
Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Molecular Epidemiology of EGFR Mutations
1.1.1. Common EGFR Mutations: Deletion in Exon 19 (Del19) and Exon 21 Point Mutation, L858R
1.1.2. Rare EGFR Mutations
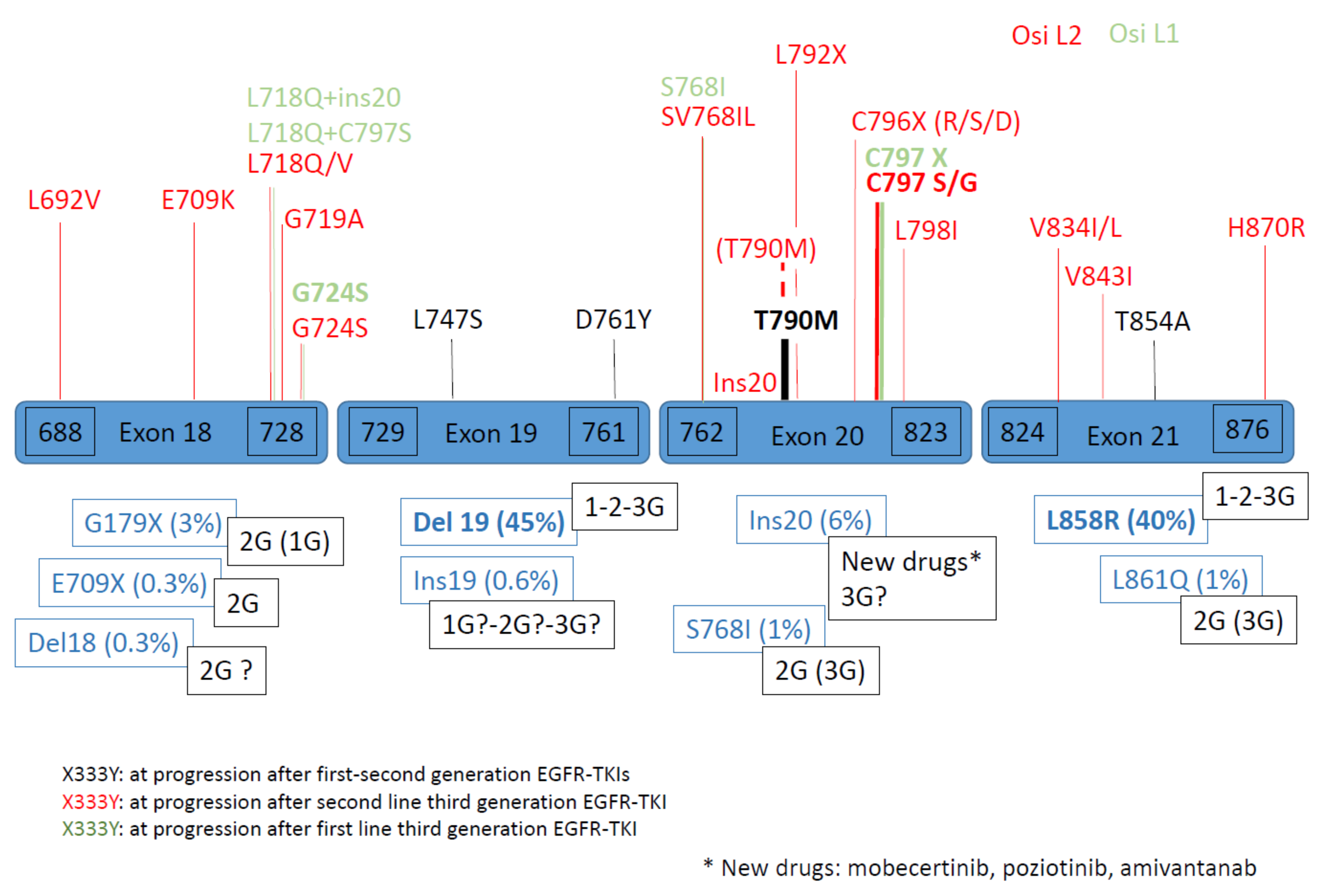
1.2. Secondary and Tertiary EGFR Mutations
1.3. Complex EGFR Mutations
1.4. Co-Mutations
1.5. Sub-Clonal Mutations
2. Mechanisms of Resistance to EGFR-TKIs
2.1. Clinical Trials Results
2.1.1. First Line Therapy of NSCLC with EGFR Mutation
First-Generation EGFR TKI
Second-Generation EGFR-TKIs
Third-Generation EGFR-TKIs
EGFR Ins20 Specific Inhibitors
EGFR-TKI Treatments Combinations
2.1.2. Second-Line with Third-Generation EGFR-TKIs
2.2. Mechanisms of Resistance
2.2.1. Primary Resistance
Primary Resistance to First–Second-Generation EGFR-TKIs
Primary Resistance to Third-Generation EGFR-TKIs
2.2.2. Secondary Resistance
EGFR-Dependent Mechanisms of Resistance
- A.
- In second line
- B. In first line
EGFR-Independent Mechanisms of Resistance
- A.
- In second line
- B. In first line
2.2.3. Particular Points
Comparison Second/First Line Osimertinib
Other Rare Mechanisms
Variability of Resistance Mechanism Depends on Type of EGFR Mutation
Resistance Treatments
Gene Silencing
Other Situations of Resistance
2.2.4. Perspectives
3. Technical Aspects of Molecular Testing
3.1. Nucleic Acids DNA, RNA and TNA
3.2. Molecular Analysis
3.2.1. Next-Generation Sequencing (NGS)
3.2.2. Non-NGS Multiplex Targeted Methods
3.2.3. Sensitive PCR-Based Methods
4. Molecular Testing and Resistance to EGFR-TKIs
4.1. Anticipation of Resistance Mechanism at Diagnosis
4.1.1. Sub-Clonal Alterations at Diagnosis
EGFR T790M Mutations
Other Alterations
4.1.2. Histological Transformation
4.1.3. Molecular Characterization of Residual Tumor Cells after Partial Response or Stable Disease
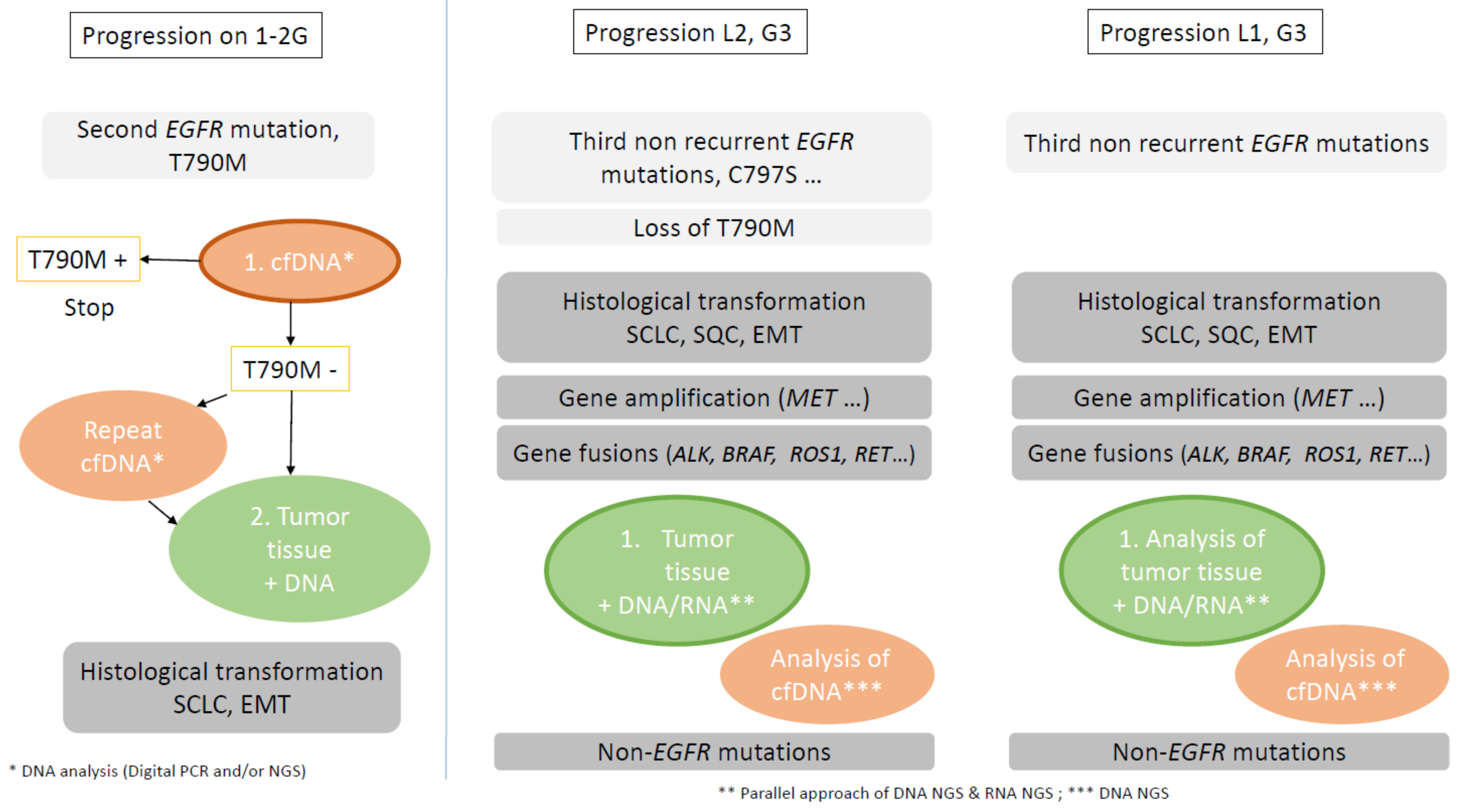
4.2. Molecular and Histological Characterization Identification at Time of Resistance
4.2.1. Plasma Cell-Free DNA
Physiopathology
Technical Considerations
Place of cfDNA Analysis at Progression
4.2.2. Tissue Analysis
5. Perspectives
5.1. Biomarker Driven Approaches
5.1.1. EGFR-Dependent Mechanisms
5.1.2. EGFR-Independent Mechanisms
MET Amplification
HER2 Amplification
Other TKIs
Fusions
5.2. No Biomarker-Driven Approach
5.2.1. Chemotherapy, with or without EGFR-TKI Continuation
5.2.2. Immunotherapy
5.2.3. Other Treatments
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Biomarkers France contributors. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung. Cancer Res. 2015, 4, 36–54. [Google Scholar] [PubMed]
- Leduc, C.; Merlio, J.; Besse, B.; Blons, H.; Debieuvre, D.; Bringuier, P.; Monnet, I.; Rouquette, I.; Fraboulet-Moreau, S.; Lemoine, A.; et al. French Cooperative Thoracic Intergroup (IFCT). Clinical and molecular characteristics of non-small-cell lung cancer (NSCLC) harboring EGFR mutation: Results of the nationwide French Cooperative Thoracic Intergroup (IFCT) program. Ann. Oncol. 2017, 28, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in this Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gristina, V.; Malapelle, U.; Galvano, A.; Pisapia, P.; Pepe, F.; Rolfo, C.; Tortorici, S.; Bazan, V.; Troncone, G.; Russo, A. The significance of epidermal growth factor receptor uncommon mutations in non-small cell lung cancer: A systematic review and critical appraisal. Cancer Treat. Rev. 2020, 85, 101994. [Google Scholar] [CrossRef]
- Zhang, T.; Wan, B.; Zhao, Y.; Li, C.; Liu, H.; Lv, T.; Zhan, P.; Song, Y. Treatment of uncommon EGFR mutations in non-small cell lung cancer: New evidence and treatment. Transl. Lung Cancer Res. 2019, 8, 302–316. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. FLAURA Investigators. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. ADAURA Investigators.Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Janjigian, Y.Y.; Ahye, N.; Riely, G.J.; Chaft, J.E.; Sima, C.S.; Shen, R.; Zheng, J.; Dycoco, J.; Kris, M.G.; et al. Distinct clinical course of EGFR-mutant resected lung cancers: Results of testing of 1118 surgical specimens and effects of adjuvant gefitinib and erlotinib. J. Thorac. Oncol. 2012, 7, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, S.; Seitlinger, J.; Guerrera, F.; Reeb, J.; Beau-Faller, M.; Voegeli, A.C.; Siat, J.; Clément-Duchêne, C.; Tiotiu, A.; Santelmo, N.; et al. Prognostic Value of Exon 19 Versus 21 EGFR Mutations Varies According to Disease Stage in Surgically Resected Non-small Cell Lung Cancer Adenocarcinoma. Ann. Surg. Oncol. 2018, 25, 1069–1078. [Google Scholar] [CrossRef]
- Suda, K.; Mitsudomi, T.; Shintani, Y.; Okami, J.; Ito, H.; Ohtsuka, T.; Toyooka, S.; Mori, T.; Watanabe, S.I.; Asamura, H.; et al. Japanese Joint Committee of Lung Cancer Registry. Clinical Impacts of EGFR Mutation Status: Analysis of 5780 Surgically Resected Lung Cancer Cases. Ann. Thorac. Surg. 2021, 111, 269–276. [Google Scholar] [CrossRef]
- Kohsaka, S.; Nagano, M.; Ueno, T.; Suehara, Y.; Hayashi, T.; Shimada, N.; Takahashi, K.; Suzuki, K.; Takamochi, K.; Takahashi, F.; et al. A method for high throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Title. Sci. Transl. Med. 2017, 9, eaan6566. [Google Scholar] [CrossRef] [Green Version]
- Beau-Faller, M.; Prim, N.; Ruppert, A.M.; Nanni-Metéllus, I.; Lacave, R.; Lacroix, L.; Escande, F.; Lizard, S.; Pretet, J.L.; Rouquette, I.; et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: A multicentre observational study by the French ERMETIC-IFCT network. Ann. Oncol. 2014, 25, 126–131. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, J.; Tang, D.; Ye, X.; Li, S.; Mu, N.; Li, Z.; Liu, R.; Xiang, L.; Huang, C.; et al. Tyrosine Kinase Inhibitors Could Be Effective Against Non-small Cell Lung Cancer Brain Metastases Harboring Uncommon EGFR Mutations. Front. Oncol. 2020, 10, 224. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.E.; Nafa, K.; Chaft, J.E.; Rekhtman, N.; Lau, C.; Reva, B.A.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. EGFR exon 20 insertion mutations in lung adenocarcinomas: Prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol. Cancer Ther. 2013, 12, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaux, J.P.; Elamin, Y.Y.; Tan, Z.; Carter, B.W.; Zhang, S.; Liu, S.; Li, S.; Chen, T.; Poteete, A.; Estrada-Bernal, A.; et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat. Med. 2018, 24, 638–646. [Google Scholar] [CrossRef]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef] [Green Version]
