
At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention

 ,
,  , and
, and 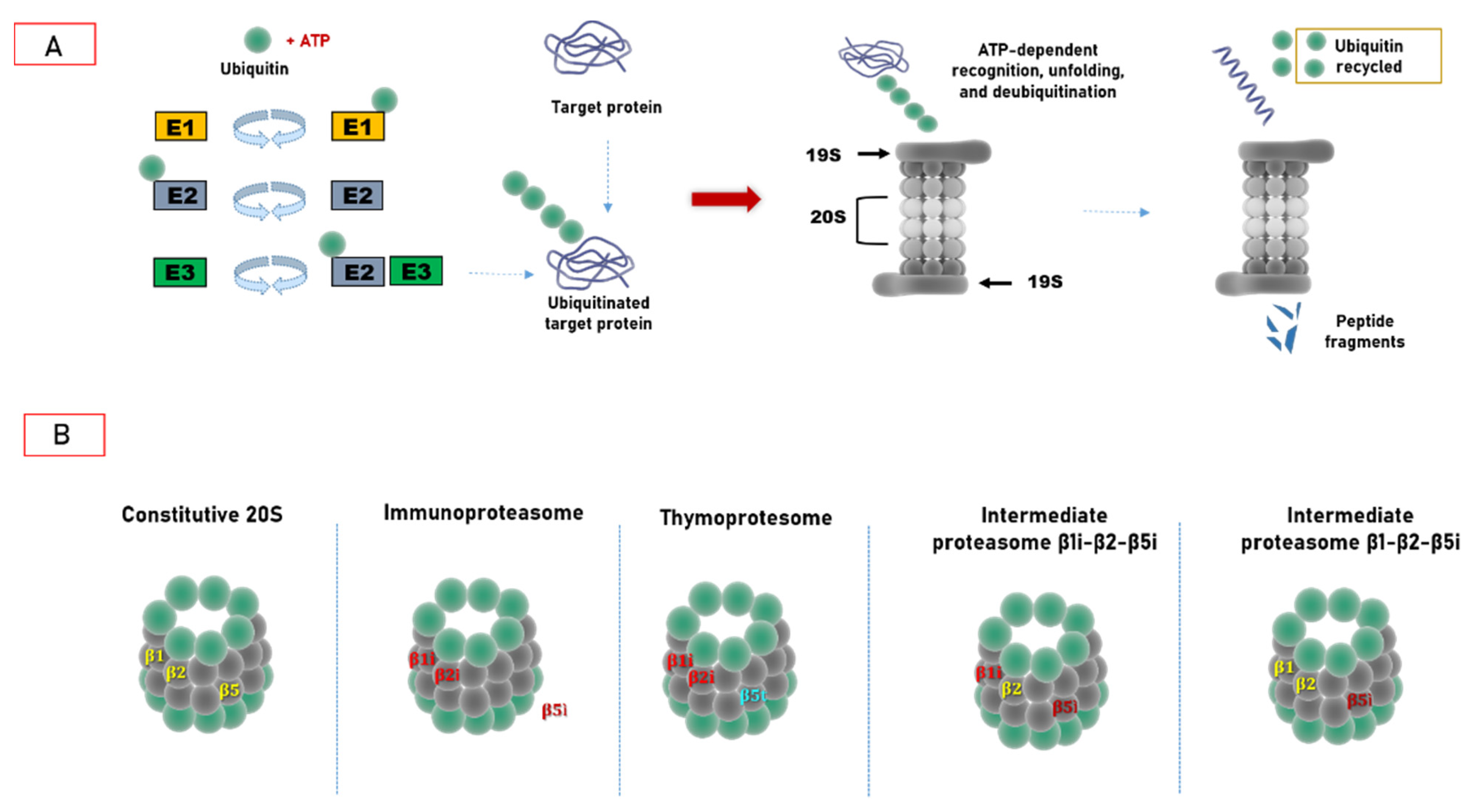{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
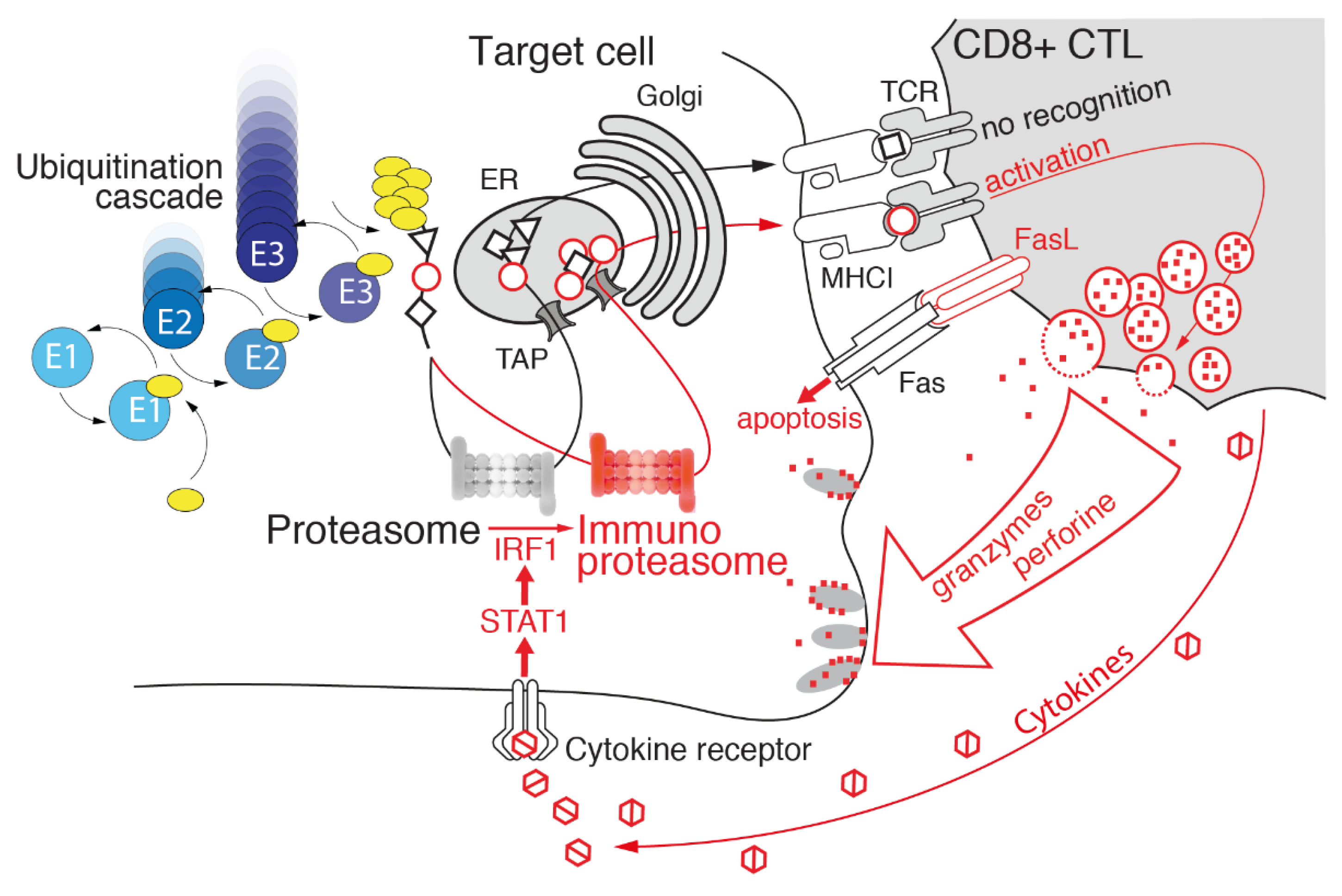
2. Ubiquitin–Proteasome System: Cellular Biocomputing Machinery
3. Hats off to Proteasome Variability
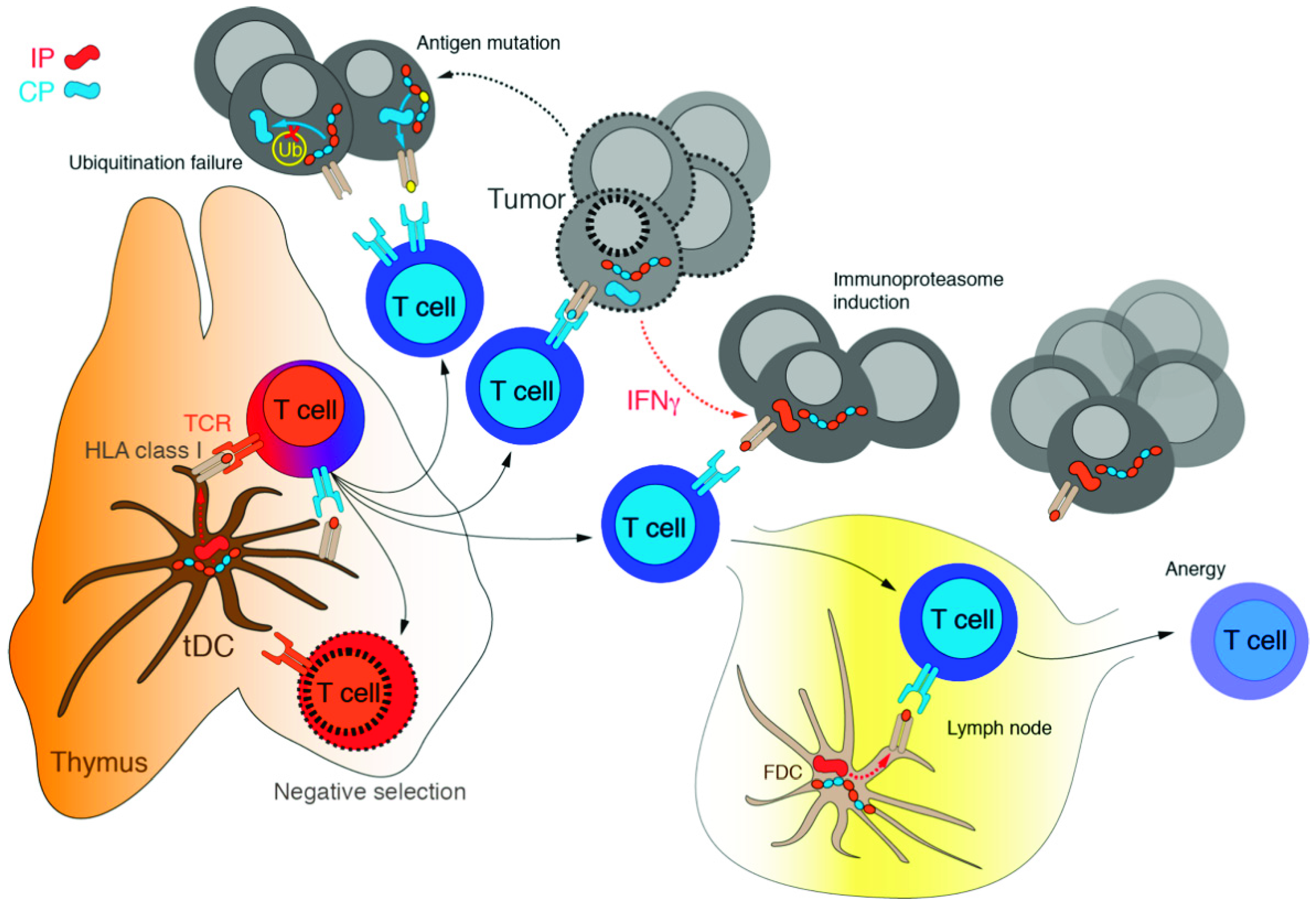
3.1. Immunoproteasome as a Specialized Apparatus of Self-Target Designation
3.2. Regulatory Particles Which Associate with Immunoproteasome
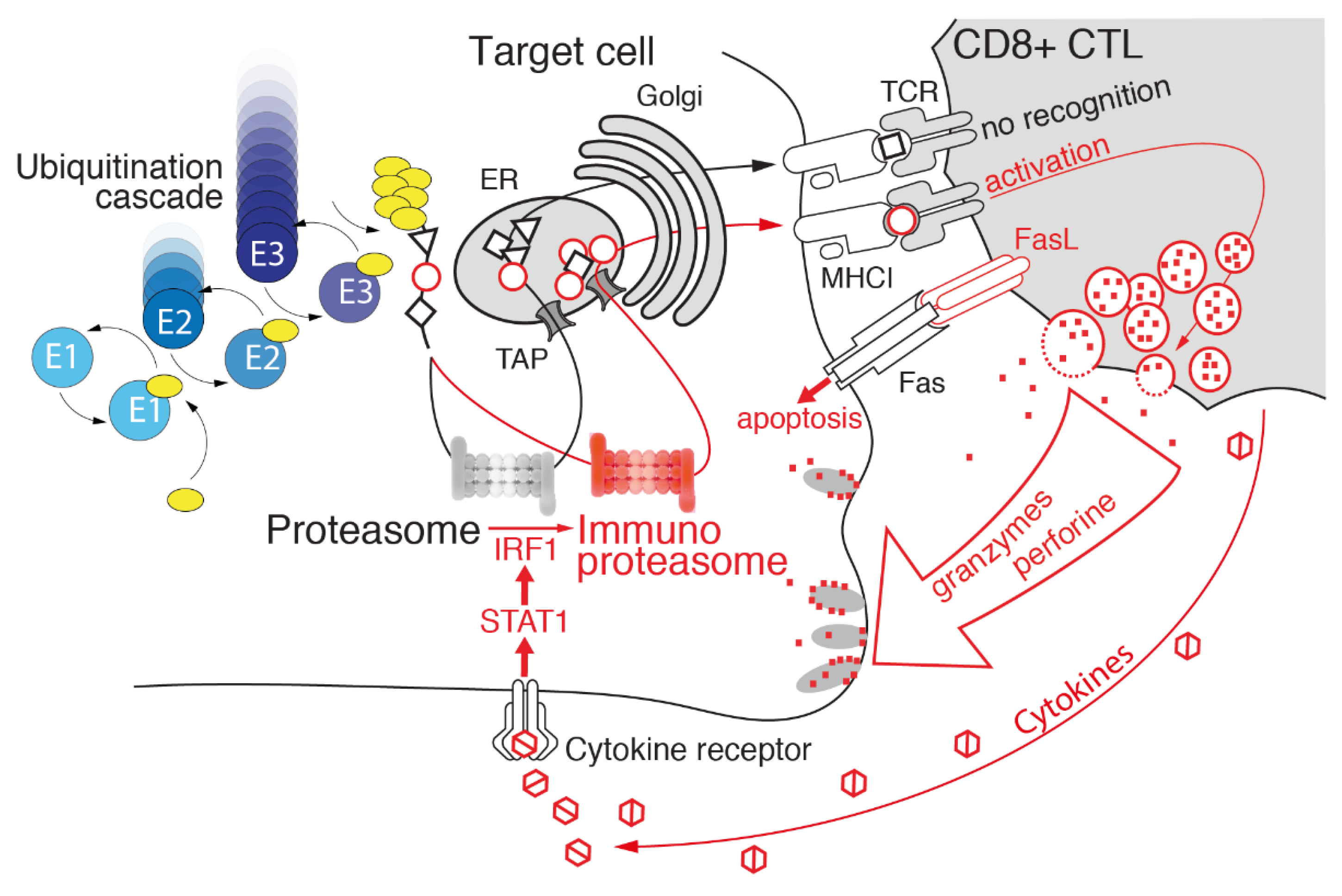
3.3. Immunoproteasome: Beyond Antigen Processing
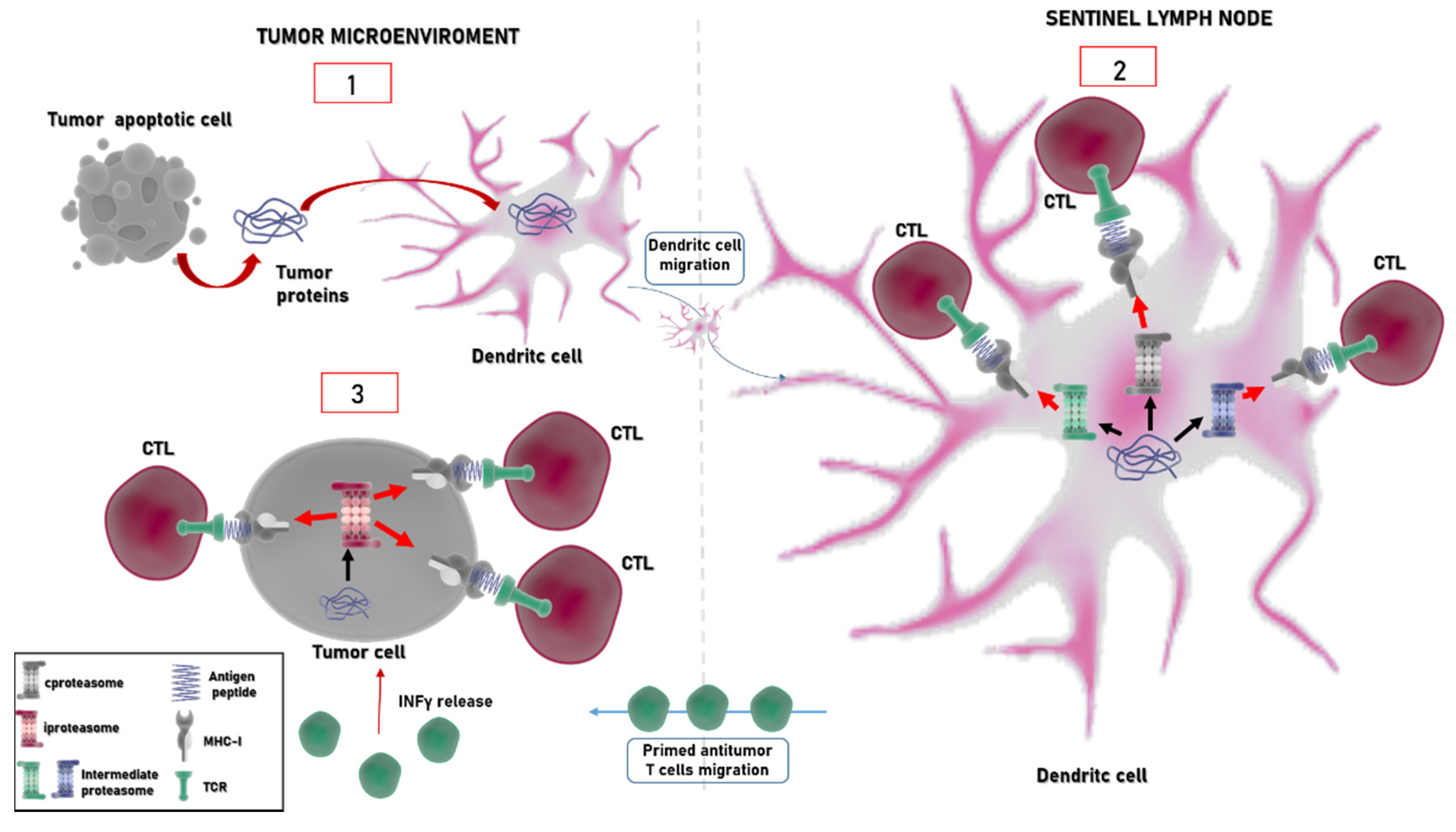
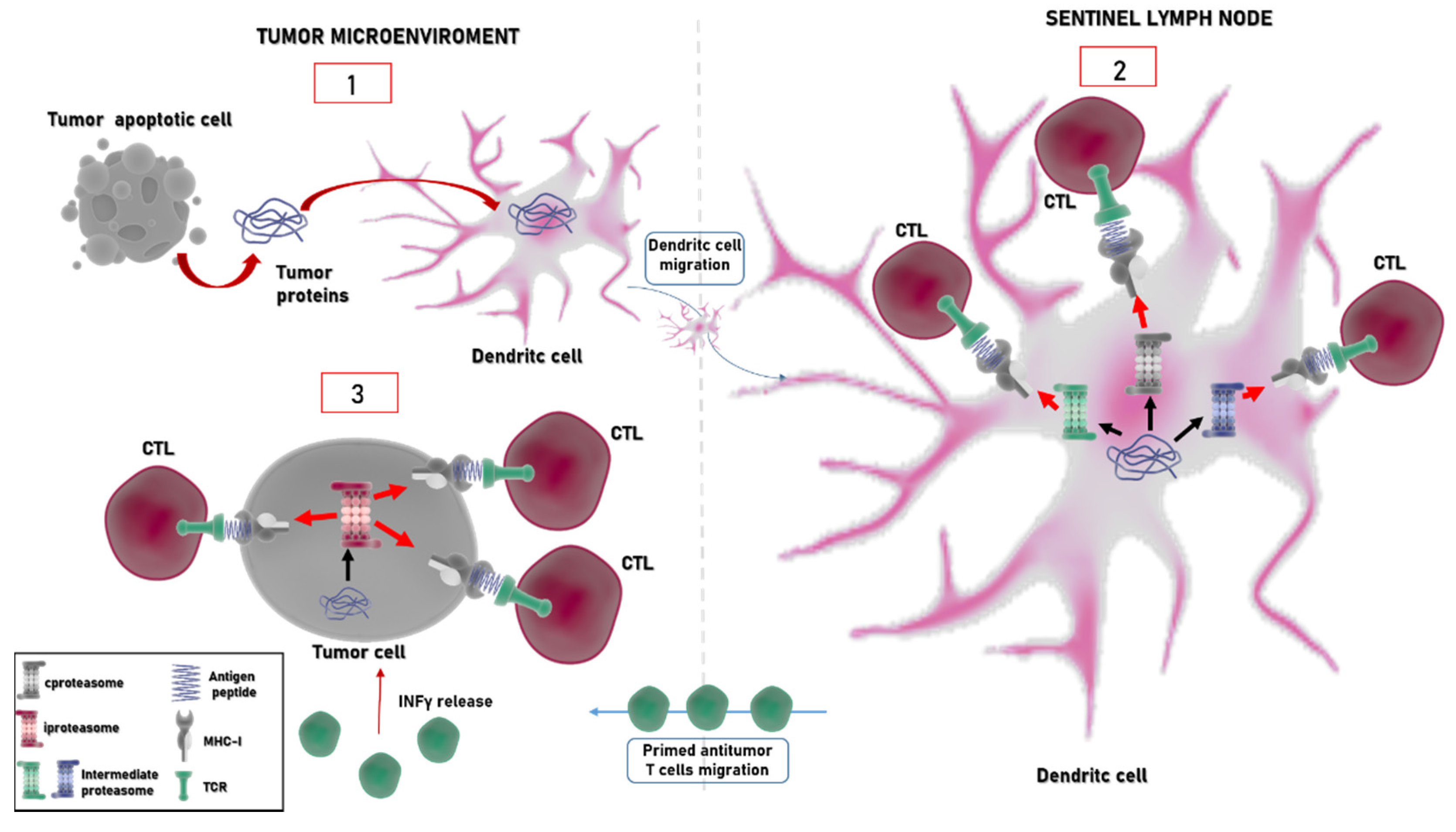
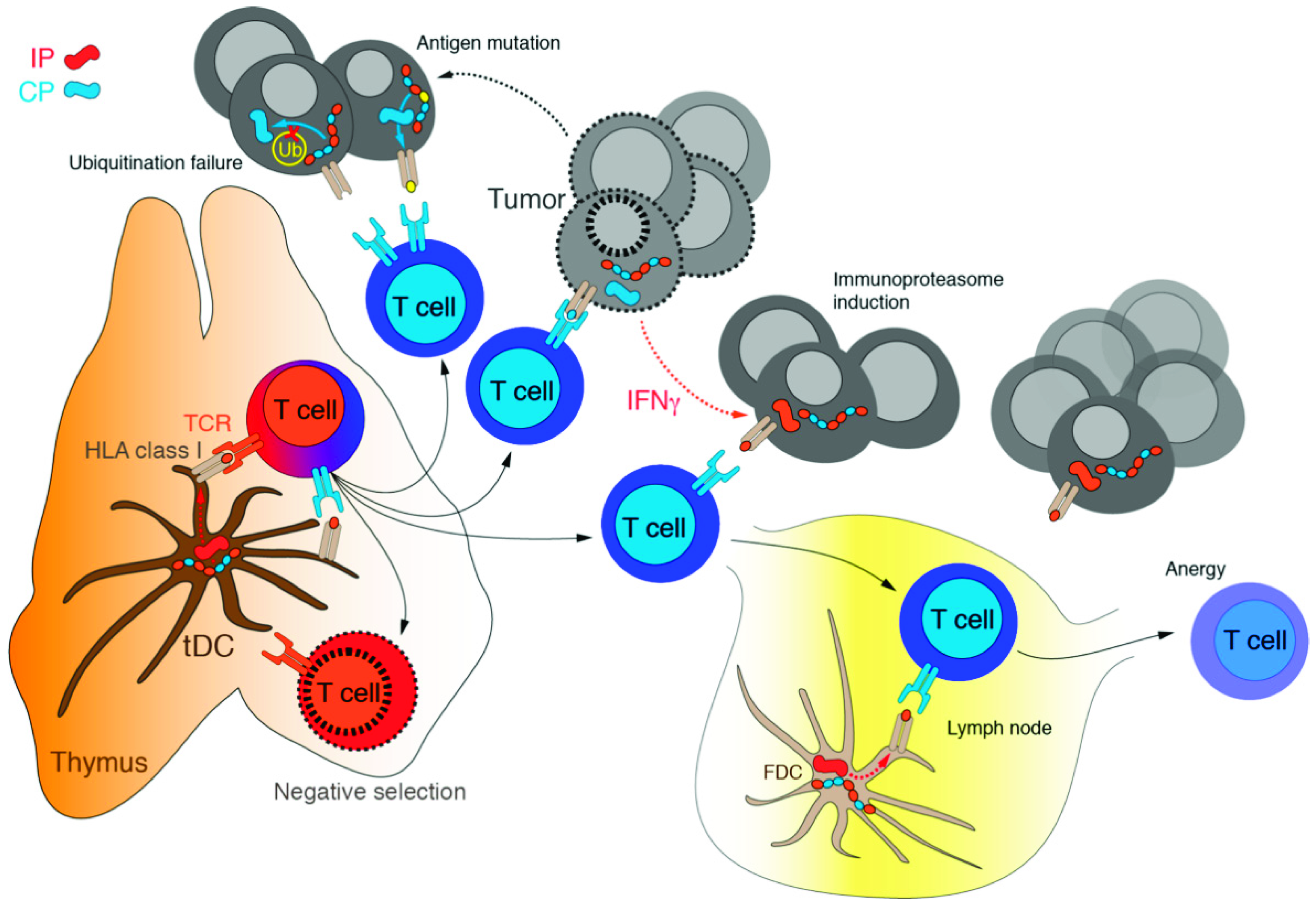
4. Immunoproteasome: An Emerging Target in Cancer
