
Molecular Stratification of Childhood Ependymomas as a Basis for Personalized Diagnostics and Treatment


Abstract
Simple Summary
Abstract
1. Introduction
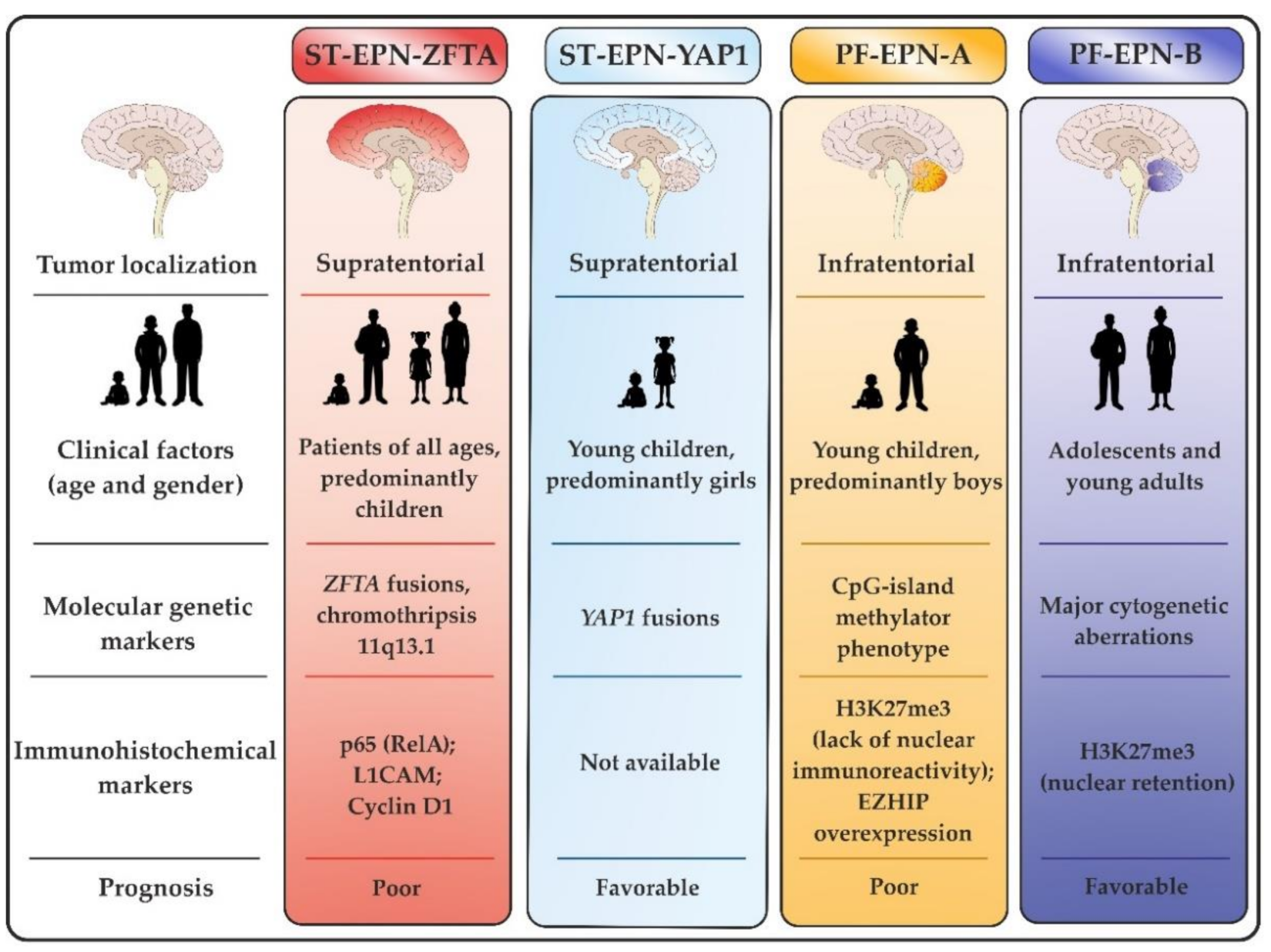
2. Molecular Profiles of ST-EPNs
2.1. ST-EPN-ZFTA Group
2.2. ST-EPN-YAP1 Group
2.3. Non-ZFTA/Non-YAP1 ST-EPNs
3. Molecular Profiles of PF-EPNs
3.1. PF-EPN-A Group
3.2. PF-EPN-B Group
3.3. ST-EPN-ZFTA-like PF-EPNs
4. Molecular Profiles of Sp-EPNs
5. Molecular Profiles of Subependymomas
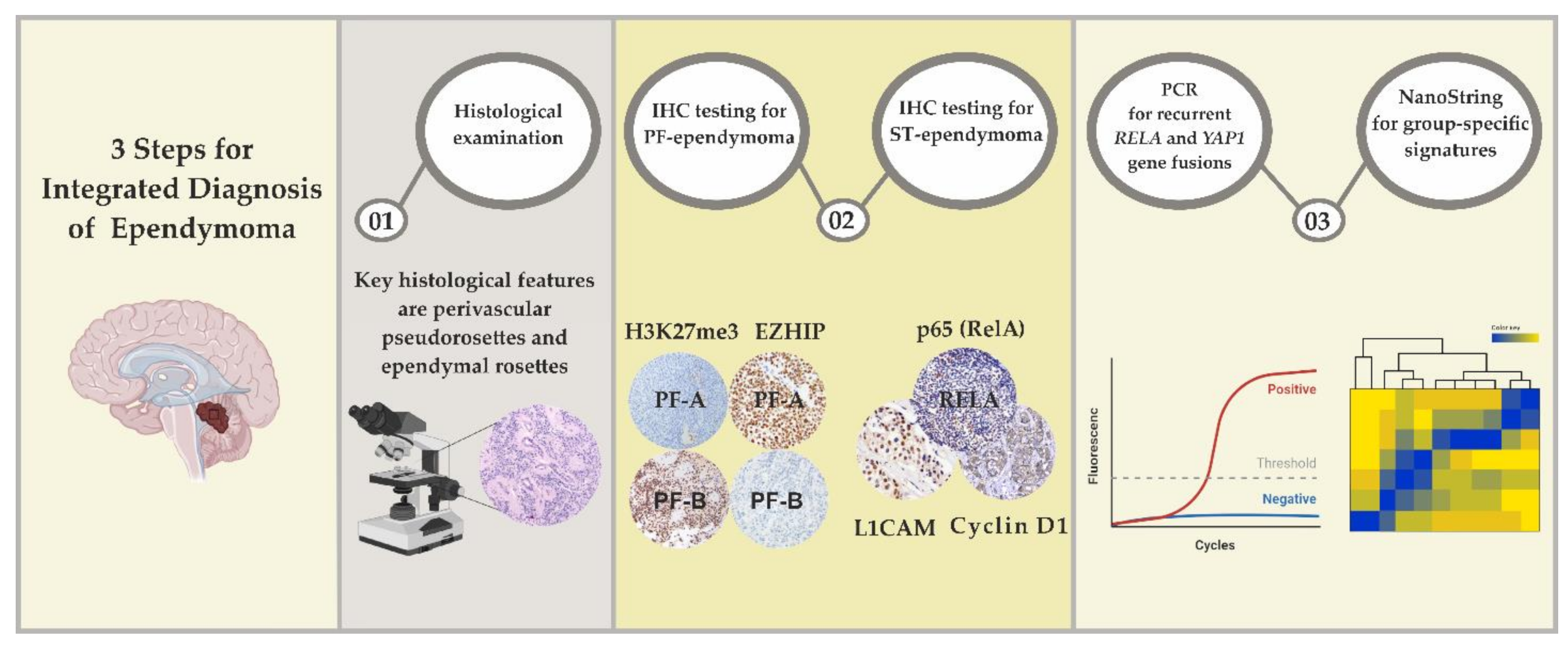
6. Laboratory Approaches for EPN Diagnostics
6.1. Differential Diagnosis of ST-EPNs
6.2. Differential Diagnosis of PF-EPNs
7. Therapeutic Targeting of EPNs
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncol. 2020, 22 (Suppl. 1), iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Jünger, S.T.; Mynarek, M.; Wohlers, I.; Dörner, E.; Mühlen, A.Z.; Velez-Char, N.; von Hoff, K.; Rutkowski, S.; Warmuth-Metz, M.; Kortmann, R.D.; et al. Improved Risk-Stratification for Posterior Fossa Ependymoma of Childhood Considering Clinical, Histological and Genetic Features—A Retrospective Analysis of the HIT Ependymoma Trial Cohort. Acta Neuropathol. Commun. 2019, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Zapotocky, M.; Beera, K.; Adamski, J.; Laperierre, N.; Guger, S.; Janzen, L.; Lassaletta, A.; Figueiredo Nobre, L.; Bartels, U.; Tabori, U.; et al. Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer 2019, 125, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Barretta, F.; Modena, P.; Witt, H.; Minasi, S.; Pfister, S.M.; Pajtler, K.W.; Antonelli, M.; Gandola, L.; Luisa Garrè, M.; et al. Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: An integrated molecular and clinical characterization with a long-term follow-up. Neuro Oncol. 2021, 23, 848–857. [Google Scholar] [CrossRef]
- Ritzmann, T.A.; Rogers, H.A.; Paine, S.M.L.; Storer, L.C.D.; Jacques, T.S.; Chapman, R.J.; Ellison, D.; Donson, A.M.; Foreman, N.K.; Grundy, R.G. A Retrospective Analysis of Recurrent Pediatric Ependymoma Reveals Extremely Poor Survival and Ineffectiveness of Current Treatments across Central Nervous System Locations and Molecular Subgroups. Pediatr. Blood Cancer 2020, 67, e28426. [Google Scholar] [CrossRef]
- Ellison, D.W.; Kocak, M.; Figarella-Branger, D.; Felice, G.; Catherine, G.; Pietsch, T.; Frappaz, D.; Massimino, M.; Grill, J.; Boyett, J.M.; et al. Histopathological Grading of Pediatric Ependymoma: Reproducibility and Clinical Relevance in European Trial Cohorts. J. Negat. Results BioMed 2011, 10, 7. [Google Scholar] [CrossRef]
- Upadhyaya, S.A.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.A.; Billups, C.A.; Bowers, D.C.; Bendel, A.E.; Hassall, T.; Crawford, J.R.; Partap, S.; et al. Molecular Grouping and Outcomes of Young Children with Newly Diagnosed Ependymoma Treated on the Multi-Institutional SJYC07 Trial. Neuro-Oncology 2019, 21, 1319–1330. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Leu, S.; von Felten, S.; Frank, S.; Boulay, J.L.; Mariani, L. IDH Mutation Is Associated with Higher Risk of Malignant Transformation in Low-Grade Glioma. J. Neurooncol. 2016, 127, 363–372. [Google Scholar] [CrossRef]
- Jairam, V.; Kann, B.H.; Park, H.S.; Miccio, J.A.; Beckta, J.M.; Yu, J.B.; Prabhu, R.S.; Gao, S.J.; Mehta, M.P.; Curran, W.J.; et al. Defining an Intermediate-Risk Group for Low-Grade Glioma: A National Cancer Database Analysis. Anticancer Res. 2019, 39, 2911–2918. [Google Scholar] [CrossRef]