- Floc’h, N.; Martin, M.J.; Riess, J.W.; Orme, J.P.; Staniszewska, A.D.; Ménard, L.; Cuomo, M.E.; O’Neill, D.J.; Ward, R.A.; Finlay, M.R.V.; et al. Antitumor Activity of Osimertinib, an Irreversible Mutant-Selective EGFRTyrosine Kinase Inhibitor, in NSCLC Harboring EGFR Exon 20 Insertions. Mol. Cancer Ther. 2018, 17, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banno, E.; Togashi, Y.; Nakamura, Y.; Chiba, M.; Kobayashi, Y.; Hayashi, H.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; et al. Sensitivities to various epidermal growth factor receptor-tyrosine kinase inhibitors of uncommon epidermal growth factor receptor mutations L861Q and S768I: What is the optimal epidermal growth factor receptor-tyrosine kinase inhibitor? Cancer Sci. 2016, 107, 1134–1140. [Google Scholar]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Bronte, G.; Chiadini, E.; Ludovini, V.; Dubini, A.; Papi, M.; Baglivo, S.; De Luigi, N.; et al. Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J. Clin. Med. 2020, 9, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blons, H.; Oudart, J.B.; Merlio, J.P.; Debieuvre, D.; de Fraipont, F.; Audigier-Valette, C.; Escande, F.; Hominal, S.; Bringuier, P.P.; Fraboulet-Moreau, S.; et al. French Cooperative Thoracic Intergroup (IFCT). PTEN, ATM, IDH1 mutations and MAPK pathway activation as modulators of PFS and OS in patients treated by first line EGFR TKI, an ancillary study of the French Cooperative Thoracic Intergroup (IFCT) Biomarkers France project. Lung Cancer 2021, 151, 69–75. [Google Scholar] [PubMed]
- Kohsaka, S.; Petronczki, M.; Solca, F.; Maemondon, M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: Implications for therapeutic sequencing. Future Oncol. 2019, 15, 637–652. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment with Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Vaclova, T.; Grazini, U.; Ward, L.; O’Neill, D.; Markovets, A.; Huang, X.; Chmielecki, J.; Hartmaier, R.; Thress, K.S.; Smith, P.D.; et al. Clinical impact of subclonal EGFR T790M mutations in advanced-stage EGFR-mutant non-small-cell lung cancers. Nat. Commun. 2021, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Beau-Faller, M.; Pencreach, E.; Leduc, C.; Blons, H.; Merlio, J.P.; Bringuier, P.P.; de Fraipont, F.; Escande, F.; Lemoine, A.; Ouafik, L.; et al. French Cooperative Thoracic Intergroup (IFCT). Independent prognostic value of ultra-sensitive quantification of tumor pre-treatment T790M subclones in EGFR mutated non-small cell lung cancer (NSCLC) treated by first/second generation TKI, depends on variant allele frequency (VAF): Results of the French cooperative thoracic intergroup (IFCT) biomarkers France project. Lung Cancer 2020, 140, 19–26. [Google Scholar]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Liam, C.-K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Fukuoka, M.; Wu, Y.L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.S.; Sriuranpong, V.; Chao, T.-Y.; Nakagawa, K.; Chu, D.-T.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Shi, Y.K.; Wang, L.; Han, B.H.; Li, W.; Yu, P.; Liu, Y.P.; Ding, C.M.; Song, X.; Ma, Z.Y.; Ren, X.L.; et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE): A phase 3, open-label, randomized study. Ann. Oncol. 2017, 28, 2443–2450. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; Obyrne, K.J.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Riely, G.J.; Neal, J.W.; Camidge, D.R.; Spira, A.I.; Piotrowska, Z.; Costa, D.B.; Tsao, A.S.; Patel, J.D.; Gadgeel, S.M.; Bazhenova, L.; et al. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non-Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations from a Phase I/II Trial. Cancer Discov. 2021, 11, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Lee, S.-H.; Kim, S.-Y.; Jeong, S.-Y.; Kim, J.-H.; Pyo, K.-H.; Park, C.-W.; Heo, S.G.; Yun, M.R.; Lim, S.; et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion-Driven NSCLC. Cancer Discov. 2020, 10, 1194–1209. [Google Scholar] [PubMed]
- Yang, J.C.H.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.-H.; Sequist, L.V.; Geater, S.L.; Tsai, C.-M.; Mok, T.; Schuler, M.; Yamamoto, N.; Yu, C.-J.; I Ou, S.-H.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Blackhall, F.; Krzakowski, M.; Barrios, C.H.; Park, K.; Bover, I.; Heo, D.S.; Rosell, R.; Talbot, D.C.; Frank, R.; et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3337–3344. [Google Scholar] [CrossRef] [Green Version]
- Prelaj, A.; Bottiglieri, A.; Proto, C.; Russo, G.L.; Signorelli, D.; Ferrara, R.; Galli, G.; De Toma, A.; Viscardi, G.; Brambilla, M.; et al. Poziotinib for EGFR and HER2 exon 20 insertion mutation in advanced NSCLC: Results from the expanded access program. Eur. J. Cancer 2021, 149, 235–248. [Google Scholar] [CrossRef]
- Cortot, A.B.; Madroszyk-Flandin, A.; Giroux-Leprieur, E.; Molinier, O.; Quoix, E.; Bérard, H.; Otto, J.; Rault, I.; Moro-Sibilot, D.; Raimbourg, J.; et al. First-Line Afatinib plus Cetuximab for EGFR-Mutant Non–Small Cell Lung Cancer: Results from the Randomized Phase II IFCT-1503 ACE-Lung Study. Clin. Cancer. Res. 2021, 27, 4168–4176. [Google Scholar] [CrossRef]
- Rosell, R.; Dafni, U.; Felip, E.; Curioni-Fontecedro, A.; Gautschi, O.; Peters, S.; Massuti, B.; Palmero, R.; Aix, S.P.; Carcereny, E.; et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): An international, multicentre, single-arm, phase 2 trial. Lancet Respir. Med. 2017, 5, 435–444. [Google Scholar] [CrossRef]