Immunoproteasome Inhibitors
5. Immunoproteasome and Immune Checkpoint Inhibitors: A Glance to the Future?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Spotlights on Immune Checkpoints
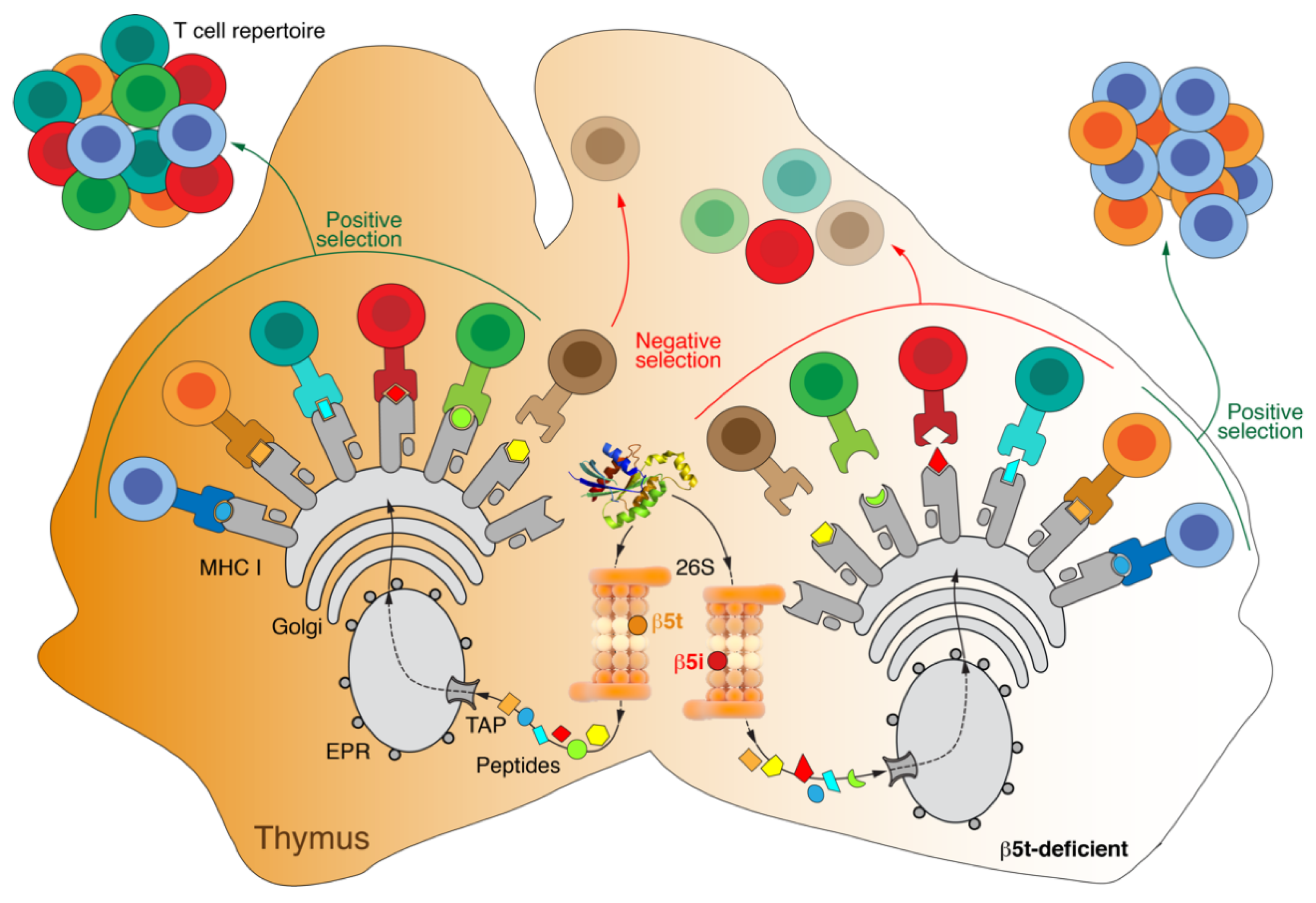
Appendix B. Thymoproteasome

References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer Immunotherapy: Opportunities and Challenges in the Rapidly Evolving Clinical Landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of Peripheral T-Cell Tolerance and Autoimmunity via the CTLA-4 and PD-1 Pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Decker, W.K.; da Silva, R.F.; Sanabria, M.H.; Angelo, L.S.; Guimarães, F.; Burt, B.M.; Kheradmand, F.; Paust, S. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front. Immunol. 2017, 8, 829. [Google Scholar] [CrossRef] [Green Version]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.M.; Lenardo, M.J. Development of Immune Checkpoint Therapy for Cancer. J. Exp. Med. 2019, 216, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chaudhary, N.; Garg, M.; Floudas, C.S.; Soni, P.; Chandra, A.B. Current Diagnosis and Management of Immune Related Adverse Events (IrAEs) Induced by Immune Checkpoint Inhibitor Therapy. Front. Pharm. 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.-S. Antigen Presentation and Tumor Immunogenicity in Cancer Immunotherapy Response Prediction. Elife 2019, 8, e49020. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1604958 (accessed on 21 June 2021).