- Yang, R.R.; Aibaidula, A.; Wang, W.W.; Chan, A.K.; Shi, Z.F.; Zhang, Z.Y.; Chan, D.; Poon, W.S.; Liu, X.Z.; Li, W.C.; et al. Pediatric low-grade gliomas can be molecularly stratified for risk. Acta Neuropathol. 2018, 136, 641–655. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel Molecular Subgroups for Clinical Classification and Outcome Prediction in Childhood Medulloblastoma: A Cohort Study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.W.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Hielscher, T.; Mack, S.C.; Lassaletta, A.; Lin, T.; Pajtler, K.W.; Jones, D.T.W.; Luu, B.; Cavalli, F.M.G.; Aldape, K.; et al. Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J. Clin. Oncol. 2016, 34, 2468–2477. [Google Scholar] [CrossRef]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.; Venneti, S.; et al. CIMPACT-NOW Update 7: Advancing the Molecular Classification of Ependymal Tumors. Brain Pathol. 2020, 30, 863–866. [Google Scholar]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95–RELA Fusions Drive Oncogenic NF-ΚB Signalling in Ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef]
- Malgulwar, P.B.; Nambirajan, A.; Pathak, P.; Faruq, M.; Rajeshwari, M.; Singh, M.; Suri, V.; Sarkar, C.; Sharma, M.C. C11orf95-RELA Fusions and Upregulated NF-KB Signalling Characterise a Subset of Aggressive Supratentorial Ependymomas That Express L1CAM and Nestin. J. Neurooncol. 2018, 138, 29–39. [Google Scholar] [CrossRef]
- Pagès, M.; Pajtler, K.W.; Puget, S.; Castel, D.; Boddaert, N.; Tauziède-Espariat, A.; Picot, S.; Debily, M.; Kool, M.; Capper, D.; et al. Diagnostics of Pediatric Supratentorial RELA Ependymomas: Integration of Information from Histopathology, Genetics, DNA Methylation and Imaging. Brain Pathol. 2019, 29, 325–335. [Google Scholar] [CrossRef]
- Torre, M.; Alexandrescu, S.; Dubuc, A.M.; Ligon, A.H.; Hornick, J.L.; Meredith, D.M. Characterization of Molecular Signatures of Supratentorial Ependymomas. Mod. Pathol. 2020, 33, 47–56. [Google Scholar] [CrossRef]
- Fukuoka, K.; Kanemura, Y.; Shofuda, T.; Fukushima, S.; Yamashita, S.; Narushima, D.; Kato, M.; Honda-Kitahara, M.; Ichikawa, H.; Kohno, T.; et al. Significance of Molecular Classification of Ependymomas: C11orf95-RELA Fusion-Negative Supratentorial Ependymomas Are a Heterogeneous Group of Tumors. Acta Neuropathol. Commun. 2018, 6, 134. [Google Scholar] [CrossRef]
- Gessi, M.; Giagnacovo, M.; Modena, P.; Elefante, G.; Gianno, F.; Buttarelli, F.R.; Arcella, A.; Donofrio, V.; Diomedi Camassei, F.; Nozza, P.; et al. Role of Immunohistochemistry in the Identification of Supratentorial C11ORF95-RELA Fused Ependymoma in Routine Neuropathology. Am. J. Surg. Pathol. 2019, 43, 56–63. [Google Scholar] [CrossRef]
- Pietsch, T.; Wohlers, I.; Goschzik, T.; Dreschmann, V.; Denkhaus, D.; Dörner, E.; Rahmann, S.; Klein-Hitpass, L. Supratentorial Ependymomas of Childhood Carry C11orf95–RELA Fusions Leading to Pathological Activation of the NF-ΚB Signaling Pathway. Acta Neuropathol. 2014, 127, 609–611. [Google Scholar] [CrossRef]
- Zschernack, V.; Jünger, S.T.; Mynarek, M.; Rutkowski, S.; Garre, M.L.; Ebinger, M.; Neu, M.; Faber, J.; Erdlenbruch, B.; Claviez, A.; et al. Supratentorial Ependymoma in Childhood: More than Just RELA or YAP. Acta Neuropathol. 2021, 141, 455–466. [Google Scholar] [CrossRef]
- Tamai, S.; Nakano, Y.; Kinoshita, M.; Sabit, H.; Nobusawa, S.; Arai, Y.; Hama, N.; Totoki, Y.; Shibata, T.; Ichimura, K.; et al. Ependymoma with C11orf95-MAML2 Fusion: Presenting with Granular Cell and Ganglion Cell Features. Brain Tumor Pathol. 2021, 38, 64–70. [Google Scholar] [CrossRef]
- Tauziède-Espariat, A.; Siegfried, A.; Nicaise, Y.; Kergrohen, T.; Sievers, P.; Vasiljevic, A.; Roux, A.; Dezamis, E.; Benevello, C.; Machet, M.C.; et al. Supratentorial non-RELA, ZFTA-fused ependymomas: A comprehensive phenotype genotype correlation highlighting the number of zinc fingers in ZFTA-NCOA1/2 fusions. Acta Neuropathol. Commun. 2021, 9, 135. [Google Scholar] [CrossRef]
- Zheng, T.; Ghasemi, D.R.; Okonechnikov, K.; Korshunov, A.; Sill, M.; Maass, K.K.; Goncalves, B.; da Silva, P.; Ryzhova, M.; Gojo, J.; et al. Cross-species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion-positive supratentorial ependymomas. Cancer Discov. 2021, 11, 2230–2247. [Google Scholar] [CrossRef]
- Tomomasa, R.; Arai, Y.; Kawabata-Iwakawa, R.; Fukuoka, K.; Nakano, Y.; Hama, N.; Nakata, S.; Suzuki, N.; Ishi, Y.; Tanaka, S.; et al. Ependymoma-like tumor with mesenchymal differentiation harboring C11orf95-NCOA1/2 or -RELA fusion: A hitherto unclassified tumor related to ependymoma. Brain Pathol. 2021, 31, e12943. [Google Scholar] [CrossRef]
