features
2.7. Network Pre-Trained on Biological Images Find More Relevant Regions
3. Discussion
4. Materials and Methods
4.1. Prostatectomy Sections with Spatially-Resolved Gene Expression
4.2. Image Acquisition and Registration
4.3. Deep Morphological Feature Extraction
4.4. Visualization and Clustering
4.5. Comparing Morphological Sub-Regions to Manual Annotations
4.6. Gene Factor Analysis and Morphological Sub-Regions
4.7. Gene Expression Correlation
4.8. Comparison with Other Clustering Algorithms and ImageNet Pre-Trained Network
4.9. Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| H&E | Hematoxylin and eosin |
| ST | Spatial transcriptomics |
| ISUP | International Society of Urological Pathology |
| CNN | Convolutional neural network |
| AI | Artificial intelligence |
| RGB | Red, green, and blue |
| FDR | False discovery rate |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef]
- Singh, D.; Febbo, P.G.; Ross, K.; Jackson, D.G.; Manola, J.; Ladd, C.; Tamayo, P.; Renshaw, A.A.; D’Amico, A.V.; Richie, J.P.; et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 2002, 1, 203–209. [Google Scholar] [CrossRef]
- Gleason, D.F. Histologic grading of prostate cancer: A perspective. Hum. Pathol. 1992, 23, 273–279. [Google Scholar] [CrossRef]
- Rubin, M.A.; Girelli, G.; Demichelis, F. Genomic correlates to the newly proposed grading prognostic groups for prostate cancer. Eur. Urol. 2016, 69, 557–560. [Google Scholar] [CrossRef]
- Van Leenders, G.J.; Van Der Kwast, T.H.; Grignon, D.J.; Evans, A.J.; Kristiansen, G.; Kweldam, C.F.; Litjens, G.; McKenney, J.K.; Melamed, J.; Mottet, N.; et al. The 2019 International Society of Urological Pathology (ISUP) consensus conference on grading of prostatic carcinoma. Am. J. Surg. Pathol. 2020, 44, e87. [Google Scholar] [CrossRef]
- Stahl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstrahle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.; Berglund, E.; He, M.; Marklund, M.; Mirzazadeh, R.; Schultz, N.; Bergenstrahle, L.; Kvastad, L.; Andersson, A.; Bergenstrahle, J.; et al. The spatial landscape of clonal somatic mutations in benign and malignant tissue. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yoosuf, N.; Navarro, J.F.; Salmén, F.; Stahl, P.L.; Daub, C.O. Identification and transfer of spatial transcriptomics signatures for cancer diagnosis. Breast Cancer Res. 2020, 22, 6. [Google Scholar] [CrossRef] [PubMed]
- Acs, B.; Rantalainen, M.; Hartman, J. Artificial intelligence as the next step towards precision pathology. J. Intern. Med. 2020, 288, 62–81. [Google Scholar] [CrossRef]
- Tan, X.; Su, A.; Tran, M.; Nguyen, Q. SpaCell: Integrating tissue morphology and spatial gene expression to predict disease cells. Bioinformatics 2019, 36, 2293–2294. [Google Scholar] [CrossRef]
- Pham, D.T.; Tan, X.; Xu, J.; Grice, L.F.; Lam, P.Y.; Raghubar, A.; Vukovic, J.; Ruitenberg, M.J.; Nguyen, Q.H. stLearn: Integrating spatial location, tissue morphology and gene expression to find cell types, cell–cell interactions and spatial trajectories within undissociated tissues. bioRxiv 2020. [Google Scholar] [CrossRef]
- He, B.; Bergenstrahle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, A.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef]
- Wang, Y.; Kartasalo, K.; Valkonen, M.; Larsson, C.; Ruusuvuori, P.; Hartman, J.; Rantalainen, M. Predicting molecular phenotypes from histopathology images: A transcriptome-wide expression-morphology analysis in breast cancer. arXiv 2020, arXiv:2009.08917. [Google Scholar]
- Ström, P.; Kartasalo, K.; Olsson, H.; Solorzano, L.; Delahunt, B.; Berney, D.M.; Bostwick, D.G.; Evans, A.J.; Grignon, D.J.; Humphrey, P.A.; et al. Artificial intelligence for diagnosis and grading of prostate cancer in biopsies: A population-based, diagnostic study. Lancet Oncol. 2020, 21, 222–232. [Google Scholar] [CrossRef]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat.-Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Liu, X.; Chen, L.; Huang, H.; Lv, J.M.; Chen, M.; Qu, F.J.; Pan, X.W.; Li, L.; Yin, L.; Cui, X.G.; et al. High expression of PDLIM5 facilitates cell tumorigenesis and migration by maintaining AMPK activation in prostate cancer. Oncotarget 2017, 8, 98117. [Google Scholar] [CrossRef]