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. Antigen-Processing Machinery Breakdown and Tumor Growth. Immunol. Today 2000, 21, 455–464. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Kubiniok, P.; Kovalchik, K.A.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project: A Roadmap to Predict and Treat Immune Diseases. Mol. Cell Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The Proteasome as a Druggable Target with Multiple Therapeutic Potentialities: Cutting and Non-Cutting Edges. Pharm. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef] [PubMed]
- Cascio, P. PA28γ: New Insights on an Ancient Proteasome Activator. Biomolecules 2021, 11, 228. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping Proteostasis at the Cellular, Tissue, and Organismal Level. J. Cell Biol. 2017, 216, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Kudriaeva, A.A.; Sokolov, A.V.; Belogurov, A.A.J. Stochastics of Degradation: The Autophagic-Lysosomal System of the Cell. Acta Nat. 2020, 12, 18–32. [Google Scholar] [CrossRef]
- Kudriaeva, A.; Kuzina, E.S.; Zubenko, O.; Smirnov, I.V.; Belogurov, A. Charge-Mediated Proteasome Targeting. FASEB J. 2019, 33, 6852–6866. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Kunjappu, M.J.; Hochstrasser, M. Assembly of the 20S Proteasome. Biochim. Biophys. Acta 2014, 1843, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete Subunit Architecture of the Proteasome Regulatory Particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein Ubiquitination Involving an E1-E2-E3 Enzyme Ubiquitin Thioester Cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Leestemaker, Y.; Ovaa, H. Tools to Investigate the Ubiquitin Proteasome System. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Windheim, M.; Peggie, M.; Cohen, P. Two Different Classes of E2 Ubiquitin-Conjugating Enzymes Are Required for the Mono-Ubiquitination of Proteins and Elongation by Polyubiquitin Chains with a Specific Topology. Biochem. J. 2008, 409, 723–729. [Google Scholar] [CrossRef]
- Bellia, F.; Lanza, V.; García-Viñuales, S.; Ahmed, I.M.M.; Pietropaolo, A.; Iacobucci, C.; Malgieri, G.; D’Abrosca, G.; Fattorusso, R.; Nicoletti, V.G.; et al. Ubiquitin Binds the Amyloid β Peptide and Interferes with Its Clearance Pathways. Chem. Sci. 2019, 10, 2732–2742. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A. The Unravelling of the Ubiquitin System. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New Insights into Ubiquitin E3 Ligase Mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Kudriaeva, A.A.; Livneh, I.; Baranov, M.S.; Ziganshin, R.H.; Tupikin, A.E.; Zaitseva, S.O.; Kabilov, M.R.; Ciechanover, A.; Belogurov, A.A., Jr. In-depth characterization of ubiquitin turnover in mammalian cells by fluorescence tracking. Cell Chem. Biol. 2021, 28, 1192–1205.e9. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharm. 2018, 9, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, D.; Sato, Y.; Ito, H.; Tokunaga, F. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. Int. J. Mol. Sci 2020, 21, 3381. [Google Scholar] [CrossRef] [PubMed]
- Pfoh, R.; Lacdao, I.K.; Saridakis, V. Deubiquitinases and the New Therapeutic Opportunities Offered to Cancer. Endocr. Relat. Cancer 2015, 22, T35–T54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S Proteasome from Yeast at 2.4 A Resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A Gated Channel into the Proteasome Core Particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Tanaka, K. The Proteasome: Overview of Structure and Functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Huber, R. Substrate Access and Processing by the 20S Proteasome Core Particle. Int. J. Biochem. Cell Biol. 2003, 35, 606–616. [Google Scholar] [CrossRef]
- Bajorek, M.; Glickman, M.H. Keepers at the Final Gates: Regulatory Complexes and Gating of the Proteasome Channel. Cell Mol. Life Sci. 2004, 61, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Religa, T.L.; Breuer, S.; Witt, S.; Kay, L.E. The Proteasome Antechamber Maintains Substrates in an Unfolded State. Nature 2010, 467, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Prado, M.A. The Proteasome and Its Network: Engineering for Adaptability. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of Proteasome Activity in Health and Disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.H.; de Poot, S.A.H.; Lee, J.H.; Kim, J.H.; Han, D.H.; Kim, Y.K.; Finley, D.; Lee, M.J. Open-Gate Mutants of the Mammalian Proteasome Show Enhanced Ubiquitin-Conjugate Degradation. Nat. Commun. 2016, 7, 10963. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Pathare, G.R.; Nagy, I.; Bohn, S.; Unverdorben, P.; Hubert, A.; Körner, R.; Nickell, S.; Lasker, K.; Sali, A.; Tamura, T.; et al. The Proteasomal Subunit Rpn6 Is a Molecular Clamp Holding the Core and Regulatory Subcomplexes Together. Proc. Natl. Acad. Sci. USA 2012, 109, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Pathare, G.R.; Nagy, I.; Śledź, P.; Anderson, D.J.; Zhou, H.-J.; Pardon, E.; Steyaert, J.; Förster, F.; Bracher, A.; Baumeister, W. Crystal Structure of the Proteasomal Deubiquitylation Module Rpn8-Rpn11. Proc. Natl. Acad. Sci. USA 2014, 111, 2984–2989. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The Proteasome 19S Cap and Its Ubiquitin Receptors Provide a Versatile Recognition Platform for Substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Cohen, S.; Ciechanover, A. Modification by Single Ubiquitin Moieties Rather than Polyubiquitination Is Sufficient for Proteasomal Processing of the P105 NF-KappaB Precursor. Mol. Cell 2009, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Shabek, N.; Herman-Bachinsky, Y.; Buchsbaum, S.; Lewinson, O.; Haj-Yahya, M.; Hejjaoui, M.; Lashuel, H.A.; Sommer, T.; Brik, A.; Ciechanover, A. The Size of the Proteasomal Substrate Determines Whether Its Degradation Will Be Mediated by Mono- or Polyubiquitylation. Mol. Cell 2012, 48, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Yau, R.; Rape, M. The Increasing Complexity of the Ubiquitin Code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Ju, D.; Xie, Y. Proteasomal Degradation of RPN4 via Two Distinct Mechanisms, Ubiquitin-Dependent and -Independent. J. Biol. Chem. 2004, 279, 23851–23854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, J.M.; Viktorova, E.G.; Pilipenko, E.V. Proteasomes Can Degrade a Significant Proportion of Cellular Proteins Independent of Ubiquitination. J. Mol. Biol. 2009, 386, 814–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, A.; Kuzina, E.; Kudriaeva, A.; Kononikhin, A.; Kovalchuk, S.; Surina, Y.; Smirnov, I.; Lomakin, Y.; Bacheva, A.; Stepanov, A.; et al. Ubiquitin-Independent Proteosomal Degradation of Myelin Basic Protein Contributes to Development of Neurodegenerative Autoimmunity. FASEB J. 2015, 29, 1901–1913. [Google Scholar] [CrossRef]
- Belogurov, A.; Kudriaeva, A.; Kuzina, E.; Smirnov, I.; Bobik, T.; Ponomarenko, N.; Kravtsova-Ivantsiv, Y.; Ciechanover, A.; Gabibov, A. Multiple Sclerosis Autoantigen Myelin Basic Protein Escapes Control by Ubiquitination during Proteasomal Degradation. J. Biol. Chem. 2014, 289, 17758–17766. [Google Scholar] [CrossRef] [Green Version]
- Raynes, R.; Pomatto, L.C.D.; Davies, K.J.A. Degradation of Oxidized Proteins by the Proteasome: Distinguishing between the 20S, 26S, and Immunoproteasome Proteolytic Pathways. Mol. Asp. Med. 2016, 50, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Shringarpure, R.; Grune, T.; Mehlhase, J.; Davies, K.J.A. Ubiquitin Conjugation Is Not Required for the Degradation of Oxidized Proteins by Proteasome. J. Biol. Chem. 2003, 278, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Morozov, A.V.; Karpov, V.L. Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbardella, D.; Tundo, G.R.; Sciandra, F.; Bozzi, M.; Gioia, M.; Ciaccio, C.; Tarantino, U.; Brancaccio, A.; Coletta, M.; Marini, S. Proteasome Activity Is Affected by Fluctuations in Insulin-Degrading Enzyme Distribution. PLoS ONE 2015, 10, e0132455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; et al. Multiple Functions of Insulin-Degrading Enzyme: A Metabolic Crosslight? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Tundo, G.R.; Coletta, A.; Marcoux, J.; Koufogeorgou, E.I.; Ciaccio, C.; Santoro, A.M.; Milardi, D.; Grasso, G.; Cozza, P.; et al. The Insulin-Degrading Enzyme Is an Allosteric Modulator of the 20S Proteasome and a Potential Competitor of the 19S. Cell Mol. Life Sci. 2018, 75, 3441–3456. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef]
- Dahlmann, B. Mammalian Proteasome Subtypes: Their Diversity in Structure and Function. Arch. Biochem. Biophys. 2016, 591, 132–140. [Google Scholar] [CrossRef]
- Kniepert, A.; Groettrup, M. The Unique Functions of Tissue-Specific Proteasomes. Trends. Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.-X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.-Y.; Ji, D.-Y.; Wang, G.-F.; Zhu, Q.-Q.; Song, W.; Yu, Y.; et al. Acetylation-Mediated Proteasomal Degradation of Core Histones during DNA Repair and Spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar] [CrossRef] [Green Version]
- Uechi, H.; Hamazaki, J.; Murata, S. Characterization of the Testis-Specific Proteasome Subunit A4s in Mammals. J. Biol. Chem. 2014, 289, 12365–12374. [Google Scholar] [CrossRef] [Green Version]
- Khor, B.; Bredemeyer, A.L.; Huang, C.-Y.; Turnbull, I.R.; Evans, R.; Maggi, L.B.; White, J.M.; Walker, L.M.; Carnes, K.; Hess, R.A.; et al. Proteasome Activator PA200 Is Required for Normal Spermatogenesis. Mol. Cell Biol. 2006, 26, 2999–3007. [Google Scholar] [CrossRef] [Green Version]
- Ustrell, V.; Pratt, G.; Gorbea, C.; Rechsteiner, M. Purification and Assay of Proteasome Activator PA200. Methods Enzym. 2005, 398, 321–329. [Google Scholar] [CrossRef]
- Toste Rêgo, A.; da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.; Wang, Y.; Yu, T.; Huang, Y.; Li, M.; Saeed, A.F.U.H.; Perčulija, V.; Li, D.; Xiao, J.; Wang, D.; et al. Cryo-EM Structures of the Human PA200 and PA200–20S Complex Reveal Regulation of Proteasome Gate Opening and Two PA200 Apertures. PLoS Biol. 2020, 18, e3000654. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Meul, T.; Meiners, S. Exploring the Proteasome System: A Novel Concept of Proteasome Inhibition and Regulation. Pharm. Ther. 2020, 211, 107526. [Google Scholar] [CrossRef]