- de Sousa, G.R.; Marie, S.; Oba-Shinjo, S.M.; Ramalho, L.; Tone, L.G.; Valera, E.T. A novel type of C11orf95-LOC-RELA fusion in a grade II supratentorial ependymoma: Report of a case with literature review. Childs Nerv. Syst. 2019, 35, 689–694. [Google Scholar] [CrossRef]
- Zhu, J.J.; Jillette, N.; Li, X.N.; Cheng, A.W.; Lau, C.C. C11orf95-RELA reprograms 3D epigenome in supratentorial ependymoma. Acta Neuropathol. 2020, 140, 951–960. [Google Scholar] [CrossRef]
- Arabzade, A.; Zhao, Y.; Varadharajan, S.; Chen, H.-C.; Jessa, S.; Rivas, B.; Stuckert, A.J.; Solis, M.; Kardian, A.; Tlais, D.; et al. ZFTA-RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma. Cancer Discov. 2021, 11, 2200–2215. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Kaneko, S.; Szulzewsky, F.; Qiao, Z.; Takadera, M.; Narita, Y.; Kondo, T.; Holland, E.C.; Hamamoto, R.; Ichimura, K. C11orf95-RELA Fusion Drives Aberrant Gene Expression through the Unique Epigenetic Regulation for Ependymoma Formation. Acta Neuropathol. Commun. 2021, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; Bendel, A.E.; Sabin, N.D.; Burger, P.C.; Shaw, D.W.; Chang, E.; Wu, S.; Zhou, T.; Eisenstat, D.D.; Foreman, N.K.; et al. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J. Clin. Oncol. 2019, 37, 974–983. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Wei, Y.; Okonechnikov, K.; Silva, P.B.G.; Vouri, M.; Zhang, L.; Brabetz, S.; Sieber, L.; Gulley, M.; Mauermann, M.; et al. YAP1 Subgroup Supratentorial Ependymoma Requires TEAD and Nuclear Factor I-Mediated Transcriptional Programmes for Tumorigenesis. Nat. Commun. 2019, 10, 3914. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Fu, L.; Li, Q.C.; Qiu, X.S.; Wang, E.H.; Yu, J.H. Supratentorial ependymoma with YAP1:FAM118B fusion: A case report. Neuropathology 2021, 41, 133–138. [Google Scholar] [CrossRef]
- Andreiuolo, F.; Varlet, P.; Tauziède-Espariat, A.; Jünger, S.T.; Dörner, E.; Dreschmann, V.; Kuchelmeister, K.; Waha, A.; Haberler, C.; Slavc, I.; et al. Childhood Supratentorial Ependymomas with YAP1-MAMLD1 Fusion: An Entity with Characteristic Clinical, Radiological, Cytogenetic and Histopathological Features: YAP1-MAMLD1 Ependymoma. Brain Pathol. 2019, 29, 205–216. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Mack, S.C.; Ramaswamy, V.; Smith, C.A.; Witt, H.; Smith, A.; Hansford, J.R.; von Hoff, K.; Wright, K.D.; Hwang, E.; et al. The Current Consensus on the Clinical Management of Intracranial Ependymoma and Its Distinct Molecular Variants. Acta Neuropathol. 2017, 133, 5–12. [Google Scholar] [CrossRef]
- Sievers, P.; Henneken, S.C.; Blume, C.; Sill, M.; Schrimpf, D.; Stichel, D.; Okonechnikov, K.; Reuss, D.E.; Benzel, J.; Maaß, K.K.; et al. Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol. 2021. [Google Scholar] [CrossRef]
- Lopez-Nunez, O.; Cafferata, B.; Santi, M.; Ranganathan, S.; Pearce, T.M.; Kulich, S.M.; Bailey, K.M.; Broniscer, A.; Rossi, S.; Zin, A.; et al. The spectrum of rare central nervous system (CNS) tumors with EWSR1-non-ETS fusions: Experience from three pediatric institutions with review of the literature. Brain Pathol. 2021, 31, 70–83. [Google Scholar] [CrossRef]
- Siegfried, A.; Rousseau, A.; Maurage, C.A.; Pericart, S.; Nicaise, Y.; Escudie, F.; Grand, D.; Delrieu, A.; Gomez-Brouchet, A.; Le Guellec, S.; et al. EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 2019, 29, 53–62. [Google Scholar] [CrossRef]
- Olsen, T.K.; Panagopoulos, I.; Meling, T.R.; Micci, F.; Gorunova, L.; Thorsen, J.; Due-Tønnessen, B.; Scheie, D.; Lund-Iversen, M.; Krossnes, B.; et al. Fusion Genes with ALK as Recurrent Partner in Ependymoma-like Gliomas: A New Brain Tumor Entity? Neuro Oncol. 2015, 17, 1365–1373. [Google Scholar] [CrossRef]
- Baroni, L.V.; Sundaresan, L.; Heled, A.; Coltin, H.; Pajtler, K.W.; Lin, T.; Merchant, T.E.; McLendon, R.; Faria, C.; Buntine, M.; et al. Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol. 2021, 23, 1360–1370. [Google Scholar] [CrossRef]