- Munkley, J.; McClurg, U.L.; Livermore, K.E.; Ehrmann, I.; Knight, B.; Mccullagh, P.; Mcgrath, J.; Crundwell, M.; Harries, L.W.; Leung, H.Y.; et al. The cancer-associated cell migration protein TSPAN1 is under control of androgens and its upregulation increases prostate cancer cell migration. Sci. Rep. 2017, 7, 5249. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Weremowicz, S.; Feng, B.; Gentleman, R.C.; Marks, J.R.; Gelman, R.; Brennan, C.; Polyak, K. Combined cDNA array comparative genomic hybridization and serial analysis of gene expression analysis of breast tumor progression. Cancer Res. 2006, 66, 4065–4078. [Google Scholar] [CrossRef] [PubMed]
- Acosta, N.; Zabaleta, J.; Varela, R.; Mesa, J.; Serrano, S.; Garay, J.; Baddoo, M.; Nataly, C.; Combita, A.L.; Sanabria-Salas, M.C. Abstract B69: Aggressiveness and Tumor Biology in Prostate Cancer Patients with and without Biochemical Recurrence. Tumor Biol. 2018, 27, B69. [Google Scholar]
- Bu, H.; Bormann, S.; Schäfer, G.; Horninger, W.; Massoner, P.; Neeb, A.; Lakshmanan, V.K.; Maddalo, D.; Nestl, A.; Sültmann, H.; et al. The anterior gradient 2 (AGR2) gene is overexpressed in prostate cancer and may be useful as a urine sediment marker for prostate cancer detection. Prostate 2011, 71, 575–587. [Google Scholar] [CrossRef]
- Huang, L.; Chen, K.; Cai, Z.P.; Chen, F.C.; Shen, H.Y.; Zhao, W.H.; Yang, S.J.; Chen, X.B.; Tang, G.X.; Lin, X. DEPDC1 promotes cell proliferation and tumor growth via activation of E2F signaling in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 490, 707–712. [Google Scholar] [CrossRef]
- Rauber, P.E.; Falcao, A.X.; Telea, A.C. Projections as visual aids for classification system design. Inf. Vis. 2018, 17, 282–305. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.R.; Beyan, O.; Zappa, A.; Costa, I.G.; Rebholz-Schuhmann, D.; Cochez, M.; Decker, S. Deep learning-based clustering approaches for bioinformatics. Briefings Bioinform. 2021, 22, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Bergenstråhle, J.; Asp, M.; Bergenstråhle, L.; Jurek, A.; Navarro, J.F.; Lundeberg, J. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 2020, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ciompi, F.; Geessink, O.; Bejnordi, B.E.; De Souza, G.S.; Baidoshvili, A.; Litjens, G.; Van Ginneken, B.; Nagtegaal, I.; Van Der Laak, J. The importance of stain normalization in colorectal tissue classification with convolutional networks. In Proceedings of the 2017 IEEE 14th International Symposium on Biomedical Imaging (ISBI 2017), Melbourne, VIC, Australia, 18–21 April 2017; pp. 160–163. [Google Scholar]
- Szegedy, C.; Vanhoucke, V.; Ioffe, S.; Shlens, J.; Wojna, Z. Rethinking the inception architecture for computer vision. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 2818–2826. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Goode, A.; Gilbert, B.; Harkes, J.; Jukic, D.; Satyanarayanan, M. OpenSlide: A vendor-neutral software foundation for digital pathology. J. Pathol. Inform. 2013, 4, 27. [Google Scholar] [PubMed]
- van der Walt, S.; Schönberger, J.L.; Nunez-Iglesias, J.; Boulogne, F.; Warner, J.D.; Yager, N.; Gouillart, E.; Yu, T.; The Scikit-Image Contributors. scikit-image: Image processing in Python. PeerJ 2014, 2, e453. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Solorzano, L.; Partel, G.; Wählby, C. TissUUmaps: Interactive visualization of large-scale spatial gene expression and tissue morphology data. Bioinformatics 2020, 36, 4363–4365. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chelebian, E.; Avenel, C.; Kartasalo, K.; Marklund, M.; Tanoglidi, A.; Mirtti, T.; Colling, R.; Erickson, A.; Lamb, A.D.; Lundeberg, J.; et al. Morphological Features Extracted by AI Associated with Spatial Transcriptomics in Prostate Cancer. Cancers 2021, 13, 4837. https://doi.org/10.3390/cancers13194837
Chelebian E, Avenel C, Kartasalo K, Marklund M, Tanoglidi A, Mirtti T, Colling R, Erickson A, Lamb AD, Lundeberg J, et al. Morphological Features Extracted by AI Associated with Spatial Transcriptomics in Prostate Cancer. Cancers. 2021; 13(19):4837. https://doi.org/10.3390/cancers13194837
Chicago/Turabian StyleChelebian, Eduard, Christophe Avenel, Kimmo Kartasalo, Maja Marklund, Anna Tanoglidi, Tuomas Mirtti, Richard Colling, Andrew Erickson, Alastair D. Lamb, Joakim Lundeberg, and et al. 2021. "Morphological Features Extracted by AI Associated with Spatial Transcriptomics in Prostate Cancer" Cancers 13, no. 19: 4837. https://doi.org/10.3390/cancers13194837
APA StyleChelebian, E., Avenel, C., Kartasalo, K., Marklund, M., Tanoglidi, A., Mirtti, T., Colling, R., Erickson, A., Lamb, A. D., Lundeberg, J., & Wählby, C. (2021). Morphological Features Extracted by AI Associated with Spatial Transcriptomics in Prostate Cancer. Cancers, 13(19), 4837. https://doi.org/10.3390/cancers13194837