
Organ-Specific Uptake of Extracellular Vesicles Secreted by Urological Cancer Cells


 , ,
, ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
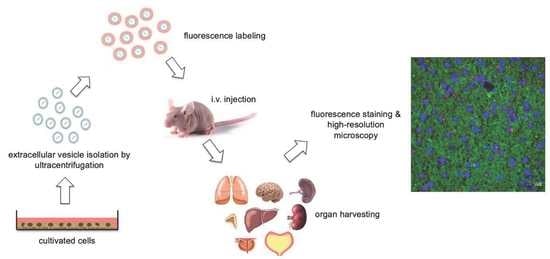
2. Materials and Methods
3. Results
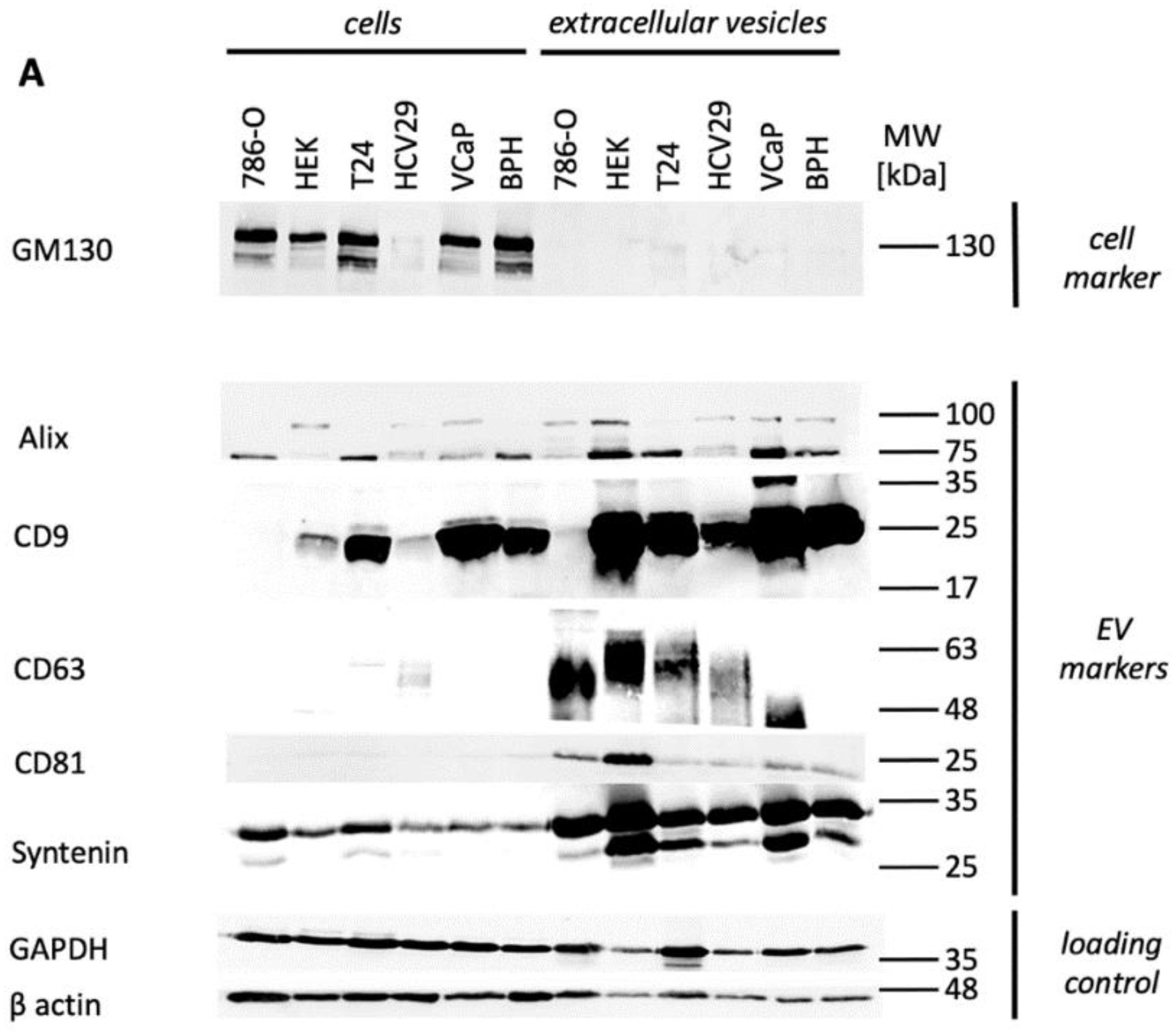
3.1. EV Characterization
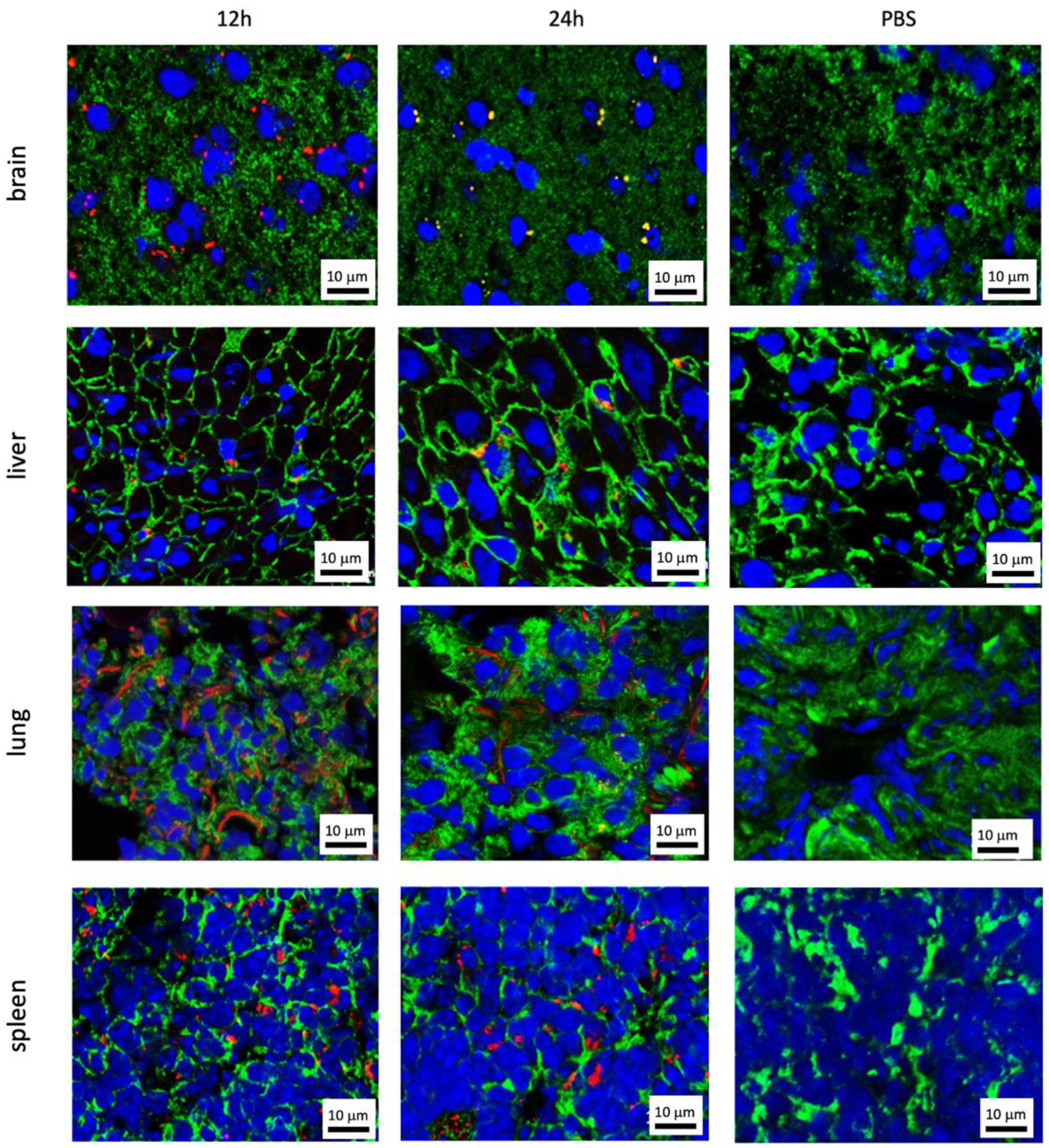
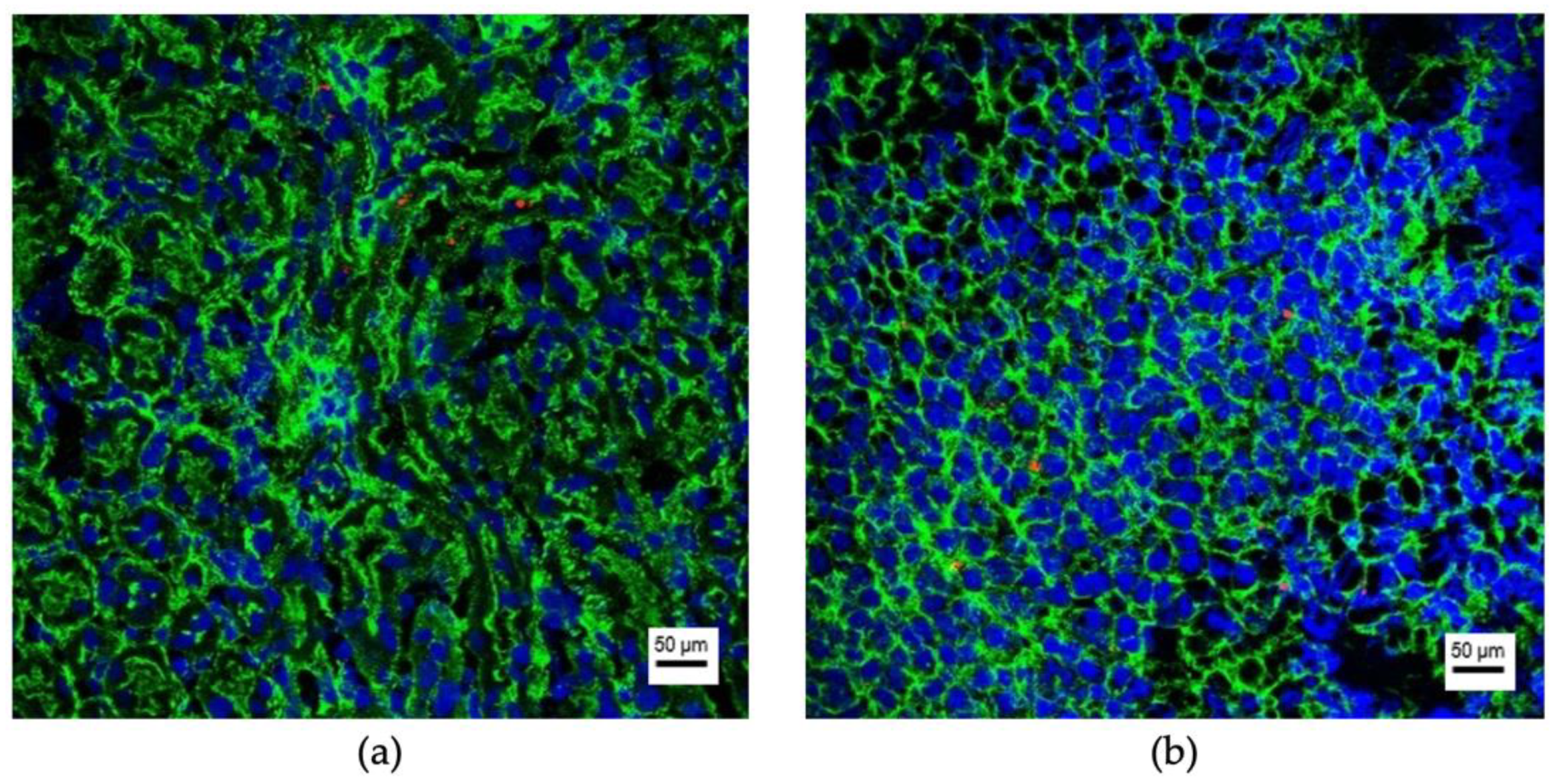
3.2. Organ-Specific Uptake of EVs
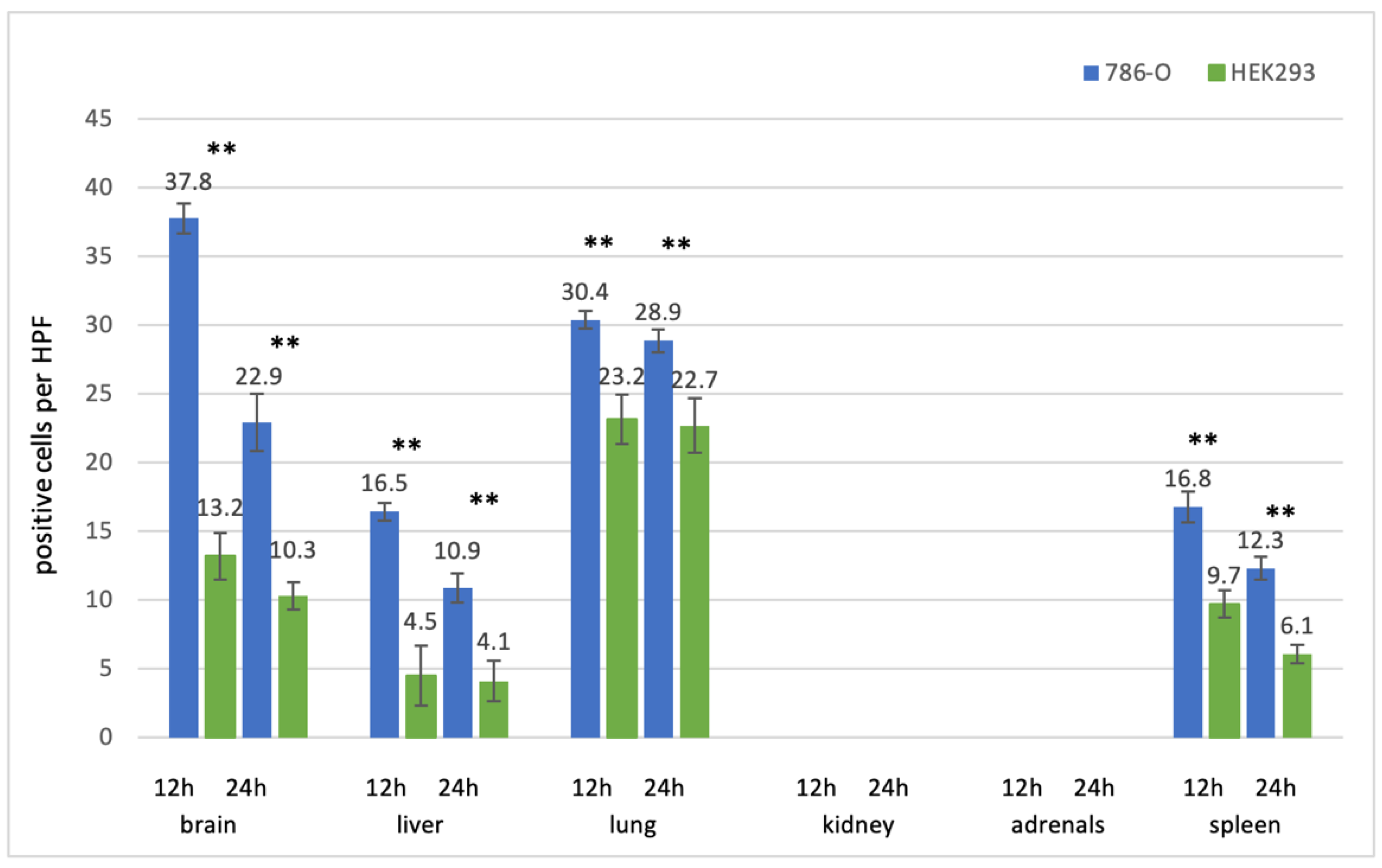
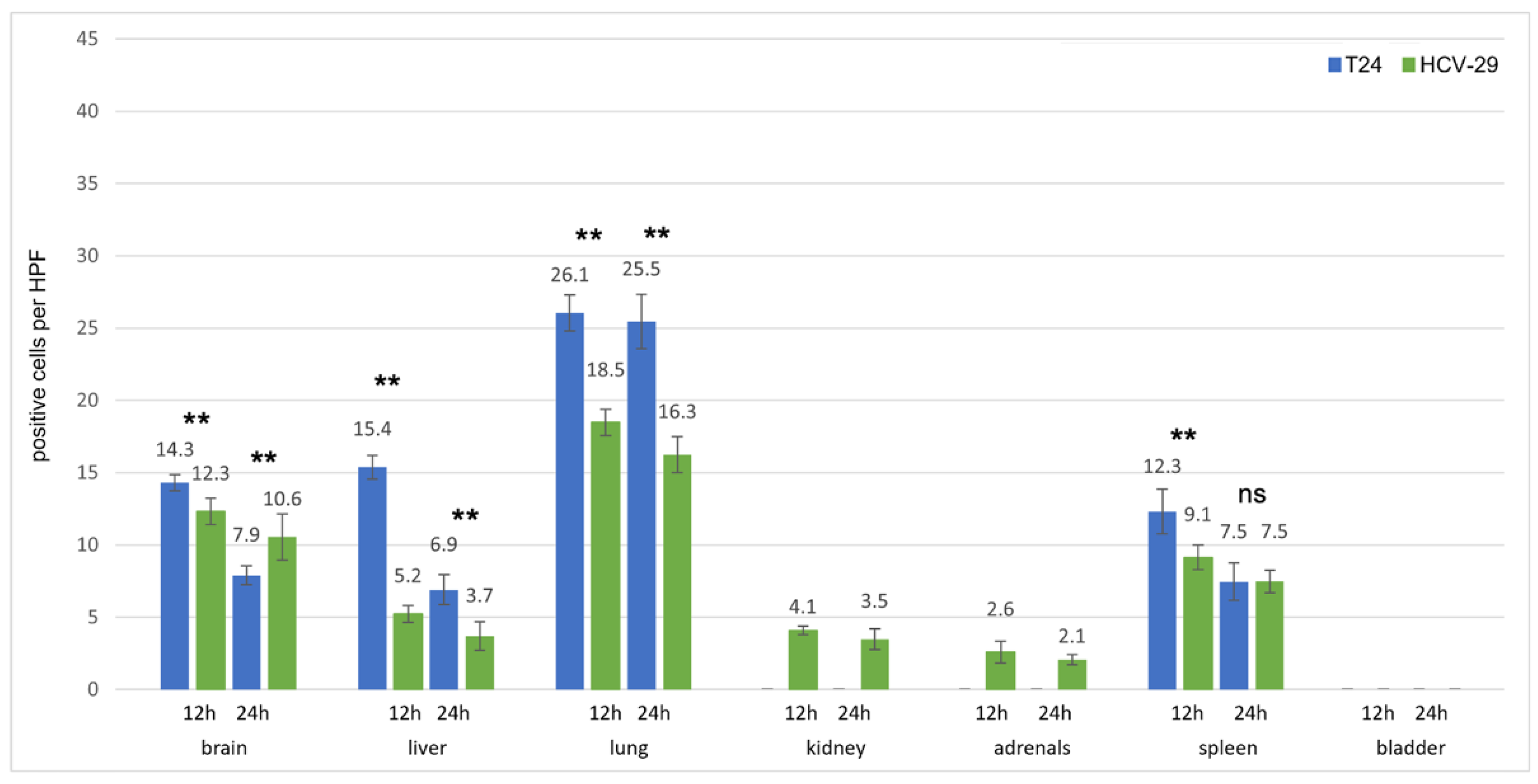
3.2.1. Prostate Cell Lines
3.2.2. Kidney Cell Lines
3.2.3. Bladder Cell Lines
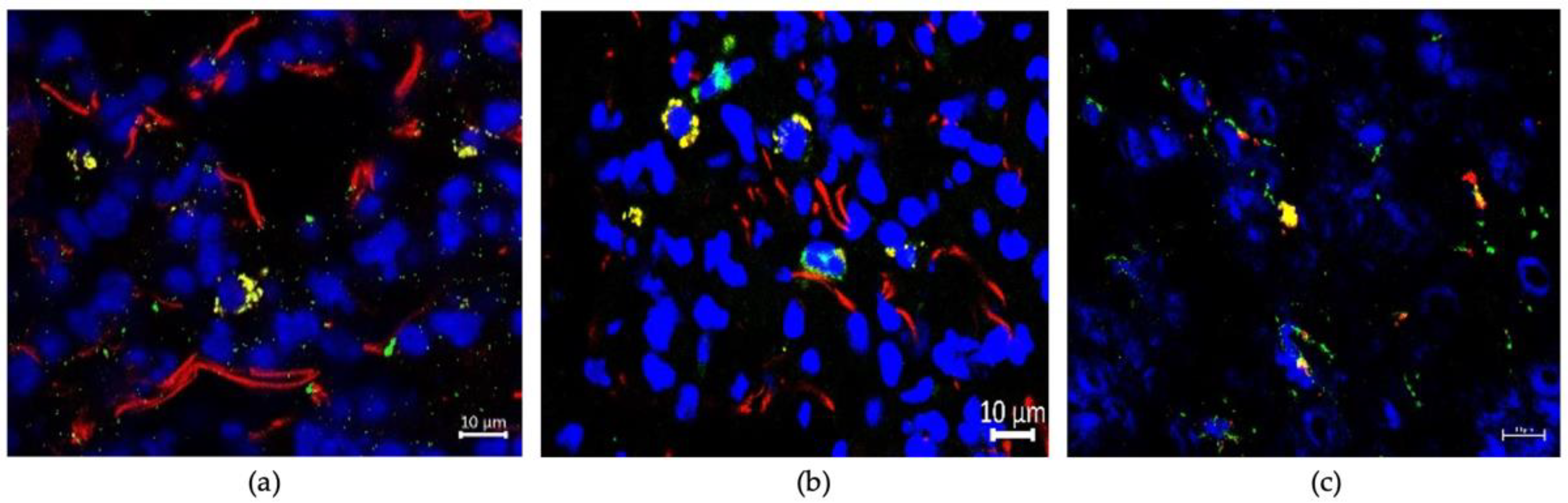
3.3. Uptaking Cell Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bianchi, M.; Sun, M.; Jeldres, C.; Shariat, S.F.; Trinh, Q.-D.; Briganti, A.; Tian, Z.; Schmitges, J.; Graefen, M.; Perrotte, P.; et al. Distribution of metastatic sites in renal cell carcinoma: A population-based analysis. Ann. Oncol. 2011, 23, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.-D.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2013, 74, 210–216. [Google Scholar] [CrossRef]