- Thomas, C.; Thierfelder, F.; Träger, M.; Soschinski, P.; Müther, M.; Edelmann, D.; Förster, A.; Geiler, C.; Kim, H.Y.; Filipski, K.; et al. TERT promoter mutation and chromosome 6 loss define a high-risk subtype of ependymoma evolving from posterior fossa subependymoma. Acta Neuropathol. 2021, 141, 959–970. [Google Scholar] [CrossRef]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic Alterations Define Lethal CIMP-Positive Ependymomas of Infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef]
- Bayliss, J.; Mukherjee, P.; Lu, C.; Jain, S.U.; Chung, C.; Martinez, D.; Sabari, B.; Margol, A.S.; Panwalkar, P.; Parolia, A.; et al. Lowered H3K27me3 and DNA Hypomethylation Define Poorly Prognostic Pediatric Posterior Fossa Ependymomas. Sci. Transl. Med. 2016, 8, 366ra161. [Google Scholar] [CrossRef]
- Beringer, M.; Pisano, P.; Di Carlo, V.; Blanco, E.; Chammas, P.; Vizán, P.; Gutiérrez, A.; Aranda, S.; Payer, B.; Wierer, M.; et al. EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol. Cell 2016, 64, 645–658. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Wen, J.; Sill, M.; Lin, T.; Orisme, W.; Tang, B.; Hübner, J.-M.; Ramaswamy, V.; Jia, S.; Dalton, J.D.; et al. Molecular Heterogeneity and CXorf67 Alterations in Posterior Fossa Group A (PFA) Ependymomas. Acta Neuropathol. 2018, 136, 211–226. [Google Scholar] [CrossRef]
- Michealraj, K.A.; Kumar, S.A.; Kim, L.; Cavalli, F.; Przelicki, D.; Wojcik, J.B.; Delaidelli, A.; Bajic, A.; Saulnier, O.; MacLeod, G.; et al. Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 2020, 181, 1329–1345.e24. [Google Scholar] [CrossRef]
- Hübner, J.M.; Müller, T.; Papageorgiou, D.N.; Mauermann, M.; Krijgsveld, J.; Russell, R.B.; Ellison, D.W.; Pfister, S.M.; Pajtler, K.W.; Kool, M. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 2019, 21, 878–889. [Google Scholar] [CrossRef]
- Jain, S.U.; Do, T.J.; Lund, P.J.; Rashoff, A.Q.; Diehl, K.L.; Cieslik, M.; Bajic, A.; Juretic, N.; Deshmukh, S.; Venneti, S.; et al. PFA Ependymoma-Associated Protein EZHIP Inhibits PRC2 Activity through a H3 K27M-like Mechanism. Nat. Commun. 2019, 10, 2146. [Google Scholar] [CrossRef]
- Dewaele, B.; Przybyl, J.; Quattrone, A.; Finalet Ferreiro, J.; Vanspauwen, V.; Geerdens, E.; Gianfelici, V.; Kalender, Z.; Wozniak, A.; Moerman, P.; et al. Identification of a Novel, Recurrent MBTD1-CXorf67 Fusion in Low-grade Endometrial Stromal Sarcoma. Int. J. Cancer 2014, 134, 1112–1122. [Google Scholar] [CrossRef]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M Mutation in Histone H3.3 Defines Clinically and Biologically Distinct Subgroups of Pediatric Diffuse Intrinsic Pontine Gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Karremann, M.; Gielen, G.H.; Hoffmann, M.; Wiese, M.; Colditz, N.; Warmuth-Metz, M.; Bison, B.; Claviez, A.; van Vuurden, D.G.; von Bueren, A.O.; et al. Diffuse High-Grade Gliomas with H3 K27M Mutations Carry a Dismal Prognosis Independent of Tumor Location. Neuro Oncol. 2018, 20, 123–131. [Google Scholar] [CrossRef]
- Zaytseva, M.A.; Shekhtman, A.P.; Papusha, L.I.; Valiakhmetova, E.F.; Yasko, L.A.; Druy, A.E. Analysis of genetic aberrations in pediatric high-grade gliomas. Advances in Molecular Oncology 2020, 7, 37–47. (In Russian) [Google Scholar] [CrossRef]
- Infinger, L.; Stevenson, C. Re-Examining the Need for Tissue Diagnosis in Pediatric Diffuse Intrinsic Pontine Gliomas: A Review. Curr. Neuropharmacol. 2016, 15, 129–133. [Google Scholar] [CrossRef][Green Version]
- Lu, V.M.; Alvi, M.A.; McDonald, K.L.; Daniels, D.J. Impact of the H3K27M mutation on survival in pediatric high-grade glioma: A systematic review and meta-analysis. J. Neurosurg. Pediatr. 2018, 23, 308–316. [Google Scholar] [CrossRef]
- Mosaab, A.; El-Ayadi, M.; Khorshed, E.N.; Amer, N.; Refaat, A.; El-Beltagy, M.; Hassan, Z.; Soror, S.H.; Zaghloul, M.S.; El-Naggar, S. Histone H3K27M Mutation Overrides Histological Grading in Pediatric Gliomas. Sci. Rep. 2020, 10, 8368. [Google Scholar] [CrossRef]