- Seto, T.; Kato, T.; Nishio, M.; Goto, K.; Atagi, S.; Hosomi, Y.; Yamamoto, N.; Hida, T.; Maemondo, M.; Nakagawa, K.; et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): An open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014, 15, 1236–1244. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Aix, S.P.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Luo, W.; Li, W.; Wang, T.; Huang, L.; Xu, F. First-Generation EGFR-TKI Plus Chemotherapy Versus EGFR-TKI Alone as First-Line Treatment in Advanced NSCLC With EGFR Activating Mutation: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Oncol. 2021, 11, 598265. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-H.; Cheng, Y.; Murakami, H.; Yang, P.-C.; He, J.; Nakagawa, K.; Kang, J.; Kim, J.-H.; Wnag, X.; Enatsu, S.; et al. Gefitinib with or without pemetrexed in nonsquamous (NS) non-small cell lung cancer (NSCLC) with EGFR mutation (mut): Final overall survival (OS) results from a randomized phase II study. Ann. Oncol. 2018, 29, viii495–viii496. [Google Scholar] [CrossRef]
- La Monica, S.; Minari, R.; Cretella, D.; Flammini, L.; Fumarola, C.; Bonelli, M.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Madeddu, D.; et al. Third generation EGFR inhibitor osimertinib combined with pemetrexed or cisplatin exerts long-lasting anti-tumor effect in EGFR-mutated pre-clinical models of NSCLC. J. Exp. Clin. Cancer. Res. 2019, 38, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebuzzi, S.E.; Alfieri, R.; La Monica, S.; Minari, R.; Petronini, P.G.; Tiseo, M. Combination of EGFR-TKIs and chemotherapy in advanced EGFR mutated NSCLC: Review of the literature and future perspectives. Crit. Rev. Oncol. Hematol. 2020, 146, 102820. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, Y.; Xu, Z.; Yang, Q.; Zhu, G.; Liao, X.; Chen, X.; Zhu, B.; Duan, Y.; Sun, J. Concurrent EGFR-TKI and Thoracic Radiotherapy as First-Line Treatment for Stage IV Non-Small Cell Lung Cancer Harboring EGFR Active Mutations. Oncologist 2019, 24, 1031. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Xia, L.; Niu, K.; Chen, X.; Lu, D.; Kong, R.; Chen, Z.; Sun, J. Continued EGFR-TKI with concurrent radiotherapy to improve time to progression (TTP) in patients with locally progressive non-small cell lung cancer (NSCLC) after front-line EGFR-TKI treatment. Clin. Transl. Oncol. 2018, 20, 366–373. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Chen, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. LBA50 Mechanisms of acquired resistance to first-line osimertinib: Preliminary dat from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.-M.; A Shepherd, F.; Bazhenova, L.; Lee, J.S.; Chang, G.-C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. AURA3 Investigators. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [Green Version]
- Michels, S.; Heydt, C.; van Veggel, B.; Deschler-Baier, B.; Pardo, N.; Monkhorst, K.; Rüsseler, V.; Stratmann, J.; Griesinger, F.; Steinhauser, S.; et al. Genomic Profiling Identifies Outcome-Relevant Mechanisms of Innate and Acquired Resistance to Third-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy in Lung Cancer. JCO Precis. Oncol. 2019, 3, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mehlman, C.; Cadranel, J.; Rousseau-Bussac, G.; Lacave, R.; Pujals, A.; Girard, N.; Callens, C.; Gounant, V.; Théou-Anton, N.; Friard, S.; et al. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: A multicentric retrospective French study. Lung Cancer 2019, 137, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Wu, Y.L.; Han, J.Y.; Ahn, M.F.; Ramalingam, S.S.; John, T. LBA51 Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations as Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Passiglia, F.; Rizzo, S.; Di Maio, M.; Galvano, A.; Badalamenti, G.; Listì, A.; Gulotta, L.; Castiglia, M.; Fulfaro, F.; Bazan, V.; et al. The diagnostic accuracy of circulating tumor DNA for the detection of EGFR-T790M mutation in NSCLC: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 13379. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Schaller, A.; Beau-Faller, M.; Mennecier, B.; Renaud-Picard, B.; Weingertner, N.; Massard, G.; Quoix, E. Lung Adenocarcinoma with Pulmonary Miliary Metastases and Complex Somatic Heterozygous EGFR Mutation. Case Rep. Oncol. 2014, 7, 769–773. [Google Scholar] [CrossRef]
- Attili, I.; Karachaliou, N.; Conte, P.; Bonanno, L.; Rosell, R. Therapeutic approaches for T790M mutation positive non-small-cell lung cancer. Expert. Rev. Anticancer. Ther. 2018, 18, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Molina-Vila, M.A.; Ruan, S.Y.; Su, K.Y.; Liao, W.Y.; Yu, K.L.; Ho, C.C.; Shih, J.Y.; Yu, C.J.; Yang, J.C.; et al. Coexistence of EGFR T790M mutation and common activating mutations in pretreatment non-small cell lung cancer: A systematic review and meta-analysis. Lung Cancer 2016, 94, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tsui, D.W.Y.; Murtaza, M.; Wong, A.S.C.; Rueda, O.M.; Smith, C.G.; Chandrananda, D.; Soo, R.A.; Lim, H.L.; Goh, B.C.; Caldas, C.; et al. Dynamics of multiple resistance mechanisms in plasma DNA during EGFR-targeted therapies in non-small cell lung cancer. EMBO Mol. Med. 2018, 10, e7945. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011, 1, 352–365. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walthe, Z.; Wurtz, A.; Heynen, G.J.; et al. Downward JReduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Calvayrac, O.; Mazières, J.; Figarol, S.; Marty-Detraves, C.; Raymond-Letron, I.; Bousquet, E.; Farella, M.; Clermont-Taranchon, E.; Milia, J.; Rouquette, I.; et al. The RAS-related GTPase RHOB confers resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer via an AKT-dependent mechanism. EMBO Mol. Med. 2017, 9, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Chaib, I.; Cardona, A.F.; Berenguer, J.; Bracht, J.W.P.; Yang, J.; Cai, X.; Wang, Z.; Hu, C.; Drozdowskyj, A.; et al. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive Non-small Cell Lung Cancer Associated with Poor Prognosis. EBioMedicine 2018, 29, 112–127. [Google Scholar] [CrossRef]