- Bianchi, M.; Roghmann, F.; Becker, A.; Sukumar, S.; Briganti, A.; Menon, M.; Karakiewicz, P.I.; Sun, M.; Noldus, J.; Trinh, Q.-D. Age-stratified distribution of metastatic sites in bladder cancer: A population-based analysis. Can. Urol. Assoc. J. 2014, 8, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.R.; Yuan, W.; Wagner, S.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 Destroys Vascular Endothelial Barriers to Promote Metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Rumney, R.; Schoof, E.M.; Perryman, L.; Høye, A.; Agrawal, A.; Bird, D.; Ab.Latif, N.B.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Silva, B.; Aiello, N.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nature 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Linxweiler, J.; Junker, K. Extracellular vesicles in urological malignancies: An update. Nat. Rev. Urol. 2019, 17, 11–27. [Google Scholar] [CrossRef]
- Möller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]
- Zeuschner, P.; Linxweiler, J.; Junker, K. Non-coding RNAs as biomarkers in liquid biopsies with a special emphasis on extracellular vesicles in urological malignancies. Expert Rev. Mol. Diagn. 2019, 20, 151–167. [Google Scholar] [CrossRef]
- Himbert, D.; Zeuschner, P.; Ayoubian, H.; Heinzelmann, J.; Stöckle, M.; Junker, K. Characterization of CD147, CA9, and CD70 as Tumor-Specific Markers on Extracellular Vesicles in Clear Cell Renal Cell Carcinoma. Diagnostics 2020, 10, 1034. [Google Scholar] [CrossRef]