- Rudà, R.; Reifenberger, G.; Frappaz, D.; Pfister, S.M.; Laprie, A.; Santarius, T.; Roth, P.; Tonn, J.C.; Soffietti, R.; Weller, M.; et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018, 20, 445–456. [Google Scholar] [CrossRef]
- Merchant, T.E.; Boop, F.A.; Kun, L.E.; Sanford, R.A. A retrospective study of surgery and reirradiation for recurrent ependymoma. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 87–97. [Google Scholar] [CrossRef]
- Han, J.; Yu, M.; Bai, Y.; Yu, J.; Jin, F.; Li, C.; Zeng, R.; Peng, J.; Li, A.; Song, X.; et al. Elevated CXorf67 Expression in PFA Ependymomas Suppresses DNA Repair and Sensitizes to PARP Inhibitors. Cancer Cell 2020, 38, 844–856.e7. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.W.; Manoharan, N.; Alexandrescu, S.; Zimmerman, M.A.; Scully, J.; Chordas, C.; Clymer, J.; Wright, K.D.; Filbin, M.; Ullrich, N.J.; et al. Outcomes after first relapse of childhood intracranial ependymoma. Pediatr. Blood Cancer 2021, 68, e28930. [Google Scholar] [CrossRef] [PubMed]
- Adolph, J.E.; Fleischhack, G.; Mikasch, R.; Zeller, J.; Warmuth-Metz, M.; Bison, B.; Mynarek, M.; Rutkowski, S.; Schüller, U.; von Hoff, K.; et al. Local and systemic therapy of recurrent ependymoma in children and adolescents: Short- and long-term results of the E-HIT-REZ 2005 study. Neuro Oncol. 2021, 23, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.M.G.; Hübner, J.M.; Sharma, T.; Luu, B.; Sill, M.; Zapotocky, M.; Mack, S.C.; Witt, H.; Lin, T.; Shih, D.J.H.; et al. Heterogeneity within the PF-B ependymoma subgroup. Acta Neuropathol. 2018, 136, 227–237. [Google Scholar] [CrossRef]
- Keenan, C.; Graham, R.T.; Harreld, J.H.; Lucas, J.T., Jr.; Finkelstein, D.; Wheeler, D.; Li, X.; Dalton, J.; Upadhyaya, S.A.; Raimondi, S.C.; et al. Infratentorial C11orf95-fused gliomas share histologic, immunophenotypic, and molecular characteristics of supratentorial RELA-fused ependymoma. Acta Neuropathol. 2020, 140, 963–965. [Google Scholar] [CrossRef]
- Abdallah, A.; Emel, E.; Gündüz, H.B.; Sofuoğlu, Ö.E.; Asiltürk, M.; Abdallah, B.G. Long-Term Surgical Resection Outcomes of Pediatric Myxopapillary Ependymoma: Experience of Two Centers and Brief Literature Review. World Neurosurg. 2020, 136, e245–e261. [Google Scholar] [CrossRef]
- Bagley, C.A.; Wilson, S.; Kothbauer, K.F.; Bookland, M.J.; Epstein, F.; Jallo, G.I. Long term outcomes following surgical resection of myxopapillary ependymomas. Neurosurg. Rev. 2009, 32, 321–334. [Google Scholar] [CrossRef]
- Ahmad, O.; Chapman, R.; Storer, L.C.; Luo, L.; Heath, P.R.; Resar, L.; Cohen, K.J.; Grundy, R.G.; Lourdusamy, A. Integrative molecular characterization of pediatric spinal ependymoma: The UK Children’s Cancer and Leukaemia Group study. Neurooncol. Adv. 2021, 3, vdab043. [Google Scholar]
- Ghasemi, D.R.; Sill, M.; Okonechnikov, K.; Korshunov, A.; Yip, S.; Schutz, P.W.; Scheie, D.; Kruse, A.; Harter, P.N.; Kastelan, M.; et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019, 138, 1075–1089. [Google Scholar] [CrossRef]
- Swanson, A.A.; Raghunathan, A.; Jenkins, R.B.; Messing-Jünger, M.; Pietsch, T.; Clarke, M.J.; Kaufmann, T.J.; Giannini, C. Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J. Neuropathol. Exp. Neurol. 2019, 78, 791–797. [Google Scholar] [CrossRef]
- Raffeld, M.; Abdullaev, Z.; Pack, S.D.; Xi, L.; Nagaraj, S.; Briceno, N.; Vera, E.; Pittaluga, S.; Lopes Abath Neto, O.; Quezado, M.; et al. High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol. Commun. 2020, 8, 101. [Google Scholar] [CrossRef]
- D’Amico, R.S.; Praver, M.; Zanazzi, G.J.; Englander, Z.K.; Sims, J.S.; Samanamud, J.L.; Ogden, A.T.; McCormick, P.C.; Feldstein, N.A.; McKhann, G.M.; et al. Subependymomas Are Low-Grade Heterogeneous Glial Neoplasms Defined by Subventricular Zone Lineage Markers. World Neurosurg. 2017, 107, 451–463. [Google Scholar] [CrossRef]
- Kweh, B.; Rosenfeld, J.V.; Hunn, M.; Tee, J.W. Tumor characteristics and surgical outcomes of intracranial subependymomas: A systematic review and meta-analysis. J. Neurosurg. 2021, 1, 1–13. [Google Scholar] [CrossRef]