- Bruggeman, J.W.; Koster, J.; Lodder, P.; Repping, S.; Hamer, G. Massive Expression of Germ Cell-Specific Genes Is a Hallmark of Cancer and a Potential Target for Novel Treatment Development. Oncogene 2018, 37, 5694–5700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-H, L.; Felipe-Medina, N.; Condezo, Y.B.; Garcia-Valiente, R.; Ramos, I.; Suja, J.A.; Barbero, J.L.; Roig, I.; Sánchez-Martín, M.; de Rooij, D.G.; et al. The PSMA8 Subunit of the Spermatoproteasome Is Essential for Proper Meiotic Exit and Mouse Fertility. PLoS Genet. 2019, 15, e1008316. [Google Scholar] [CrossRef] [Green Version]
- Cascio, P. PA28αβ: The Enigmatic Magic Ring of the Proteasome? Biomolecules 2014, 4, 566–584. [Google Scholar] [CrossRef] [Green Version]
- Morozov, A.V.; Karpov, V.L. Biological Consequences of Structural and Functional Proteasome Diversity. Heliyon 2018, 4, e00894. [Google Scholar] [CrossRef] [Green Version]
- Cascio, P.; Call, M.; Petre, B.M.; Walz, T.; Goldberg, A.L. Properties of the Hybrid Form of the 26S Proteasome Containing Both 19S and PA28 Complexes. EMBO J. 2002, 21, 2636–2645. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharm. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Martinez, C.K.; Monaco, J.J. Homology of Proteasome Subunits to a Major Histocompatibility Complex-Linked LMP Gene. Nature 1991, 353, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the Proteasome Block the Degradation of Most Cell Proteins and the Generation of Peptides Presented on MHC Class I Molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-Gamma Induces Different Subunit Organizations and Functional Diversity of Proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Boes, B.; Hengel, H.; Ruppert, T.; Multhaup, G.; Koszinowski, U.H.; Kloetzel, P.M. Interferon Gamma Stimulation Modulates the Proteolytic Activity and Cleavage Site Preference of 20S Mouse Proteasomes. J. Exp. Med. 1994, 179, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, J.; Brown, M.G.; Finley, D.; Monaco, J.J. MHC-Linked LMP Gene Products Specifically Alter Peptidase Activities of the Proteasome. Nature 1993, 365, 262–264. [Google Scholar] [CrossRef]
- Gaczynska, M.; Rock, K.L.; Goldberg, A.L. Role of Proteasomes in Antigen Presentation. Enzym. Protein 1993, 47, 354–369. [Google Scholar] [CrossRef]
- Griffin, T.A.; Nandi, D.; Cruz, M.; Fehling, H.J.; Kaer, L.V.; Monaco, J.J.; Colbert, R.A. Immunoproteasome Assembly: Cooperative Incorporation of Interferon Gamma (IFN-Gamma)-Inducible Subunits. J. Exp. Med. 1998, 187, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groettrup, M.; Standera, S.; Stohwasser, R.; Kloetzel, P.M. The Subunits MECL-1 and LMP2 Are Mutually Required for Incorporation into the 20S Proteasome. Proc. Natl. Acad. Sci. USA 1997, 94, 8970–8975. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K. Role of Proteasomes Modified by Interferon-Gamma in Antigen Processing. J. Leukoc Biol. 1994, 56, 571–575. [Google Scholar] [CrossRef]
- Heink, S.; Ludwig, D.; Kloetzel, P.-M.; Krüger, E. IFN-Gamma-Induced Immune Adaptation of the Proteasome System Is an Accelerated and Transient Response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Van den Eynde, B.J. Two Abundant Proteasome Subtypes That Uniquely Process Some Antigens Presented by HLA Class I Molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; van den Broek, M.; Schwarz, K.; de Giuli, R.; Diener, P.A.; Groettrup, M. Immunoproteasomes Largely Replace Constitutive Proteasomes during an Antiviral and Antibacterial Immune Response in the Liver. J. Immunol. 2001, 167, 6859–6868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groettrup, M.; Kirk, C.J.; Basler, M. Proteasomes in Immune Cells: More than Peptide Producers? Nat. Rev. Immunol. 2010, 10, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, J.; Guan, X.Q.; Kisselev, A.F.; Papasian, C.J.; Qureshi, A.A.; Morrison, D.C.; Van Way, C.W.; Vogel, S.N.; Qureshi, N. LPS-Induced Formation of Immunoproteasomes: TNF-α and Nitric Oxide Production Are Regulated by Altered Composition of Proteasome-Active Sites. Cell Biochem. Biophys. 2011, 60, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.-C.; Seifert, U.; Kato, T.; Rice, C.M.; Feinstone, S.M.; Kloetzel, P.-M.; Rehermann, B. Virus-Induced Type I IFN Stimulates Generation of Immunoproteasomes at the Site of Infection. J. Clin. Investig. 2006, 116, 3006–3014. [Google Scholar] [CrossRef] [Green Version]
- Johnston-Carey, H.K.; Pomatto, L.C.D.; Davies, K.J.A. The Immunoproteasome in Oxidative Stress, Aging, and Disease. Crit Rev. Biochem Mol. Biol. 2015, 51, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Höhn, T.J.A.; Grune, T. The Proteasome and the Degradation of Oxidized Proteins: Part III-Redox Regulation of the Proteasomal System. Redox Biol. 2014, 2, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, N.; Abi Habib, J.; Van den Eynde, B.J. Learning from the Proteasome How to Fine-Tune Cancer Immunotherapy. Trends Cancer 2017, 3, 726–741. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, K.; Kagawa, S.; Tamura, T.; Shimbara, N.; Takashina, M.; Kristensen, P.; Hendil, K.B.; Tanaka, K.; Ichihara, A. Replacement of Proteasome Subunits X and Y by LMP7 and LMP2 Induced by Interferon-Gamma for Acquirement of the Functional Diversity Responsible for Antigen Processing. FEBS Lett. 1994, 343, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Platteel, A.C.M.; Liepe, J.; van Eden, W.; Mishto, M.; Sijts, A.J.A.M. An Unexpected Major Role for Proteasome-Catalyzed Peptide Splicing in Generation of T Cell Epitopes: Is There Relevance for Vaccine Development? Front. Immunol. 2017, 8, 1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, N.; Ferrari, V.; Stroobant, V.; Abi Habib, J.; Van den Eynde, B.J. Peptide Splicing by the Proteasome. J. Biol. Chem. 2017, 292, 21170–21179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, N.; Stroobant, V.; Chapiro, J.; Ooms, A.; Degiovanni, G.; Morel, S.; van der Bruggen, P.; Boon, T.; Van den Eynde, B.J. An Antigenic Peptide Produced by Peptide Splicing in the Proteasome. Science 2004, 304, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A Large Fraction of HLA Class I Ligands Are Proteasome-Generated Spliced Peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, S.; Lévy, F.; Burlet-Schiltz, O.; Brasseur, F.; Probst-Kepper, M.; Peitrequin, A.L.; Monsarrat, B.; Van Velthoven, R.; Cerottini, J.C.; Boon, T.; et al. Processing of Some Antigens by the Standard Proteasome but Not by the Immunoproteasome Results in Poor Presentation by Dendritic Cells. Immunity 2000, 12, 107–117. [Google Scholar] [CrossRef]
- Chapiro, J.; Claverol, S.; Piette, F.; Ma, W.; Stroobant, V.; Guillaume, B.; Gairin, J.-E.; Morel, S.; Burlet-Schiltz, O.; Monsarrat, B.; et al. Destructive Cleavage of Antigenic Peptides Either by the Immunoproteasome or by the Standard Proteasome Results in Differential Antigen Presentation. J. Immunol. 2006, 176, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice Completely Lacking Immunoproteasomes Show Major Changes in Antigen Presentation. Nat. Immunol. 2011, 13, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kaer, L.; Ashton-Rickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered Peptidase and Viral-Specific T Cell Response in LMP2 Mutant Mice. Immunity 1994, 1, 533–541. [Google Scholar] [CrossRef]
- de Graaf, N.; van Helden, M.J.G.; Textoris-Taube, K.; Chiba, T.; Topham, D.J.; Kloetzel, P.-M.; Zaiss, D.M.W.; Sijts, A.J.A.M. PA28 and the Proteasome Immunosubunits Play a Central and Independent Role in the Production of MHC Class I-Binding Peptides in Vivo. Eur. J. Immunol. 2011, 41, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Chapatte, L.; Ayyoub, M.; Morel, S.; Peitrequin, A.-L.; Lévy, N.; Servis, C.; Van den Eynde, B.J.; Valmori, D.; Lévy, F. Processing of Tumor-Associated Antigen by the Proteasomes of Dendritic Cells Controls in Vivo T-Cell Responses. Cancer Res. 2006, 66, 5461–5468. [Google Scholar] [CrossRef] [Green Version]
- Borissenko, L.; Groll, M. Diversity of proteasomal missions: Fine tuning of the immune response. Biol. Chem. 2007, 388, 947–955. [Google Scholar] [CrossRef]
- Çetin, G.; Klafack, S.; Studencka-Turski, M.; Krüger, E.; Ebstein, F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. [Google Scholar] [CrossRef]
- Basler, M.; Kirk, C.J.; Groettrup, M. The Immunoproteasome in Antigen Processing and Other Immunological Functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Fehling, H.J.; Swat, W.; Laplace, C.; Kühn, R.; Rajewsky, K.; Müller, U.; von Boehmer, H. MHC Class I Expression in Mice Lacking the Proteasome Subunit LMP-7. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.K.; Weinberg, J.B. The Immunoproteasome and Viral Infection: A Complex Regulator of Inflammation. Front. Microbiol. 2015, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- de Verteuil, D.; Muratore-Schroeder, T.L.; Granados, D.P.; Fortier, M.-H.; Hardy, M.-P.; Bramoullé, A.; Caron, E.; Vincent, K.; Mader, S.; Lemieux, S.; et al. Deletion of Immunoproteasome Subunits Imprints on the Transcriptome and Has a Broad Impact on Peptides Presented by Major Histocompatibility Complex I Molecules. Mol. Cell Proteom. 2010, 9, 2034–2047. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Youhnovski, N.; Van Den Broek, M.; Przybylski, M.; Groettrup, M. Immunoproteasomes Down-Regulate Presentation of a Subdominant T Cell Epitope from Lymphocytic Choriomeningitis Virus. J. Immunol. 2004, 173, 3925–3934. [Google Scholar] [CrossRef] [Green Version]
- Zanker, D.; Waithman, J.; Yewdell, J.W.; Chen, W. Mixed Proteasomes Function to Increase Viral Peptide Diversity and Broaden Antiviral CD8+ T Cell Responses. J. Immunol. 2013, 191, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2016, 2, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Tubío-Santamaría, N.; Ebstein, F.; Heidel, F.H.; Krüger, E. Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells 2021, 10, 1577. [Google Scholar] [CrossRef]
- Mishto, M.; Liepe, J.; Textoris-Taube, K.; Keller, C.; Henklein, P.; Weberruß, M.; Dahlmann, B.; Enenkel, C.; Voigt, A.; Kuckelkorn, U.; et al. Proteasome Isoforms Exhibit Only Quantitative Differences in Cleavage and Epitope Generation. Eur. J. Immunol. 2014, 44, 3508–3521. [Google Scholar] [CrossRef]