- Guibert, N.; Barlesi, F.; Descourt, R.; Léna, H.; Besse, B.; Beau-Faller, M.; Mosser, J.; Pichon, E.; Merlio, J.P.; Ouafik, L.; et al. Characteristics and Outcomes of Patients with Lung Cancer Harboring Multiple Molecular Alterations: Results from the IFCT Study Biomarkers France. J. Thorac. Oncol. 2017, 12, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Eng, J.; Woo, K.M.; Sima, C.S.; Plodkowski, A.; Hellmann, M.D.; Chaft, J.E.; Kris, M.G.; Arcila, M.E.; Ladanyi, M.; Drilon, A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 1713–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minari, R.; Bordi, P.; Del Re, M.; Facchinetti, F.; Mazzoni, F.; Barbieri, F.; Camerini, A.; Comin, C.E.; Gnetti, L.; Azzoni, C.; et al. Primary resistance to osimertinib due to SCLC transformation: Issue of T790M determination on liquid re-biopsy. Lung Cancer 2018, 115, 21–27. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [Green Version]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; Delmonte, A.; Hsia, T.C.; Laskin, J.; Kim, S.W.; He, Y.; Tsai, C.M.; et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: Osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non-small cell lung cancer. Cancer 2020, 126, 373–380. [Google Scholar] [CrossRef]
- Lu, X.; Yu, L.; Zhang, Z.; Ren, X.; Smaill, J.B.; Ding, K. Targeting EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) resistance mutations in NSCLC: Current developments in medicinal chemistry. Med. Res. Rev. 2018, 38, 1550–1581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.; Müller, J.; Schneider, P.; Lakis, S.; Thress, K.; Wolf, J.; Heukamp, L.; Heuckmann, J.M.; Griesinger, F. A Novel EGFR(C797) Variant Detected in a Pleural Biopsy Specimen from an Osimertinib-Treated Patient Using a Comprehensive Hybrid Capture-Based Next-Generation Sequencing Assay. J. Thorac. Oncol. 2016, 11, e105–e107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, X.C.; Yang, J.J.; Yang, Z.F.; Bai, Y.; Su, J.; Wang, Z.; Zhang, Z.; Shao, Y.; Zhou, Q.; et al. EGFR L792H and G796R: Two Novel Mutations Mediating Resistance to the Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib. J. Thorac. Oncol. 2018, 13, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. 2017, 108, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Gan, J.; Huang, Y.; Zhou, H.; Zhang, L. Acquired EGFR L718V Mutation and Loss of T790M-Mediated Resistance to Osimertinib in a Patient with NSCLC Who Responded to Afatinib. J. Thorac. Oncol. 2019, 14, e274–e275. [Google Scholar] [CrossRef]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFRInhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; He, B.; Zhou, D.; Li, M.; Hu, C. Newly emergent acquired EGFR exon 18 G724S mutation after resistance of a T790M specific EGFR inhibitor osimertinib in non-small-cell lung cancer: A case report. OncoTargets Ther. 2018, 12, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Oztan, A.; Fischer, S.; Schrock, A.; Erlich, R.; Lovly, C.; Stephens, P.; Ross, J.; Miller, V.; Ali, S.; Ou, S.-H.; et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 2017, 111, 84–87. [Google Scholar] [CrossRef]
- Brown, B.P.; Zhang, Y.K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation. Clin. Cancer Res. 2019, 52, 3341–3351. [Google Scholar] [CrossRef] [Green Version]
- Knebel, F.H.; Bettoni, F.; Shimada, A.K.; Cruz, M.; Alessi, J.V.; Negrão, M.V.; Reis, L.F.L.; Katz, A.; Camargo, A.A. Sequential liquid biopsies reveal dynamic alterations of EGFR driver mutations and indicate EGFR amplification as a new mechanism of resistance to osimertinib in NSCLC. Lung Cancer 2017, 108, 238–241. [Google Scholar] [CrossRef]
- Lin, C.C.; Shih, J.Y.; Yu, C.J.; Ho, C.C.; Liao, W.Y.; Lee, J.H.; Tsai, T.H.; Su, K.Y.; Hsieh, M.S.; Chang, Y.L.; et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: A genomic study. Lancet Respir. Med. 2018, 6, 107–116. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Zhao, C.; Jiang, T.; Jia, Y.; Shi, J.; He, Y.; Li, J.; Zhou, F.; Gao, G.; et al. Loss of T790M mutation is associated with early progression to osimertinib in Chinese patients with advanced NSCLC who are harboring EGFR T790M. Lung Cancer 2019, 128, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Hata, A.N. Resistance to First-line Osimertinib in EGFR-mutant NSCLC: Tissue is the Issue. Clin. Cancer. Res. 2020, 26, 2441–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Jänne, P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 2895–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingertner, N.; Meyer, N.; Voegeli, A.C.; Guenot, D.; Renaud, S.; Massard, G.; Falcoz, P.E.; Olland, A.; Mennecier, B.; Gaub, M.P.; et al. Correlation between MET protein expression and MET gene copy number in a Caucasian cohort of non-small cell lung cancers according to the new IASLC/ATS/ERS classification. Pathology 2015, 47, 320–328. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Vojnic, M.; Kubota, D.; Kurzatkowski, C.; Offin, M.; Suzawa, K.; Benayed, R.; Schoenfeld, A.J.; Plodkowski, A.J.; Poirier, J.; Rudin, C.M.; et al. Acquired BRAF Rearrangements Induce Secondary Resistance to EGFR therapy in EGFR-Mutated Lung Cancers. J. Thorac. Oncol. 2019, 14, 802–815. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; et al. Clonal History and Genetic Predictors of Transformation into Small-Cell Carcinomas from Lung Adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Aquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: Distinct natural history of patients with tumors harboring the T790M mutation. Clin. Cancer Res. 2011, 17, 1616–1622. [Google Scholar] [CrossRef] [Green Version]