- Korshunov, A.; Okonechnikov, K.; Schmitt-Hoffner, F.; Ryzhova, M.; Sahm, F.; Stichel, D.; Schrimpf, D.; Reuss, D.E.; Sievers, P.; Suwala, A.K.; et al. Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol. Commun. 2021, 9, 20. [Google Scholar] [CrossRef]
- de Sousa, G.R.; Lira, R.C.P.; de Almeida Magalhães, T.; da Silva, K.R.; Nagano, L.F.P.; Saggioro, F.P.; Baroni, M.; Marie, S.K.N.; Oba-Shinjo, S.M.; Brandelise, S.; et al. A coordinated approach for the assessment of molecular subgroups in pediatric ependymomas using low-cost methods. J. Mol. Med. 2021, 99, 1101–1113. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Ichikawa, T.; Kurozumi, K.; Otani, Y.; Date, I. Clinicopathological and Genetic Features of Supratentorial Cortical Ependymomas. World Neurosurg. 2019, 129, e417–e428. [Google Scholar] [CrossRef]
- Panwalkar, P.; Clark, J.; Ramaswamy, V.; Hawes, D.; Yang, F.; Dunham, C.; Yip, S.; Hukin, J.; Sun, Y.; Schipper, M.J.; et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017, 134, 705–714. [Google Scholar] [CrossRef]
- Antin, C.; Tauziède-Espariat, A.; Debily, M.A.; Castel, D.; Grill, J.; Pagès, M.; Ayrault, O.; Chrétien, F.; Gareton, A.; Andreiuolo, F.; et al. EZHIP is a specific diagnostic biomarker for posterior fossa ependymomas, group PFA and diffuse midline gliomas H3-WT with EZHIP overexpression. Acta Neuropathol. Commun. 2020, 8, 183. [Google Scholar] [CrossRef]
- Nambirajan, A.; Sharma, A.; Rajeshwari, M.; Boorgula, M.T.; Doddamani, R.; Garg, A.; Suri, V.; Sarkar, C.; Sharma, M.C. EZH2 inhibitory protein (EZHIP/Cxorf67) expression correlates strongly with H3K27me3 loss in posterior fossa ependymomas and is mutually exclusive with H3K27M mutations. Brain Tumor Pathol. 2021, 38, 30–40. [Google Scholar] [CrossRef]
- Łastowska, M.; Matyja, E.; Sobocińska, A.; Wojtaś, B.; Niemira, M.; Szałkowska, A.; Krętowski, A.; Karkucińska-Więckowska, A.; Kaleta, M.; Ejmont, M.; et al. Transcriptional profiling of paediatric ependymomas identifies prognostically significant groups. J. Pathol. Clin. Res. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Remke, M.; Cho, Y.J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012, 123, 615–626. [Google Scholar] [CrossRef]
- Druy, A.E.; Yasko, L.A.; Konovalov, D.M.; Ektova, A.P.; Valiakhmetova, E.F.; Olshanskaya, Y.V.; Maschan, A.A.; Novichkova, G.A.; Papusha, L.I. Identification of medulloblastoma molecular subgroups by gene expression profiling. Pediatr. Hematol./Oncol. Immunopathol. 2017, 16, 85–89. (In Russian) [Google Scholar] [CrossRef]
- Łastowska, M.; Trubicka, J.; Sobocińska, A.; Wojtas, B.; Niemira, M.; Szałkowska, A.; Krętowski, A.; Karkucińska-Więckowska, A.; Kaleta, M.; Ejmontet, M.; et al. Molecular identification of CNS NB-FOXR2, CNS EFT-CIC, CNS HGNET-MN1 and CNS HGNET-BCOR pediatric brain tumors using tumor-specific signature genes. Acta Neuropathol. Commun. 2020, 8, 105. [Google Scholar] [CrossRef]
- Gururangan, S.; Fangusaro, J.; Young Poussaint, T.; Onar-Thomas, A.; Gilbertson, R.J.; Vajapeyam, S.; Gajjar, A.; Goldman, S.; Friedman, H.S.; Packer, R.J.; et al. Lack of efficacy of bevacizumab + irinotecan in cases of pediatric recurrent ependymoma--a Pediatric Brain Tumor Consortium study. Neuro Oncol. 2012, 14, 1404–1412. [Google Scholar] [CrossRef]
- DeWire, M.; Fouladi, M.; Turner, D.C.; Wetmore, C.; Hawkins, C.; Jacobs, C.; Yuan, Y.; Liu, D.; Goldman, S.; Fisher, P.; et al. An open-label, two-stage, phase II study of bevacizumab and lapatinib in children with recurrent or refractory ependymoma: A collaborative ependymoma research network study (CERN). J. Neurooncol. 2015, 123, 85–91. [Google Scholar] [CrossRef]
- Wetmore, C.; Daryani, V.M.; Billups, C.A.; Boyett, J.M.; Leary, S.; Tanos, R.; Goldsmith, K.C.; Stewart, C.F.; Blaney, S.M.; Gajjar, A. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: A children’s Oncology Group Study ACNS1021. Cancer Med. 2016, 5, 1416–1424. [Google Scholar] [CrossRef]