- Trentini, D.B.; Pecoraro, M.; Tiwary, S.; Cox, J.; Mann, M.; Hipp, M.S.; Hartl, F.U. Role for Ribosome-Associated Quality Control in Sampling Proteins for MHC Class I-Mediated Antigen Presentation. Proc. Natl. Acad. Sci. USA 2020, 117, 4099–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The Immunoproteasome and Thymoproteasome: Functions, Evolution and Human Disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef]
- Yewdell, J.W. DRiPs Solidify: Progress in Understanding Endogenous MHC Class I Antigen Processing. Trends Immunol. 2011, 32, 548–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, K.L.; Farfán-Arribas, D.J.; Colbert, J.D.; Goldberg, A.L. Re-Examining Class-I Presentation and the DRiP Hypothesis. Trends Immunol. 2014, 35, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Cascio, P.; Goldberg, A.L. Preparation of Hybrid (19S-20S-PA28) Proteasome Complexes and Analysis of Peptides Generated during Protein Degradation. Methods Enzym. 2005, 398, 336–352. [Google Scholar] [CrossRef]
- Raule, M.; Cerruti, F.; Benaroudj, N.; Migotti, R.; Kikuchi, J.; Bachi, A.; Navon, A.; Dittmar, G.; Cascio, P. PA28αβ Reduces Size and Increases Hydrophilicity of 20S Immunoproteasome Peptide Products. Chem. Biol. 2014, 21, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Rechsteiner, M.; Realini, C.; Ustrell, V. The Proteasome Activator 11 S REG (PA28) and Class I Antigen Presentation. Biochem. J. 2000, 345 Pt 1, 1–15. [Google Scholar] [CrossRef]
- Kopp, F.; Dahlmann, B.; Kuehn, L. Reconstitution of Hybrid Proteasomes from Purified PA700–20 S Complexes and PA28alphabeta Activator: Ultrastructure and Peptidase Activities. J. Mol. Biol. 2001, 313, 465–471. [Google Scholar] [CrossRef]
- Fort, P.; Kajava, A.V.; Delsuc, F.; Coux, O. Evolution of Proteasome Regulators in Eukaryotes. Genome Biol. Evol 2015, 7, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Apcher, S.; Daskalogianni, C.; Fåhraeus, R. Pioneer Translation Products as an Alternative Source for MHC-I Antigenic Peptides. Mol. Immunol. 2015, 68, 68–71. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Cascio, P.; Saric, T.; Rock, K.L. The Importance of the Proteasome and Subsequent Proteolytic Steps in the Generation of Antigenic Peptides. Mol. Immunol. 2002, 39, 147–164. [Google Scholar] [CrossRef]
- Yamano, T.; Sugahara, H.; Mizukami, S.; Murata, S.; Chiba, T.; Tanaka, K.; Yui, K.; Udono, H. Allele-Selective Effect of PA28 in MHC Class I Antigen Processing. J. Immunol. 2008, 181, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, S.K.; Seelen, M.a.J.; Lin, G.; Azzi, J.R. The Immunoproteasome: An Old Player with a Novel and Emerging Role in Alloimmunity. Am. J. Transpl. 2017, 17, 3033–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Caturegli, P.; Takahashi, M.; Suzuki, K. New Insights into the Function of the Immunoproteasome in Immune and Nonimmune Cells. J. Immunol. Res. 2015, 2015, 541984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moebius, J.; van den Broek, M.; Groettrup, M.; Basler, M. Immunoproteasomes Are Essential for Survival and Expansion of T Cells in Virus-Infected Mice. Eur. J. Immunol. 2010, 40, 3439–3449. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, D.M.W.; de Graaf, N.; Sijts, A.J.A.M. The Proteasome Immunosubunit Multicatalytic Endopeptidase Complex-like 1 Is a T-Cell-Intrinsic Factor Influencing Homeostatic Expansion. Infect. Immun. 2008, 76, 1207–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalim, K.W.; Basler, M.; Kirk, C.J.; Groettrup, M. Immunoproteasome Subunit LMP7 Deficiency and Inhibition Suppresses Th1 and Th17 but Enhances Regulatory T Cell Differentiation. J. Immunol. 2012, 189, 4182–4193. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial Effect of Novel Proteasome Inhibitors in Murine Lupus via Dual Inhibition of Type I Interferon and Autoantibody-Secreting Cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef]
- Li, J.; Basler, M.; Alvarez, G.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome Inhibition Prevents Chronic Antibody-Mediated Allograft Rejection in Renal Transplantation. Kidney Int. 2018, 93, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, K.E.; McCormack, N.M.; Abera, M.B.; Viollet, C.; Yauger, Y.J.; Sukumar, G.; Dalgard, C.L.; Burnett, B.G. The Role of the Immunoproteasome in Interferon-γ-Mediated Microglial Activation. Sci. Rep. 2017, 7, 9365. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of Colitis-Associated Cancer by Selective Targeting of Immunoproteasome Subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef] [PubMed]
- Althof, N.; Goetzke, C.C.; Kespohl, M.; Voss, K.; Heuser, A.; Pinkert, S.; Kaya, Z.; Klingel, K.; Beling, A. The Immunoproteasome-Specific Inhibitor ONX 0914 Reverses Susceptibility to Acute Viral Myocarditis. EMBO Mol. Med. 2018, 10, 200–218. [Google Scholar] [CrossRef]
- Basler, M.; Li, J.; Groettrup, M. On the Role of the Immunoproteasome in Transplant Rejection. Immunogenetics 2019, 71, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Ebstein, F.; Lange, N.; Urban, S.; Seifert, U.; Krüger, E.; Kloetzel, P.-M. Maturation of Human Dendritic Cells Is Accompanied by Functional Remodelling of the Ubiquitin-Proteasome System. Int. J. Biochem. Cell Biol. 2009, 41, 1205–1215. [Google Scholar] [CrossRef]
- Studencka-Turski, M.; Çetin, G.; Junker, H.; Ebstein, F.; Krüger, E. Molecular Insight into the IRE1α-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front. Immunol. 2019, 10, 2900. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.E.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-Mediated Degradation of IkappaBalpha and Processing of P105 in Crohn Disease and Ulcerative Colitis. J. Clin. Investig. 2006, 116, 3195–3203. [Google Scholar] [CrossRef]
- Mitchell, S.; Mercado, E.L.; Adelaja, A.; Ho, J.Q.; Cheng, Q.J.; Ghosh, G.; Hoffmann, A. An NFκB Activity Calculator to Delineate Signaling Crosstalk: Type I and II Interferons Enhance NFκB via Distinct Mechanisms. Front. Immunol. 2019, 10, 1425. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, M.; Kapphahn, R.J.; Terluk, M.R.; Heuss, N.D.; Yuan, C.; Gregerson, D.S.; Ferrington, D.A. Immunoproteasome Deficiency Modifies the Alternative Pathway of NFκB Signaling. PLoS ONE 2013, 8, e56187. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Faustman, D. Essential Role of Human Leukocyte Antigen-Encoded Proteasome Subunits in NF-ΚB Activation and Prevention of Tumor Necrosis Factor-α-Induced Apoptosis*. J. Biol. Chem. 2000, 275, 5238–5247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellerhoff, B.; Gu, S.; Laumbacher, B.; Nerlich, A.G.; Weiss, E.H.; Glas, J.; Kopp, R.; Johnson, J.P.; Wank, R. The LMP7-K Allele of the Immunoproteasome Exhibits Reduced Transcript Stability and Predicts High Risk of Colon Cancer. Cancer Res. 2011, 71, 7145–7154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-W.; Wang, P.; Liu, J.-Q.; Zhang, H.; Xi, W.-D.; Jia, X.-H.; Wang, K.-K. Coordinated Regulation of the Immunoproteasome Subunits by PML/RARα and PU.1 in Acute Promyelocytic Leukemia. Oncogene 2014, 33, 2700–2708. [Google Scholar] [CrossRef] [Green Version]
- Rouette, A.; Trofimov, A.; Haberl, D.; Boucher, G.; Lavallée, V.-P.; D’Angelo, G.; Hébert, J.; Sauvageau, G.; Lemieux, S.; Perreault, C. Expression of Immunoproteasome Genes Is Regulated by Cell-Intrinsic and -Extrinsic Factors in Human Cancers. Sci. Rep. 2016, 6, 34019. [Google Scholar] [CrossRef] [Green Version]
- Bitzer, A.; Basler, M.; Krappmann, D.; Groettrup, M. Immunoproteasome Subunit Deficiency Has No Influence on the Canonical Pathway of NF-ΚB Activation. Mol. Immunol. 2017, 83, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Hussong, S.A.; Roehrich, H.; Kapphahn, R.J.; Maldonado, M.; Pardue, M.T.; Ferrington, D.A. A Novel Role for the Immunoproteasome in Retinal Function. Investig. Ophthalmol. Vis. Sci. 2011, 52, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, S.P.; Collin, J.; Irina, N.; Anyfantis, G.; Kyung, B.K.; Lako, M.; Armstrong, L. A Putative Role for the Immunoproteasome in the Maintenance of Pluripotency in Human Embryonic Stem Cells. Stem Cells 2012, 30, 1373–1384. [Google Scholar] [CrossRef]
- Cui, Z.; Hwang, S.M.; Gomes, A.V. Identification of the Immunoproteasome as a Novel Regulator of Skeletal Muscle Differentiation. Mol. Cell Biol. 2014, 34, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orre, M.; Kamphuis, W.; Dooves, S.; Kooijman, L.; Chan, E.T.; Kirk, C.J.; Dimayuga Smith, V.; Koot, S.; Mamber, C.; Jansen, A.H.; et al. Reactive Glia Show Increased Immunoproteasome Activity in Alzheimer’s Disease. Brain 2013, 136, 1415–1431. [Google Scholar] [CrossRef] [Green Version]
- Campello, L.; Esteve-Rudd, J.; Cuenca, N.; Martín-Nieto, J. The Ubiquitin-Proteasome System in Retinal Health and Disease. Mol. Neurobiol. 2013, 47, 790–810. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Roehrich, H.; Chang, A.A.; Huang, C.W.; Maldonado, M.; Bratten, W.; Rageh, A.A.; Heuss, N.D.; Gregerson, D.S.; Nelson, E.F.; et al. Corneal Wound Healing Is Compromised by Immunoproteasome Deficiency. PLoS ONE 2013, 8, e54347. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Awasthi, N.; Egwuagu, C.E.; Wagner, B.J. Immunoproteasome Expression in a Nonimmune Tissue, the Ocular Lens. Arch. Biochem. Biophys. 2002, 405, 147–153. [Google Scholar] [CrossRef]
- Choi, J.-H.; Jo, H.S.; Lim, S.; Kim, H.-T.; Lee, K.W.; Moon, K.H.; Ha, T.; Kwak, S.S.; Kim, Y.; Lee, E.J.; et al. MTORC1 Accelerates Retinal Development via the Immunoproteasome. Nat. Commun. 2018, 9, 2502. [Google Scholar] [CrossRef]
- Schuld, N.J.; Hussong, S.A.; Kapphahn, R.J.; Lehmann, U.; Roehrich, H.; Rageh, A.A.; Heuss, N.D.; Bratten, W.; Gregerson, D.S.; Ferrington, D.A. Immunoproteasome Deficiency Protects in the Retina after Optic Nerve Crush. PLoS ONE 2015, 10, e0126768. [Google Scholar] [CrossRef]
- Sbardella, D.; Coletta, A.; Tundo, G.R.; Ahmed, I.M.M.; Bellia, F.; Oddone, F.; Manni, G.; Coletta, M. Structural and Functional Evidence for Citicoline Binding and Modulation of 20S Proteasome Activity: Novel Insights into Its pro-Proteostatic Effect. Biochem. Pharm. 2020, 177, 113977. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Guo, X. Proteasome Dysregulation in Human Cancer: Implications for Clinical Therapies. Cancer Metastasis Rev. 2017, 36, 703–716. [Google Scholar] [CrossRef]