- Schildhaus, H.U.; Schultheis, A.M.; Rüschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.D.; Schlesinger, A.; Bos, M.; et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res. 2015, 21, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.; Ahmed, A.; Xu, X.; Nuttalli, B.; Luninski, T.; Johnson, J.; Barett, J.; Dougherty, B. Orthgonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar]
- Paweletz, C.P.; Lau, C.J.; Oxnard, G.R. Does testing error underlie liquid biopsy discordance? JCO Precis. Oncol. 2019, 3, 1–2. [Google Scholar] [CrossRef]
- Guibert, N.; Hu, Y.; Feeney, N.; Kuang, Y.; Plagnol, V.; Jones, G.; Howarth, K.; Beeler, J.F.; Paweletz, C.P.; Oxnard, G.R. Amplicon-based next-generation sequencing of plasma cell-free DNA for detection of driver and resistance mutations in advanced non-small cell lung cancer. Ann. Oncol. 2018, 29, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Offin, M.; Stephens, D.; Ni, A.; Lee, A.; Pavlakis, N.; Clarke, S.; Diakos, C.I.; Datta, S.; Tandon, N.; et al. A Prospective Study of Circulating Tumor DNA to Guide Matched Targeted Therapy in Lung Cancers. J. Natl. Cancer Inst. 2019, 111, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Supplee, J.G.; Milan, M.S.D.; Lim, L.P.; Potts, K.T.; Sholl, L.M.; Oxnard, G.R.; Paweletz, C.P. Sensitivity of next-generation sequencing assays detecting oncogenic fusions in plasma cell-free DNA. Lung Cancer 2019, 134, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, M.; Liu, H.; Yin, W. AZD9291 promotes autophagy and inhibits PI3K/Akt pathway in NSCLC cancer cells. J. Cell. Biochem. 2019, 120, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, H.S.; Lee, B.; Kim, H.K.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Lee, S.H. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer 2020, 126, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Offin, M.; Somwar, R.; Rekhtman, N.; Benayed, R.; Chang, J.C.; Plodkowski, A.; Lui, A.J.W.; Eng, J.; Rosenblum, M.; Li, B.T.; et al. Acquired ALK and RET Gene Fusions as Mechanisms of Resistance to Osimertinib in EGFR-Mutant Lung Cancers. JCO Precis. Oncol. 2018, 2, PO.18.00126. [Google Scholar] [CrossRef]
- Nelson, A.W.; Schrock, A.B.; Pavlick, D.C.; Ali, S.M.; Atkinson, E.C.; Chachoua, A. Novel SPECC1L-MET Fusion Detected in Circulating Tumor DNA in a Patient with Lung Adenocarcinoma following Treatment with Erlotinib and Osimertinib. J. Thorac. Oncol. 2019, 14, e27–e29. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, H.; Li, C.; Wang, Z.; Zhang, P.; Yan, X. Transformation to small-cell carcinoma as an acquired resistance mechanism to AZD9291: A case report. Oncotarget 2017, 8, 18609–18614. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Oh, Y.T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef]
- Cho, J.H.; Lim, S.H.; An, H.J.; Kim, K.H.; Park, K.U.; Kang, E.J.; Choi, Y.H.; Ahn, M.S.; Lee, M.H.; Sun, J.M.; et al. Osimertinib for Patients with Non-Small-Cell Lung Cancer Harboring Uncommon EGFR Mutations: A Multicenter, Open-Label, Phase II Trial (KCSG-LU15-09). J. Clin. Oncol. 2020, 38, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Kato, S.; Parulkar, R.; Szeto, C.W.; Sanborn, J.Z.; Vaske, C.J.; Benz, S.C.; Reddy, S.K.; Kurzrock, R. Transcriptomic silencing as a potential mechanism of treatment resistance. JCI Insight 2020, 5, e134824. [Google Scholar] [CrossRef]
- Steuten, L.; Goulart, B.; Meropol, N.J.; Pritchard, D.; Ramsey, S.D. Cost Effectiveness of Multigene Panel Sequencing for Patients with Advanced Non–Small-Cell Lung Cancer. JCO Clin. Cancer Inform. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Teixidó, C.; Giménez-Capitán, A.; Molina-Vila, M.Á.; Peg, V.; Karachaliou, N.; Rodríguez-Capote, A.; Castellví, J.; Rosell, R. RNA Analysis as a Tool to Determine Clinically Relevant Gene Fusions and Splice Variants. Arch. Pathol. Lab. Med. 2018, 142, 474–479. [Google Scholar] [CrossRef] [Green Version]
- Heyer, E.E.; Blackburn, J. Sequencing Strategies for Fusion Gene Detection. BioEssays 2020, 42, 2000016. [Google Scholar] [CrossRef]
- Solomon, J.P.; Hechtman, J.F. Detection of NTRK Fusions: Merits and Limitations of Current Diagnostic Platforms. Cancer Res. 2019, 79, 3163–3168. [Google Scholar] [CrossRef]
- Haynes, B.C.; Blidner, R.A.; Cardwell, R.D.; Zeigler, R.; Gokul, S.; Thibert, J.R.; Chen, L.; Fujimoto, J.; Papadimitrakopoulou, V.A.; Wistuba, I.I.; et al. An Integrated Next-Generation Sequencing System for Analyzing DNA Mutations, Gene Fusions, and RNA Expression in Lung Cancer. Transl. Oncol. 2019, 12, 836–845. [Google Scholar] [CrossRef]
- Kresse, S.H.; Namløs, H.M.; Lorenz, S.; Berner, J.-M.; Myklebost, O.; Bjerkehagen, B.; Meza-Zepeda, L.A. Evaluation of Commercial DNA and RNA Extraction Methods for High-Throughput Sequencing of FFPE Samples. PLoS ONE 2018, 13, e0197456. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.M.; Forbes, T.P.; Lund, S.; Cole, K.D.; He, H.J.; Karlovich, C.; Paweletz, C.P.; Stetson, D.; Yee, L.M.; Connors, D.E.; et al. Validation of ctDNA Quality Control Materials Through a Precompetitive Collaboration of the Foundation for the National Institutes of Health. JCO Precis. Oncol. 2021, 5, PO.20.00528. [Google Scholar] [PubMed]
- Vaughn, C.P.; Costa, J.L.; Feilotter, H.E.; Petraroli, R.; Bagai, V.; Rachiglio, A.M.; Marino, F.Z.; Tops, B.; Kurth, H.M.; Sakai, K.; et al. Simultaneous Detection of Lung Fusions Using a Multiplex RT-PCR next Generation Sequencing-Based Approach: A Multi-Institutional Research Study. BMC Cancer 2018, 18, 828. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xu, C.; He, Y.; Li, F.; Wang, W.; Zhu, Y.; Gao, Y.; Ji, M.; Chen, M.; Lai, J.; et al. Simultaneous Detection of Gene Fusions and Base Mutations in Cancer Tissue Biopsies by Sequencing Dual Nucleic Acid Templates in Unified Reaction. Clin. Chem. 2020, 66, 178–187. [Google Scholar] [CrossRef]