- Fouladi, M.; Stewart, C.F.; Blaney, S.M.; Onar-Thomas, A.; Schaiquevich, P.; Packer, R.J.; Goldman, S.; Geyer, J.R.; Gajjar, A.; Kun, L.E.; et al. A molecular biology and phase II trial of lapatinib in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J. Neurooncol. 2013, 114, 173–179. [Google Scholar] [CrossRef]
- Milde, T.; Kleber, S.; Korshunov, A.; Witt, H.; Hielscher, T.; Koch, P.; Kopp, H.G.; Jugold, M.; Deubzer, H.E.; Oehme, I.; et al. A novel human high-risk ependymoma stem cell model reveals the differentiation-inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol. 2011, 122, 637–650. [Google Scholar] [CrossRef]
- Rahman, R.; Osteso-Ibanez, T.; Hirst, R.A.; Levesley, J.; Kilday, J.P.; Quinn, S.; Peet, A.; O’Callaghan, C.; Coyle, B.; Grundy, R.G. Histone deacetylase inhibition attenuates cell growth with associated telomerase inhibition in high-grade childhood brain tumor cells. Mol. Cancer Ther. 2010, 9, 2568–2581. [Google Scholar] [CrossRef]
- Bukowinski, A.; Chang, B.; Reid, J.M.; Liu, X.; Minard, C.G.; Trepel, J.B.; Lee, M.J.; Fox, E.; Weigel, B.J. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr. Blood Cancer 2021, 68, e28892. [Google Scholar] [CrossRef]
- Antonelli, R.; Jiménez, C.; Riley, M.; Servidei, T.; Riccardi, R.; Soriano, A.; Roma, J.; Martínez-Saez, E.; Martini, M.; Ruggiero, A.; et al. CN133, a Novel Brain-Penetrating Histone Deacetylase Inhibitor, Hampers Tumor Growth in Patient-Derived Pediatric Posterior Fossa Ependymoma Models. Cancers 2020, 12, 1922. [Google Scholar] [CrossRef] [PubMed]
- Servidei, T.; Meco, D.; Martini, M.; Battaglia, A.; Granitto, A.; Buzzonetti, A.; Babini, G.; Massimi, L.; Tamburrini, G.; Scambia, G.; et al. The BET Inhibitor OTX015 Exhibits In Vitro and In Vivo Antitumor Activity in Pediatric Ependymoma Stem Cell Models. Int. J. Mol. Sci. 2021, 22, 1877. [Google Scholar] [CrossRef] [PubMed]
- Witt, D.A.; Donson, A.M.; Amani, V.; Moreira, D.C.; Sanford, B.; Hoffman, L.M.; Handler, M.H.; Levy, J.; Jones, K.L.; Nellan, A.; et al. Specific expression of PD-L1 in RELA-fusion supratentorial ependymoma: Implications for PD-1-targeted therapy. Pediatr. Blood Cancer 2018, 65, e26960. [Google Scholar] [CrossRef] [PubMed]
- Nambirajan, A.; Malgulwar, P.B.; Sharma, A.; Boorgula, M.T.; Doddamani, R.; Singh, M.; Suri, V.; Sarkar, C.; Sharma, M.C. Clinicopathological evaluation of PD-L1 expression and cytotoxic T-lymphocyte infiltrates across intracranial molecular subgroups of ependymomas: Are these tumors potential candidates for immune check-point blockade? Brain Tumor Pathol. 2019, 36, 152–161. [Google Scholar] [CrossRef]
- Perruccio, K.; Mastronuzzi, A.; Lupattelli, M.; Arcioni, F.; Capolsini, I.; Cerri, C.; Gurdo, G.M.I.; Massei, M.S.; Mastrodicasa, E.; Caniglia, M. Targeted Therapy with Sirolimus and Nivolumab in a Child with Refractory Multifocal Anaplastic Ependymoma. Reports 2021, 4, 12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Localization | Molecular Group | Major Molecular Markers | Prevalence within the Group | Pathogenic Impact |
|---|---|---|---|---|
| Supratentorial | ST-EPN-ZFTA | ZFTA–RELA fusion, chromothripsis 11q13.1 | 90–95% | NF-kB pathway activation |
| ZFTA–MAML2 ZFTA–NCOA1 ZFTA–NCOA2 | 5–10% | EP300/CREBBP gene expression pathway | ||
| ST-EPN-YAP1 | YAP1–MAMLD1 | 95% | Hippo pathway activation | |
| YAP1–FAM118B | 5% | |||
| Infratentorial | PF-EPN-A | EZHIP overexpression | 95% | CpG-island methylator phenotype |
| HIST1H3C, HIST1H3B or H3F3A K27M substitution | <5% | |||
| PF-EPN-B | Major cytogenetic aberrations | Up to 100% | Ciliogenesis deregulation | |
| Spinal | Sp-MPE | HOXB cluster genes amplification | Up to 100% | Mitochondrial metabolism pathways activation |
| SP-EPN-MYCN | MYCN amplification | 100% | Proliferative signaling |
| Molecular Group | Implicated Gene * | Gene Name | Cytogenetic Band | Pathogenic Impact | Evidence Level ** | Evidence-Based Categorization *** | Hallmark of Cancer **** | |
|---|---|---|---|---|---|---|---|---|