- Algarra, I.; Collado, A.; Garrido, F. Altered MHC Class I Antigens in Tumors. Int. J. Clin. Lab. Res. 1997, 27, 95–102. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome Deficiency Is a Feature of Non-Small Cell Lung Cancer with a Mesenchymal Phenotype and Is Associated with a Poor Outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heink, S.; Fricke, B.; Ludwig, D.; Kloetzel, P.-M.; Krüger, E. Tumor Cell Lines Expressing the Proteasome Subunit Isoform LMP7E1 Exhibit Immunoproteasome Deficiency. Cancer Res. 2006, 66, 649–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Schmidt, M. Cutting through Complexity: The Proteolytic Properties of Alternate Immunoproteasome Complexes. Chem Biol. 2014, 21, 435–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewerth, D.; Jansen, G.; Assaraf, Y.G.; Zweegman, S.; Kaspers, G.J.L.; Cloos, J. Molecular Basis of Resistance to Proteasome Inhibitors in Hematological Malignancies. Drug Resist. Updat. 2015, 18, 18–35. [Google Scholar] [CrossRef]
- Weon, J.L.; Potts, P.R. The MAGE Protein Family and Cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Olsson, N.; Heberling, M.L.; Zhang, L.; Jhunjhunwala, S.; Phung, Q.T.; Lin, S.; Anania, V.G.; Lill, J.R.; Elias, J.E. An Integrated Genomic, Proteomic, and Immunopeptidomic Approach to Discover Treatment-Induced Neoantigens. Front. Immunol. 2021, 12, 662443. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, B.; Stroobant, V.; Bousquet-Dubouch, M.-P.; Colau, D.; Chapiro, J.; Parmentier, N.; Dalet, A.; Van den Eynde, B.J. Analysis of the Processing of Seven Human Tumor Antigens by Intermediate Proteasomes. J. Immunol. 2012, 189, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Schooten, E.; Di Maggio, A.; van Bergen En Henegouwen, P.M.P.; Kijanka, M.M. MAGE-A Antigens as Targets for Cancer Immunotherapy. Cancer Treat. Rev. 2018, 67, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Gou, Q.; Dong, C.; Xu, H.; Khan, B.; Jin, J.; Liu, Q.; Shi, J.; Hou, Y. PD-L1 Degradation Pathway and Immunotherapy for Cancer. Cell Death Dis. 2020, 11, 955. [Google Scholar] [CrossRef] [PubMed]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; de Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 Protein Regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 Kinase Destabilizes PD-L1 via Cullin 3-SPOP to Control Cancer Immune Surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Qian, G.; Zhang, S.; Zheng, H.; Fan, S.; Lesinski, G.B.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.-Y. Inhibition of MTOR Complex 1/P70 S6 Kinase Signaling Elevates PD-L1 Levels in Human Cancer Cells through Enhancing Protein Stabilization Accompanied with Enhanced β-TrCP Degradation. Oncogene 2019, 38, 6270–6282. [Google Scholar] [CrossRef]
- Narayanan, S.; Cai, C.-Y.; Assaraf, Y.G.; Guo, H.-Q.; Cui, Q.; Wei, L.; Huang, J.-J.; Ashby, C.R.; Chen, Z.-S. Targeting the Ubiquitin-Proteasome Pathway to Overcome Anti-Cancer Drug Resistance. Drug Resist. Updat. 2020, 48, 100663. [Google Scholar] [CrossRef]
- Grigoreva, T.A.; Tribulovich, V.G.; Garabadzhiu, A.V.; Melino, G.; Barlev, N.A. The 26S Proteasome Is a Multifaceted Target for Anti-Cancer Therapies. Oncotarget 2015, 6, 24733–24749. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of Therapy with Bortezomib in Solid Tumors: A Review Based on 32 Clinical Trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Xi, J.; Zhuang, R.; Kong, L.; He, R.; Zhu, H.; Zhang, J. Immunoproteasome-Selective Inhibitors: An Overview of Recent Developments as Potential Drugs for Hematologic Malignancies and Autoimmune Diseases. Eur. J. Med. Chem. 2019, 182, 111646. [Google Scholar] [CrossRef]
- Ebstein, F.; Kloetzel, P.-M.; Krüger, E.; Seifert, U. Emerging Roles of Immunoproteasomes beyond MHC Class I Antigen Processing. Cell Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Groettrup, M. Subunit Specific Inhibitors of Proteasomes and Their Potential for Immunomodulation. Curr. Opin. Chem. Biol. 2014, 23, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-Selective and Non-Selective Inhibitors: A Promising Approach for the Treatment of Multiple Myeloma. Pharm. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the Immunoproteasome Ameliorates Experimental Autoimmune Encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.; Lee, W.; Kim, K.B. The Immunoproteasome as a Therapeutic Target for Hematological Malignancies. Curr. Cancer Drug Targets 2014, 14, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2 S,3 R)- N-(( S)-3-(Cyclopent-1-En-1-Yl)-1-((R)-2-Methyloxiran-2-Yl)-1-Oxopropan-2-Yl)-3-Hydroxy-3-(4-Methoxyphenyl)-2-(( S)-2-(2-Morpholinoacetamido)Propanamido)Propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome Inhibitors—Molecular Basis and Current Perspectives in Multiple Myeloma. J. Cell Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef]
- Cengiz Seval, G.; Beksac, M. The Safety of Bortezomib for the Treatment of Multiple Myeloma. Expert. Opin. Drug. Saf. 2018, 17, 953–962. [Google Scholar] [CrossRef]
- Jenkins, T.W.; Downey-Kopyscinski, S.L.; Fields, J.L.; Rahme, G.J.; Colley, W.C.; Israel, M.A.; Maksimenko, A.V.; Fiering, S.N.; Kisselev, A.F. Activity of Immunoproteasome Inhibitor ONX-0914 in Acute Lymphoblastic Leukemia Expressing MLL–AF4 Fusion Protein. Sci. Rep. 2021, 11, 10883. [Google Scholar] [CrossRef] [PubMed]
- Downey-Kopyscinski, S.; Daily, E.W.; Gautier, M.; Bhatt, A.; Florea, B.I.; Mitsiades, C.S.; Richardson, P.G.; Driessen, C.; Overkleeft, H.S.; Kisselev, A.F. An Inhibitor of Proteasome Β2 Sites Sensitizes Myeloma Cells to Immunoproteasome Inhibitors. Blood Adv. 2018, 2, 2443–2451. [Google Scholar] [CrossRef]
- Zerfas, B.L.; Maresh, M.E.; Trader, D.J. The Immunoproteasome: An Emerging Target in Cancer and Autoimmune and Neurological Disorders. J. Med. Chem. 2020, 63, 1841–1858. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Johnson, H.; Anderl, J.L.; Muchamuel, T.; McMinn, D.; Morisseau, C.; Hammock, B.D.; Kirk, C.; Wang, J. Role of Epoxide Hydrolases and Cytochrome P450s on Metabolism of KZR-616, a First-in-Class Selective Inhibitor of the Immunoproteasome. Drug. Metab. Dispos. 2021, 49, 810–821. [Google Scholar] [CrossRef]
- Ogorevc, E.; Schiffrer, E.S.; Sosič, I.; Gobec, S. A Patent Review of Immunoproteasome Inhibitors. Expert Opin. Pat. 2018, 28, 517–540. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, H.; Shao, J.; He, R.; Xi, J.; Zhuang, R.; Zhang, J. Immunoproteasome-selective inhibitors: The future of autoimmune diseases? Future Med. Chem. 2020, 12, 269–272. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Busch, M.; Friese-Hamim, M.; Crosignani, S.; Fuchss, T.; Musil, D.; Rohdich, F.; Sanderson, M.P.; Seenisamy, J.; Walter-Bausch, G.; et al. Structure-Based Optimization and Discovery of M3258, a Specific Inhibitor of the Immunoproteasome Subunit LMP7 (Β5i). J. Med. Chem. 2021, 64, 10230–10245. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.P.; Friese-Hamim, M.; Walter-Bausch, G.; Busch, M.; Gaus, S.; Musil, D.; Rohdich, F.; Zanelli, U.; Downey-Kopyscinski, S.L.; Mitsiades, C.S.; et al. M3258 Is a Selective Inhibitor of the Immunoproteasome Subunit LMP7 (Β5i) Delivering Efficacy in Multiple Myeloma Models. Mol. Cancer 2021, 20, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, M. Role of Immunoproteasomes and Thymoproteasomes in Health and Disease. Pathol. Int. 2021, 71, 371–382. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Orlowski, R.Z. The Immunoproteasome as a Target in Hematologic Malignancies. Semin. Hematol. 2012, 49, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.L.; Gaczynska, M.; Grant, E.; Michalek, M.; Rock, K.L. Functions of the Proteasome in Antigen Presentation. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 479–490. [Google Scholar] [CrossRef]
- Fiebiger, B.M.; Pfister, H.; Behrends, U.; Mautner, J. Polyubiquitination of Lysine-48 Is an Essential but Indirect Signal for MHC Class I Antigen Processing. Eur. J. Immunol. 2015, 45, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N.; Van den Eynde, B.J. Insights into the Processing of MHC Class I Ligands Gained from the Study of Human Tumor Epitopes. Cell Mol. Life Sci. 2011, 68, 1503–1520. [Google Scholar] [CrossRef]
- Mpakali, A.; Stratikos, E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers 2021, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High Tumor Mutation Burden Fails to Predict Immune Checkpoint Blockade Response across All Cancer Types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Wu, J.; Mayer, A.T.; Li, R. Integrated Imaging and Molecular Analysis to Decipher Tumor Microenvironment in the Era of Immunotherapy. Semin Cancer Biol. 2020, S1044–579X(20)30264–9. [Google Scholar] [CrossRef]
- Wu, J.; Li, C.; Gensheimer, M.; Padda, S.; Kato, F.; Shirato, H.; Wei, Y.; Schönlieb, C.-B.; Price, S.J.; Jaffray, D.; et al. Radiological Tumour Classification across Imaging Modality and Histology. Nat. Mach. Intell. 2021, 3, 787–798. [Google Scholar] [CrossRef]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome Expression Is Associated with Better Prognosis and Response to Checkpoint Therapies in Melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Neubert, N.J.; Tillé, L.; Barras, D.; Soneson, C.; Baumgaertner, P.; Rimoldi, D.; Gfeller, D.; Delorenzi, M.; Fuertes Marraco, S.A.; Speiser, D.E. Broad and Conserved Immune Regulation by Genetically Heterogeneous Melanoma Cells. Cancer Res. 2017, 77, 1623–1636. [Google Scholar] [CrossRef] [Green Version]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC Proteins Confer Differential Sensitivity to CTLA-4 and PD-1 Blockade in Untreated Metastatic Melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.e3. [Google Scholar] [CrossRef]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; Gil, M.d.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrejon, D.Y.; Abril-Rodriguez, G.; Champhekar, A.S.; Tsoi, J.; Campbell, K.M.; Kalbasi, A.; Parisi, G.; Zaretsky, J.M.; Garcia-Diaz, A.; Puig-Saus, C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discov. 2020, 10, 1140–1157. [Google Scholar] [CrossRef]