- Linxweiler, J.; Körbel, C.; Müller, A.; Hammer, M.; Veith, C.; Bohle, R.M.; Stöckle, M.; Junker, K.; Menger, M.D.; Saar, M. A novel mouse model of human prostate cancer to study intraprostatic tumor growth and the development of lymph node metastases. Prostate 2018, 78, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Knuuttila, M.; Yatkin, E.; Kallio, J.; Savolainen, S.; Laajala, T.D.; Aittokallio, T.; Oksala, R.; Häkkinen, M.; Keski-Rahkonen, P.; Auriola, S.; et al. Castration Induces Up-Regulation of Intratumoral Androgen Biosynthesis and Androgen Receptor Expression in an Orthotopic VCaP Human Prostate Cancer Xenograft Model. Am. J. Pathol. 2014, 184, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y.; Hong, T.; Cheng, B.; Gan, S.; Chen, L.; Zhang, J.; Zuo, L.; Li, J.; Cui, X. Blocking the autocrine regulatory loop of Gankyrin/STAT3/CCL24/CCR3 impairs the progression and pazopanib resistance of clear cell renal cell carcinoma. Cell Death Dis. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strube, A.; Stepina, E.; Mumberg, D.; Scholz, A.; Hauff, P.; Käkönen, S.-M. Characterization of a new renal cell carcinoma bone metastasis mouse model. Clin. Exp. Metastasis 2010, 27, 319–330. [Google Scholar] [CrossRef]