- Aggarwal, C.; Rolfo, C.D.; Oxnard, G.R.; Gray, J.E.; Sholl, L.M.; Gandara, D.R. Strategies for the successful implementation of plasma-based NSCLC genotyping in clinical practice. Nat. Rev. Clin. Oncol. 2021, 18, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical Relevance of Blood-Based CtDNA Analysis: Mutation Detection and Beyond. Br. J. Cancer 2021, 124, 345–358. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Bruno, R.; Fontanini, G. Next Generation Sequencing for Gene Fusion Analysis in Lung Cancer: A Literature Review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef] [PubMed]
- Plan France Médecine Génomique 2025. Available online: https://pfmg2025.aviesan.fr/ (accessed on 14 July 2021).
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing Depth and Coverage: Key Considerations in Genomic Analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ballester, L.Y.; Luthra, R.; Kanagal-Shamanna, R.; Singh, R.R. Advances in Clinical Next-Generation Sequencing: Target Enrichment and Sequencing Technologies. Expert Rev. Mol. Diagn. 2016, 16, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Hondelink, L.M.; Solleveld-Westerink, N.; Uljee, S.M.; Ruano, D.; Cleton-Jansen, A.M.; Thüsen, J.H.v.d.; Ramai, S.R.S.; Postmus, P.E.; Roggen, J.F.G.v.; et al. Optimizing Mutation and Fusion Detection in NSCLC by Sequential DNA and RNA Sequencing. J. Thorac. Oncol. 2020, 15, 1000–1014. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored Multiplex PCR for Targeted Next-Generation Sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef]
- Davies, K.D.; Le, A.T.; Sheren, J.; Nijmeh, H.; Gowan, K.; Jones, K.L.; Varella-Garcia, M.; Aisner, D.L.; Doebele, R.C. Comparison of Molecular Testing Modalities for Detection of ROS1 Rearrangements in a Cohort of Positive Patient Samples. J. Thorac. Oncol. 2018, 13, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Garinet, S.; Blons, H. Nouvelles techniques en biologie moléculaire. Rev. Mal. Respir. Actual. 2021, 13, 464–472. [Google Scholar]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the Analytical Validity of Circulating Tumor DNA Sequencing Assays for Precision Oncology. Nat. Biotechnol. 2021, 39, 1–14. [Google Scholar] [CrossRef]
- McCoach, C.E.; Blakely, C.M.; Banks, K.C.; Levy, B.; Chue, B.M.; Raymond, V.M.; Le, A.T.; Lee, C.E.; Diaz, J.; Waqar, S.N.; et al. Clinical Utility of Cell-Free DNA for the Detection of ALK Fusions and Genomic Mechanisms of ALK Inhibitor Resistance in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.A.; Chung, J.H.; Kennedy, M.; Hughes, J.D.; Chennagiri, N.; Lieber, D.S.; Fendler, B.; Young, L.; Zhao, M.; Coyne, M.; et al. Analytical Validation of a Hybrid Capture–Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA. J. Mol. Diagn. 2018, 20, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Cadranel, J.; Ruppert, A.M.; Beau-Faller, M.; Wislez, M. Therapeutic strategy for advanced EGFR mutant non-small-cell lung carcinoma. Crit. Rev. Oncol. Hematol. 2013, 88, 477–493. [Google Scholar]
- Alì, G.; Bruno, R.; Savino, M.; Giannini, R.; Pelliccioni, S.; Menghi, M.; Boldrini, L.; Proietti, A.; Chella, A.; Ribechini, A.; et al. Analysis of Fusion Genes by NanoString System: A Role in Lung Cytology? Arch. Pathol. Lab. Med. 2018, 142, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.T.E.; Goytain, A.; Tucker, T.; Karsan, A.; Lee, C.-H.; Nielsen, T.O.; Ng, T.L. Development and Evaluation of a Pan-Sarcoma Fusion Gene Detection Assay Using the NanoString NCounter Platform. J. Mol. Diagn. JMD 2018, 20, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Sutton, B.C.; Birse, R.T.; Maggert, K.; Ray, T.; Hobbs, J.; Ezenekwe, A.; Kazmierczak, J.; Mosko, M.; Kish, J.; Bullock, A.; et al. Assessment of Common Somatic Mutations of EGFR, KRAS, BRAF, NRAS in Pulmonary Non-Small Cell Carcinoma Using IPLEX® HS, a New Highly Sensitive Assay for the MassARRAY® System. PLoS ONE 2017, 12, e0183715. [Google Scholar] [CrossRef] [Green Version]
- Belloum, Y.; Janning, M.; Mohme, M.; Simon, R.; Kropidlowski, J.; Sartori, A.; Irwin, D.; Westphal, M.; Lamszus, K.; Loges, S.; et al. Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay. Cells 2020, 9, 2337. [Google Scholar] [CrossRef] [PubMed]
- Lamy, P.-J.; van der Leest, P.; Lozano, N.; Becht, C.; Duboeuf, F.; Groen, H.J.M.; Hilgers, W.; Pourel, N.; Rifaela, N.; Schuuring, E.; et al. Mass Spectrometry as a Highly Sensitive Method for Specific Circulating Tumor DNA Analysis in NSCLC: A Comparison Study. Cancers 2020, 12, E3002. [Google Scholar] [CrossRef] [PubMed]
- Denis, J.; Guillerm, E.; Coulet, F.; Larsen, A.; Lacorte, J. The Role of BEAMing and Digital PCR for Multiplexed Analysis in Molecular Oncology in the Era of Next-Generation Sequencing. Mol. Diagn. Ther. 2017, 21, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.S.; Huggett, J.F.; Tzonev, S. Fundamentals of Multiplexing with Digital PCR. Biomol. Detect. Quantif. 2016, 10, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Jones, V.; Topkas, E.; Harraway, J. Reduced Sensitivity for EGFR T790M Mutations Using the Idylla EGFR Mutation Test. J. Clin. Pathol. 2021, 74, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Bocciarelli, C.; Cohen, J.; Pelletier, R.; Tran Van Nhieu, J.; Derman, J.; Favre, L.; Bourgogne, A.; Monnet, I.; Chouaid, C.; Pujals, A.; et al. Evaluation of the Idylla system to detect the EGFRT790M mutation using extracted DNA. Pathol. Res. Pract. 2020, 216, 152773. [Google Scholar] [CrossRef]
- Ricordel, C.; Friboulet, L.; Facchinetti, F.; Soria, J.C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018, 29 (Suppl. 1), i28–i37. [Google Scholar]