- Woods, K.; Knights, A.J.; Anaka, M.; Schittenhelm, R.B.; Purcell, A.W.; Behren, A.; Cebon, J. Mismatch in Epitope Specificities between IFNγ Inflamed and Uninflamed Conditions Leads to Escape from T Lymphocyte Killing in Melanoma. J. Immunother. Cancer 2016, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Lizée, G.; Hwu, P. Blockade of the PD-1 Pathway Enhances the Efficacy of Adoptive Cell Therapy against Cancer. Onco. Immunol. 2013, 2, e22691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, A.S.; Rodríguez, M.S.; Matthiesen, R. Review and Literature Mining on Proteostasis Factors and Cancer. Methods Mol. Biol. 2016, 1449, 71–84. [Google Scholar] [CrossRef]
- Vahid, S.; Thaper, D.; Zoubeidi, A. Chaperoning the Cancer: The Proteostatic Functions of the Heat Shock Proteins in Cancer. Recent Pat. Anticancer Drug Discov. 2017, 12, 35–47. [Google Scholar] [CrossRef]
- Santoro, A.M.; D’Urso, A.; Cunsolo, A.; Milardi, D.; Purrello, R.; Sbardella, D.; Tundo, G.R.; Diana, D.; Fattorusso, R.; Dato, A.D.; et al. Cooperative Binding of the Cationic Porphyrin Tris-T4 Enhances Catalytic Activity of 20S Proteasome Unveiling a Complex Distribution of Functional States. Int. J. Mol. Sci. 2020, 21, 7190. [Google Scholar] [CrossRef]
- Santoro, A.M.; Cunsolo, A.; D’Urso, A.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Coletta, M.; Diana, D.; Fattorusso, R.; Persico, M.; et al. Cationic Porphyrins Are Tunable Gatekeepers of the 20S Proteasome. Chem. Sci. 2016, 7, 1286–1297. [Google Scholar] [CrossRef] [Green Version]
- Dato, A.D.; Cunsolo, A.; Persico, M.; Santoro, A.M.; D’Urso, A.; Milardi, D.; Purrello, R.; Stefanelli, M.; Paolesse, R.; Tundo, G.R.; et al. Electrostatic Map Of Proteasome α-Rings Encodes The Design of Allosteric Porphyrin-Based Inhibitors Able To Affect 20S Conformation By Cooperative Binding. Sci. Rep. 2017, 7, 17098. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Kim, H.T.; Lee, D.; Collins, G.A. Mechanisms That Activate 26S Proteasomes and Enhance Protein Degradation. Biomolecules 2021, 11, 779. [Google Scholar] [CrossRef]
- Kudriaeva, A.A.; Saratov, G.A.; Kaminskaya, A.N.; Vladimirov, V.I.; Barzilovich, P.Y.; Belogurov, A.A. Polyamines Counteract Carbonate-Driven Proteasome Stalling in Alkaline Conditions. Biomolecules 2020, 10, 1597. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.A.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, S.; Xu, J. Proteasomal and Lysosomal Degradation for Specific and Durable Suppression of Immunotherapeutic Targets. Cancer Biol. Med. 2020, 17, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, J.; Chu, M.; Liu, Y.; Wang, Z.; Zhu, X. Emerging Role of Ubiquitination in the Regulation of PD-1/PD-L1 in Cancer Immunotherapy. Mol. Ther. 2021, 29, 908–919. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, W.; Huang, Y.; Cui, R.; Li, X.; Li, B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell Physiol. Biochem. 2018, 47, 721–734. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Saenger, Y. The Mechanism of Anti-CTLA-4 Activity and the Negative Regulation of T-Cell Activation. Oncologist 2008, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Robainas, M.; Otano, R.; Bueno, S.; Ait-Oudhia, S. Understanding the Role of PD-L1/PD1 Pathway Blockade and Autophagy in Cancer Therapy. Onco Targets 2017, 10, 1803–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-Oncology Drug Development Goes Global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Takahama, Y.; Tanaka, K. Thymoproteasome: Probable Role in Generating Positively Selecting Peptides. Curr. Opin. Immunol. 2008, 20, 192–196. [Google Scholar] [CrossRef]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen Presentation in the Thymus for Positive Selection and Central Tolerance Induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef]
- Nil, A.; Firat, E.; Sobek, V.; Eichmann, K.; Niedermann, G. Expression of Housekeeping and Immunoproteasome Subunit Genes Is Differentially Regulated in Positively and Negatively Selecting Thymic Stroma Subsets. Eur. J. Immunol. 2004, 34, 2681–2689. [Google Scholar] [CrossRef]
- Macagno, A.; Gilliet, M.; Sallusto, F.; Lanzavecchia, A.; Nestle, F.O.; Groettrup, M. Dendritic Cells Up-Regulate Immunoproteasomes and the Proteasome Regulator PA28 during Maturation. Eur. J. Immunol. 1999, 29, 4037–4042. [Google Scholar] [CrossRef]
- Nitta, T.; Murata, S.; Sasaki, K.; Fujii, H.; Ripen, A.M.; Ishimaru, N.; Koyasu, S.; Tanaka, K.; Takahama, Y. Thymoproteasome Shapes Immunocompetent Repertoire of CD8+ T Cells. Immunity 2010, 32, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Jameson, S.C.; Hogquist, K.A. Thymoproteasome Subunit-Β5T Generates Peptide-MHC Complexes Specialized for Positive Selection. Proc. Natl. Acad. Sci. USA 2013, 110, 6979–6984. [Google Scholar] [CrossRef] [Green Version]
- Frantzeskakis, M.; Takahama, Y.; Ohigashi, I. The Role of Proteasomes in the Thymus. Front. Immunol. 2021, 12, 646209. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.-I.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ T Cell Development by Thymus-Specific Proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.M.; Ohigashi, I.; Motosugi, R.; Nakayama, T.; Sakata, M.; Hamazaki, J.; Nishito, Y.; Rode, I.; Tanaka, K.; Takemoto, T.; et al. Foxn1-Β5t Transcriptional Axis Controls CD8+ T-Cell Production in the Thymus. Nat. Commun. 2017, 8, 14419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaru, U.; Ishizu, A.; Murata, S.; Miyatake, Y.; Suzuki, S.; Takahashi, S.; Kazamaki, T.; Ohara, J.; Baba, T.; Iwasaki, S.; et al. Exclusive Expression of Proteasome Subunit {beta}5t in the Human Thymic Cortex. Blood 2009, 113, 5186–5191. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.E.; Žuklys, S.; Zhanybekova, S.; Ohigashi, I.; Teh, H.-Y.; Sansom, S.N.; Shikama-Dorn, N.; Hafen, K.; Macaulay, I.C.; Deadman, M.E.; et al. Dynamic Spatio-Temporal Contribution of Single Β5t+ Cortical Epithelial Precursors to the Thymus Medulla. Eur J. Immunol. 2016, 46, 846–856. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Takada, K.; Ohte, Y.; Kondo, H.; Sorimachi, H.; Tanaka, K.; Takahama, Y.; Murata, S. Thymoproteasomes Produce Unique Peptide Motifs for Positive Selection of CD8(+) T Cells. Nat. Commun. 2015, 6, 7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasahara, M.; Flajnik, M.F. Origin and Evolution of the Specialized Forms of Proteasomes Involved in Antigen Presentation. Immunogenetics 2019, 71, 251–261. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Xing, Y. Why CD8+ T Cells Need Diversity When Growing Up. Immunity 2010, 32, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, E.Z.; Murata, S.; Tanaka, K.; Rock, K.L. Specialized Proteasome Subunits Have an Essential Role in the Thymic Selection of CD8(+) T Cells. Nat. Immunol. 2016, 17, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Tomaru, U.; Konno, S.; Miyajima, S.; Kimoto, R.; Onodera, M.; Kiuchi, S.; Murata, S.; Ishizu, A.; Kasahara, M. Restricted Expression of the Thymoproteasome Is Required for Thymic Selection and Peripheral Homeostasis of CD8+ T Cells. Cell Rep. 2019, 26, 639–651.e2. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Van Laethem, F.; Xing, Y.; Akane, K.; Suzuki, H.; Murata, S.; Tanaka, K.; Jameson, S.C.; Singer, A.; Takahama, Y. TCR Affinity for Thymoproteasome-Dependent Positively Selecting Peptides Conditions Antigen Responsiveness in CD8(+) T Cells. Nat. Immunol. 2015, 16, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Tomaru, U.; Takahashi, S.; Ishizu, A.; Miyatake, Y.; Gohda, A.; Suzuki, S.; Ono, A.; Ohara, J.; Baba, T.; Murata, S.; et al. Decreased Proteasomal Activity Causes Age-Related Phenotypes and Promotes the Development of Metabolic Abnormalities. Am. J. Pathol. 2012, 180, 963–972. [Google Scholar] [CrossRef]
- Tomaru, U.; Kasahara, M. Thymoproteasome: Role in Thymic Selection and Clinical Significance as a Diagnostic Marker for Thymic Epithelial Tumors. Arch. Immunol. Exp. 2013, 61, 357–365. [Google Scholar] [CrossRef]
- Yamada, Y.; Tomaru, U.; Ishizu, A.; Kiuchi, T.; Marukawa, K.; Matsuno, Y.; Kasahara, M. Expression of Proteasome Subunit Β5t in Thymic Epithelial Tumors. Am. J. Surg. Pathol. 2011, 35, 1296–1304. [Google Scholar] [CrossRef]
- Yamada, Y.; Tomaru, U.; Ishizu, A.; Kiuchi, T.; Kasahara, M.; Matsuno, Y. Expression of Thymoproteasome Subunit Β5t in Type AB Thymoma. J. Clin. Pathol 2014, 67, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, U.; Tsuji, T.; Kiuchi, S.; Ishizu, A.; Suzuki, A.; Otsuka, N.; Ito, T.; Ikeda, H.; Fukasawa, Y.; Kasahara, M. Decreased Expression of Thymus-Specific Proteasome Subunit Β5t in Down Syndrome Patients. Histopathology 2015, 67, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Kochi, Y.; Muro, R.; Tomofuji, Y.; Okamura, T.; Murata, S.; Suzuki, H.; Sumida, T.; Yamamoto, K.; Takayanagi, H. Human Thymoproteasome Variations Influence CD8 T Cell Selection. Sci. Immunol. 2017, 2, eaan5165. [Google Scholar] [CrossRef]
- Ohigashi, I.; Ohte, Y.; Setoh, K.; Nakase, H.; Maekawa, A.; Kiyonari, H.; Hamazaki, Y.; Sekai, M.; Sudo, T.; Tabara, Y.; et al. A Human PSMB11 Variant Affects Thymoproteasome Processing and CD8+ T Cell Production. JCI Insight 2017, 2, 93664. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tundo, G.R.; Sbardella, D.; Oddone, F.; Kudriaeva, A.A.; Lacal, P.M.; Belogurov, A.A., Jr.; Graziani, G.; Marini, S. At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention. Cancers 2021, 13, 4852. https://doi.org/10.3390/cancers13194852
Tundo GR, Sbardella D, Oddone F, Kudriaeva AA, Lacal PM, Belogurov AA Jr., Graziani G, Marini S. At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention. Cancers. 2021; 13(19):4852. https://doi.org/10.3390/cancers13194852
Chicago/Turabian StyleTundo, Grazia R., Diego Sbardella, Francesco Oddone, Anna A. Kudriaeva, Pedro M. Lacal, Alexey A. Belogurov, Jr., Grazia Graziani, and Stefano Marini. 2021. "At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention" Cancers 13, no. 19: 4852. https://doi.org/10.3390/cancers13194852
APA StyleTundo, G. R., Sbardella, D., Oddone, F., Kudriaeva, A. A., Lacal, P. M., Belogurov, A. A., Jr., Graziani, G., & Marini, S. (2021). At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention. Cancers, 13(19), 4852. https://doi.org/10.3390/cancers13194852