- Kawai, Y.; Imada, K.; Akamatsu, S.; Zhang, F.; Seiler, R.; Hayashi, T.; Leong, J.; Beraldi, E.; Saxena, N.; Kretschmer, A.; et al. Paternally Expressed Gene 10 (PEG10) Promotes Growth, Invasion, and Survival of Bladder Cancer. Mol. Cancer Ther. 2020, 19, 2210–2220. [Google Scholar] [CrossRef]
- Brito, R.B.O.; Malta, C.S.; Souza, D.M.; Matheus, L.H.G.; Matos, Y.; Silva, C.S.; Ferreira, J.M.; Nunes, V.S.; Franca, C.; Dellê, H. 1-Methyl-D-Tryptophan Potentiates TGF-β-Induced Epithelial-Mesenchymal Transition in T24 Human Bladder Cancer Cells. PLoS ONE 2015, 10, e0134858. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huyan, T.; Cai, S.; Huang, Q.; Zhang, M.; Peng, H.; Zhang, Y.; Liu, N.; Zhang, W. The role of exosomal miR-375-3p: A potential suppressor in bladder cancer via the Wnt/β-catenin pathway. FASEB J. 2020, 34, 12177–12196. [Google Scholar] [CrossRef]
- Guo, J.; Chen, Z.; Jiang, H.; Yu, Z.; Peng, J.; Xie, J.; Li, Z.; Wu, W.; Cheng, Z.; Xiao, K. The lncRNA DLX6-AS1 promoted cell proliferation, invasion, migration and epithelial-to-mesenchymal transition in bladder cancer via modulating Wnt/β-catenin signaling pathway. Cancer Cell Int. 2019, 19, 312. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhu, H.; Chen, H.; Xia, J.; Zhang, F.; Xu, R.; Lin, Q. Glucose promotes epithelial-mesenchymal transitions in bladder cancer by regulating the functions of YAP1 and TAZ. J. Cell. Mol. Med. 2020, 24, 10391–10401. [Google Scholar] [CrossRef]
- Wu, K.; Fan, J.; Zhang, L.; Ning, Z.; Zeng, J.; Zhou, J.; Li, L.; Chen, Y.; Zhang, T.; Wang, X.; et al. PI3K/Akt to GSK3β/β-catenin signaling cascade coordinates cell colonization for bladder cancer bone metastasis through regulating ZEB1 transcription. Cell. Signal. 2012, 24, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Santos, M.F.; Karbanová, J.; Kurth, T.; Rappa, G.; Lorico, A. Uptake and Fate of Extracellular Membrane Vesicles: Nucleoplasmic Reticulum-Associated Late Endosomes as a New Gate to Intercellular Communication. Cells 2020, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.; Pink, R.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Kanada, M.; Bachmann, M.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Thomsen, M.B.H.; Nordentoft, I.K.; Lamy, P.; Vang, S.; Reinert, L.; Mapendano, C.K.; Høyer, S.; Ørntoft, T.F.; Jensen, J.B.; Dyrskjøt, L. Comprehensive multiregional analysis of molecular heterogeneity in bladder cancer. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2020, 18, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.G.; Ham, S.; Shin, H.; Chai, E.P.Z.; Lek, E.S.H.; Lobb, R.J.; Müller, A.F.; Mathivanan, S.; Yeo, B.; Choi, Y.; et al. Tumor microenvironmental cytokines bound to cancer exosomes determine uptake by cytokine receptor-expressing cells and biodistribution. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Shimizu, A.; Sawada, K.; Kobayashi, M.; Yamamoto, M.; Yagi, T.; Kinose, Y.; Kodama, M.; Hashimoto, K.; Kimura, T. Exosomal CD47 Plays an Essential Role in Immune Evasion in Ovarian Cancer. Mol. Cancer Res. 2021, 19, 1583–1595. [Google Scholar] [CrossRef]
- Williams, C.; Pazos, R.; Royo, F.; González, E.; Roura-Ferrer, M.; Martinez, A.; Gamiz, J.; Reichardt, N.-C.; Falcón-Pérez, J.M. Assessing the role of surface glycans of extracellular vesicles on cellular uptake. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.-E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood–Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, G.; Hoshino, A.; Kenific, C.M.; Matei, I.R.; Steiner, L.; Freitas, D.; Kim, H.S.; Oxley, P.R.; Scandariato, I.; Casanova-Salas, I.; et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nature 2019, 21, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Liu, Y.; Wu, S.