- Vendrell, J.A.; Mazieres, J.; Senal, R.; Rouquette, I.; Quantin, X.; Pujol, J.L.; Roch, B.; Bouidioua, A.; Godreuil, S.; Coyaud, E.; et al. Ultra-sensitive EGFR T790M Detection as an Independent Prognostic Marker for Lung Cancer Patients Harboring EGFR del19 Mutations and Treated with First-generation TKIs. Clin. Cancer Res. 2019, 25, 4280–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 784–1793. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Radhakrishna, K.V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.; Xie, Y.; Lim, K.; Cejas, P.; et al. Long HW, Gray NS, Jänne PA. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Thress, K.S.; Brant, R.; Carr, T.H.; Dearden, S.; Jenkins, S.; Brown, H.; Hammett, T.; Cantarini, M.; Barrett, J.C. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer 2015, 90, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; McCormack, R.; Webster, A.; Milenkova, T. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: A phase-IV, open-label, single-arm study. Br. J. Cancer. 2014, 110, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.; et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.E.; Okamoto, I.; Sriuranpong, V.; Vansteenkiste, J.; Imamura, F.; Lee, J.S.; Pang, Y.K.; Cobo, M.; Kasahara, K.; Cheng, Y.; et al. Tissue and Plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR Tyrosine Kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6644–6652. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Jänne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Vollbrecht, C.; Lehmann, A.; Lenze, D.; Hummel, M. Validation and comparison of two NGS assays for the detection of EGFR T790M resistance mutation in liquid biopsies of NSCLC patients. Oncotarget 2018, 9, 18529–18539. [Google Scholar] [CrossRef] [Green Version]
- Li, B.T.; Janku, F.; Jung, B.; Hou, C.; Madwani, K.; Alden, R.; Razavi, P.; Reis-Filho, J.S.; Shen, R.; Isbell, J.M.; et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann. Oncol. 2019, 30, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Bordi, P.; Del Re, M.; Minari, R.; Rofi, E.; Buti, S.; Restante, G.; Squadrilli, A.; Crucitta, S.; Casartelli, C.; Gnetti, L.; et al. From the beginning to resistance: Study of plasma monitoring and resistance mechanisms in a cohort of patients treated with osimertinib for advanced T790M-positive NSCLC. Lung Cancer 2019, 131, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Gregorc, V.; Karachaliou, N.; Rosell, R.; Santarpia, M. Mechanisms of resistance to osimertinib. J. Thorac. Dis. 2020, 12, 2851–2858. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Rooney, M.; Nagy, R.J.; Lin, J.J.; Chin, E.; Ferris, L.A.; Ackil, J.; Lennerz, J.K.; Lanman, R.B.; Gainor, J.F.; et al. Molecular Analysis of Plasma from Patients with ROS1-Positive NSCLC. J. Thorac. Oncol. 2019, 14, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Brannon, A.R.; Ferris, L.A.; Campbell, C.D.; Lin, J.J.; Schultz, K.R.; Ackil, J.; Stevens, S.; Dardaei, L.; Yoda, S.; et al. Tracking the Evolution of Resistance to ALK Tyrosine Kinase Inhibitors through Longitudinal Analysis of Circulating Tumor DNA. JCO Precis. Oncol. 2018, 2, 1–14. [Google Scholar] [CrossRef]
- Mezquita, L.; Swalduz, A.; Jovelet, C.; Ortiz-Cuaran, S.; Howarth, K.; Planchard, D.; Avrillon, V.; Recondo, G.; Marteau, S.; Benitez, J.C.; et al. Clinical Relevance of an Amplicon-Based Liquid Biopsy for Detecting ALK and ROS1 Fusion and Resistance Mutations in Patients with Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2020, 4, 272–282. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Lennerz, J.K. Personalized Diagnostic Workflows: The Next Wave of Precision Medicine in NSCLC. J. Thorac. Oncol. 2020, 15, 888–890. [Google Scholar] [CrossRef]
- Labbé, C.; Cabanero, M.; Korpanty, G.J.; Tomasini, P.; Doherty, M.K.; Mascaux, C.; Jao, K.; Pitcher, B.; Wang, R.; Pintilie, M.; et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 2017, 111, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer. Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef]
- Miller, T.E.; Yang, M.; Bajor, D.; Friedman, J.D.; Chang, R.Y.C.; Dowlati, A.; Willis, J.E. Sadri N Clinical utility of reflex testing using focused next-generation sequencing for management of patients with advanced lung adenocarcinoma. J. Clin. Pathol. 2018, 71, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Dineen, T.; Lim, S.M.; Park, C.-W.; Hsieh, J.; Woessner, R.; Zhang, Z.; Wilson, K.; Eno, M.; Wilson, D.; et al. 1296P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann. Oncol. 2020, 1, S839. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, S.; Pai, K.S.R.; Nayak, Y. Advances in targeting EGFR allosteric site as anti-NSCLC therapy to overcome the drug resistance. Pharmacol. Rep. 2020, 72, 799–813. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhang, L.; Kim, D.-W.; Liu, X.; Lee, D.H.; Yang, J.C.-H.; Ahn, M.-J.; Vansteenkiste, J.F.; Su, W.-C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.-W.; Soo, R.A.; Kim, S.-W.; Pan, H.; et al. INSIGHT Investigators. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): An open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar]
- Scagliotti, G.V.; Shuster, D.; Orlov, S.; von Pawel, J.; Shepherd, F.A.; Ross, J.S.; Wang, Q.; Schwartz, B.; Akerley, W. Tivantinib in Combination with Erlotinib versus Erlotinib Alone for EGFR-Mutant NSCLC: An Exploratory Analysis of the Phase 3 MARQUEE Study. J. Thorac. Oncol. 2018, 13, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Govindan, R.; Eaton, K.D.; Otterson, G.A.; Gutierrez, M.E.; Mita, A.C.; Argiris, A.; Brega, N.M.; Usari, T.; Tan, W.; et al. Phase I Results from a Study of Crizotinib in Combination with Erlotinib in Patients with Advanced Nonsquamous Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Vijayaraghavan, S.; Lipfert, L.; Chevalier, K.; Bushey, B.S.; Henley, B.; Lenhart, R.; Sendecki, J.; Beqiri, M.; Milliar, H.J.; Packman, K.; et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/cMet Bispecific Antibody, Induces Receptor Downmodulation and Antitumor Activity by Monocyte/Macrophage Trogocytosis. Mol. Cancer Ther. 2020, 19, 2044–2056. [Google Scholar] [CrossRef]