-Y.; Wu, K.; Sharma, S.; Mo, Y.-Y.; Feng, J.; Sanders, S.; Jin, G.; Singh, R.; et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018, 78, 4316–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre–metastatic Niche. Cancer Res. 2015, 76, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; Macfarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Probert, C.; Dottorini, T.; Speakman, A.; Hunt, S.; Nafee, T.; Fazeli, A.; Wood, S.; Brown, J.E.; James, V. Communication of prostate cancer cells with bone cells via extracellular vesicle RNA; a potential mechanism of metastasis. Oncogene 2018, 38, 1751–1763. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Liu, J.; Xu, F.; Lin, X.; Zhong, J.; Wu, F.; Yuan, L.-Q. Role of tumor-derived exosomes in bone metastasis (Review). Oncol. Lett. 2019, 18, 3935–3945. [Google Scholar] [CrossRef]
- Linxweiler, J.; Körbel, C.; Müller, A.; Jüngel, E.; Blaheta, R.; Heinzelmann, J.; Stöckle, M.; Junker, K.; Menger, M.D.; Saar, M. Experimental imaging in orthotopic renal cell carcinoma xenograft models: Comparative evaluation of high-resolution 3D ultrasonography, in-vivo micro-CT and 9.4T MRI. Sci. Rep. 2017, 7, 14249. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X.; et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001–8017. [Google Scholar] [CrossRef]
- Xie, X.; Nie, H.; Zhou, Y.; Lian, S.; Mei, H.; Lu, Y.; Dong, H.; Li, F.; Li, T.; Li, B.; et al. Eliminating blood oncogenic exosomes into the small intestine with aptamer-functionalized nanoparticles. Nat. Commun. 2019, 10, 5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linxweiler, J.; Kolbinger, A.; Himbert, D.; Zeuschner, P.; Saar, M.; Stöckle, M.; Junker, K. Organ-Specific Uptake of Extracellular Vesicles Secreted by Urological Cancer Cells. Cancers 2021, 13, 4937. https://doi.org/10.3390/cancers13194937
Linxweiler J, Kolbinger A, Himbert D, Zeuschner P, Saar M, Stöckle M, Junker K. Organ-Specific Uptake of Extracellular Vesicles Secreted by Urological Cancer Cells. Cancers. 2021; 13(19):4937. https://doi.org/10.3390/cancers13194937
Chicago/Turabian StyleLinxweiler, Johannes, Anja Kolbinger, Dirk Himbert, Philip Zeuschner, Matthias Saar, Michael Stöckle, and Kerstin Junker. 2021. "Organ-Specific Uptake of Extracellular Vesicles Secreted by Urological Cancer Cells" Cancers 13, no. 19: 4937. https://doi.org/10.3390/cancers13194937