- Li, B.T.; Michelini, F.; Misale, S.; Cocco, E.; Baldino, L.; Cai, Y.; Shifman, S.; Tu, H.-Y.; Myers, M.L.; Xu, C.; et al. HER2-Mediated Internalization of Cytotoxic Agents in ERBB2 Amplified or Mutant Lung Cancers. Cancer Discov. 2020, 10, 674–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Monica, S.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Digiacomo, G.; Flammini, L.; Barocelli, E.; Minari, R.; Naldi, N.; et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J. Exp. Clin. Cancer Res. 2017, 36, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurutani, J.; Iwata, H.; Krop, I.; Jänne, P.A.; Doi, T.; Takahashi, S.; Park, H.; Redfern, C.; Tamura, K.; Wise-Draper, T.M.; et al. Targeting HER2 with Trastuzumab Deruxtecan: A Dose-Expansion, Phase I Study in Multiple Advanced Solid Tumors. Cancer Discov. 2020, 10, 688–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef]
- Yu, H. P2.01-22 ORCHARD: A phase II platform study in patients with advanced NSCLC who have progressed on first-line osimertinib therapy. J. Thorac. Oncol. 2019, 14, S647. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Kim, S.W.; Wu, Y.L.; Nakagawa, K.; Yang, J.J.; Ahn, M.J.; Wang, J.; Yang, J.C.-H.; Lu, Y.; Atagi, S.; et al. Gefitinib Plus Chemotherapy Versus Chemotherapy in Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer Resistant to First-Line Gefitinib (IMPRESS): Overall Survival and Biomarker Analyses. J. Clin. Oncol. 2017, 35, 4027–4034. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Man, J.; Lord, S.J.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C.-H. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- To, K.K.W.; Fong, W.; Cho, W.C.S. Immunotherapy in Treating EGFR-Mutant Lung Cancer: Current Challenges and New Strategies. Front. Oncol. 2021, 11, 635007. [Google Scholar] [CrossRef]
- Chen, K.; Cheng, G.; Zhang, F.; Zhu, G.; Xu, Y.; Yu, X.; Huang, Z.; Fan, Y. PD-L1 expression and T cells infiltration in patients with uncommon EGFR-mutant non-small cell lung cancer and the response to immunotherapy. Lung Cancer Amst. Neth. 2020, 142, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Yonesaka, K. HER2-/HER3-Targeting Antibody—Drug Conjugates for Treating Lung and Colorectal Cancers Resistant to EGFR Inhibitors. Cancers. 2021, 13, 1047. [Google Scholar] [CrossRef]
- Inamura, K.; Yokouchi, Y.; Kobayashi, M.; Ninomiya, H.; Sakakibara, R.; Subat, S.; Nagano, H.; Nomura, K.; Okumura, S.; Shibutani, T.; et al. Association of tumor TROP2 expression with prognosis varies among lung cancer subtypes. Oncotarget 2017, 8, 28725–28735. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer. 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Meador, C.B.; Milan, M.S.D.; Hu, E.Y.; Awad, M.M.; Rabin, M.S.; Paweletz, C.P.; Hartmaier, R.; Laus, G.; Oxnard, G.R. High Sensitivity of Plasma Cell-Free DNA Genotyping in Cases with Evidence of Adequate Tumor Content. JCO Precis. Oncol. 2021, 5, 921–930. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generation EGFR-TKI | Study | Agent | EGFR Mutation | N | Median PFS (Months) | PFS HR [CI] |
|---|---|---|---|---|---|---|
| First generation | IPASS [33] | Gefitinib | Del19/ L858R | 261 | 9.5 vs 6.3 | 0.48 (0.34–0.67) |
| NEJGSG002 [34] | 228 | 10.8 vs 5.4 | 0.30 (0.24–0.44) | |||
| WJTOG3405 [35] | 177 | 9.2 vs 6.3 | 0.49 (0.38–0.72) | |||
| OPTIMAL [36] | Erlotinib | Del19/ L858R | 154 | 13.1 vs 4.6 | 0.16 (0.10–0.26) | |
| EURTAC [37] | 173 | 9.7 vs 5.2 | 0.37 (0.25–0.54) | |||
| ENSURE [38] | 217 | 11.0 vs 5.5 | 0.34 (0.22–0.51) | |||
| CONVINCE [39] | Icotinib | Del19/ L858R | 285 | 11.2 vs 7.9 | 0.61 (0.43–0.87) | |
| Second generation | LUX-Lung 3 [40] | Afatinib | Del19/ L858R | 345 | 11.1 vs 6.9 | 0.58 (0.43–0.78) |
| LUX-Lung 6 [41] | 364 | 11.0 vs 5.6 | 0.28 (0.20–0.39) | |||
| ARCHER 1050 [42] | Dacomitinib | Del19/ L858R +/- T790M | 452 | 14.7 vs 9.2 | 0.59 (0.47–0.74) | |
| Third generation | FLAURA [10] | Osimertinib | Del19/ L858R | 556 | 18.9 vs 10.2 | 0.46 (0.37–0.57) |
| Others EGFR ins20 inhibitors | ZENITH20-cohort1 [43] | Poziotinib | EGFR/HER2 ins20 | 88 | 4.1 | |
| Phase I/II [44] | Mobocertinib | EGFR ins20 EGFR ins20 | 70 | 7.3 | ||
| CHRYSALIS [45] | Amivantamab | 50 | 8.3 |
| Technique: Multiplex Strategy | LoD | Sample | M | CNV | Trsl | Exp | Use |
|---|---|---|---|---|---|---|---|
| High throughput | |||||||
| WGS-WES | 5% | Frozen | XX | R | |||
| WTS-RNAseq | 5% | Frozen | XX | X | R | ||
| Targeted NGS | |||||||
| DNA | |||||||
| FoundationOne CDx® | 2–5% | FFPE | XX | (X) | X | R, C | |
| FoundationOne Liquid CDx® | 0.2–1% | cfDNA | XX | (X) | X | R, C | |
| Guardant Assay® | 0.2–0.4% | FFPE, cfDNA | XX | (X) | X | R, C | |
| Enlarged custom targeted panels | 2–5% | FFPE, cfDNA | XX | (X) | C | ||
| RNA | |||||||
| TruSight RNA fusion panel® (Illumina) | 10–15% | FFPE | X | XX | X | C | |
| Targeted RNAscan custom® (Qiagen) | 10–15% | FFPE | X | XX | X | C | |
| Oncomine Focus® (Thermo Fischer) | 10–15% | FFPE | X | XX | X | C | |
| FusionPlex kit® (ArcherDx) | 10–15% | FFPE | X | XX | X | C | |
| Technique, targeted methods | LoD | Sample | M | CNV | Trsl | Exp | Use |
| Nanostring® | 10000 transcript copies | FFPE | X | X | X | R | |
| Real-time PCR (Cobas®, Therascreen®) | 1–5% | FFPE, cfDNA | X | X | C | ||
| Idylla® | 5% | FFPE, cfDNA | X | C | |||
| Mass-Array® | 1–2% | FFPE, cfDNA | X | C | |||
| dPCR | 0.1–0.01% | FFPE, cfDNA | X | X | R, C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reita, D.; Pabst, L.; Pencreach, E.; Guérin, E.; Dano, L.; Rimelen, V.; Voegeli, A.-C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021, 13, 4926. https://doi.org/10.3390/cancers13194926
Reita D, Pabst L, Pencreach E, Guérin E, Dano L, Rimelen V, Voegeli A-C, Vallat L, Mascaux C, Beau-Faller M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers. 2021; 13(19):4926. https://doi.org/10.3390/cancers13194926
Chicago/Turabian StyleReita, Damien, Lucile Pabst, Erwan Pencreach, Eric Guérin, Laurent Dano, Valérie Rimelen, Anne-Claire Voegeli, Laurent Vallat, Céline Mascaux, and Michèle Beau-Faller. 2021. "Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring" Cancers 13, no. 19: 4926. https://doi.org/10.3390/cancers13194926