| Promotes | Suppresses | |||||||
| ST-EPN-ZFTA | ZFTA | Zinc finger translocation associated | 11q13.1 | 5′-partner gene in ZFTA–RELA fusion | I | Tier I, level A | Genome instability | |
| RELA | V-Rel avian reticuloendotheliosis viral oncogene homolog A | 11q13.1 | 3′-partner gene in ZFTA–RELA fusion | I | Tier I, level A | Escaping programmed cell death; tumor promoting inflammation | ||
| MAML2 | Mastermind-like transcriptional coactivator 2 | 11q21 | 3′-partner gene in ZFT–MAML2 fusion | III | Tier II, level C | Proliferative signaling; angiogenesis | ||
| NCOA1 | Nuclear receptor coactivator 1 | 2p23.3 | 3′-partner gene in ZFTA–NCOA1 fusion | III | Tier II, level C | Proliferative signaling; change of cellular energetics | ||
| NCOA2 | Nuclear receptor coactivator 2 | 8q13.3 | 3′-partner gene in ZFTA–NCOA2 fusion | III | Tier II, level C | Proliferative signaling; change of cellular energetics; escaping programmed cell death | ||
| ST-EPN-YAP1 | YAP1 | Yes1-associated transcriptional regulator | 11q22.1 | 5′-partner gene in YAP1–MAMLD1 fusion | II | Tier I, level A | Proliferative signaling; escaping programmed cell death; invasion and metastasis | Escaping programmed cell death |
| MAMLD1 | Mastermind-like domain-containing 1 | Xq28 | 3′-partner gene in YAP1–MAMLD1 fusion | II | Tier I, level A | Proliferative signaling; angiogenesis | Escaping programmed cell death | |
| FAM118B | Family with sequence similarity 118 member B | 11q24.2 | 3′-partner gene in YAP1–FAM118B fusion | IV | Tier II, level D | Unknown | ||
| Non-ZFTA/Non-YAP1 ST-EPNs | PLAGL1 | PLAG1-like zinc finger 1 | 6q24.2 | 3′-partner gene in EWSR1-PLAGL1 fusion; 5′-partner gene in PLAGL1–FOXO1 or PLAGL1–EP300 fusion | IV | Tier II, level D | Suppression of growth | Escaping immunic response to cancer; tumor promoting inflammation; invasion and metastasis; angiogenesis |
| EWSR1 | EWS RNA binding protein 1 | 22q12.2 | 5′-partner gene in EWSR1–PLAGL1 or EWSR1–PATZ1 fusion | IV | Tier II, level D | Proliferative signaling; escaping programmed cell death; angiogenesis; invasion and metastasis | Genome instability and mutations | |
| FOXO1 | Forkhead box O1 | 13q14.11 | 3′-partner gene in PLAGL1–FOXO1 fusion | IV | Tier II, level D | Change of cellular energetics | Escaping programmed cell death | |
| EP300 | E1A binding protein P300 | 22q13.2 | 3′-partner gene in PLAGL1–EP300 fusion | IV | Tier II, level D | Suppression of growth | Escaping programmed cell death | |
| PATZ1 | POZ/BTB and AT hook-containing zinc finger 1 | 22q12.2 | 3′-partner gene in EWSR1–PATZ1 or MN1–PATZ1 fusion | IV | Tier II, level D | Proliferative signaling; escaping programmed cell death | ||
| MN1 | MN1 proto-oncogene, transcriptional regulator | 22q12.1 | 5′-partner gene in MN1-PATZ1 fusion | IV | Tier II, level D | Suppression of growth | Escaping programmed cell death | |
| PF-EPN-A | EZHIP | EZH inhibitory protein | Xp11.22 | Overexpression | IV | Tier II, level D | EZH1/EZH2-mediated trimethylation of H3K27 | |
| EPOP | Elongin BC and polycomb repressive complex 2-associated protein | 17q12 | Overexpression | IV | Tier II, level D | EZH2-mediated trimethylation of H3K27 | ||
| HIST1H3C | H3 clustered histone 3 | 6p22.2 | Somatic mutation | IV | Tier II, level D | EZH2-mediated trimethylation of H3K27 | ||
| HIST1H3B | H3 clustered histone 2 | 6p22.2 | Somatic mutation | IV | Tier II, level D | EZH2-mediated trimethylation of H3K27 | ||
| H3F3A | H3.3 histone A | 1q42.12 | Somatic mutation | IV | Tier II, level D | EZH2-mediated trimethylation of H3K27 | ||
| BCL9 | BCL9 transcription coactivator | 1q21.2 | Oncogene, involved in 1q gain | V | NA | Proliferative signaling; invasion and metastasis; angiogenesis | ||
| ARNT | Aryl hydrocarbon receptor nuclear translocator | 1q21.3 | Oncogene, involved in 1q gain | V | NA | Angiogenesis; change of cellular energetics | Invasion and metastasis | |
| SETDB1 | SET domain bifurcated histone lysine methyltransferase 1 | 1q21.3 | Oncogene, involved in 1q gain | V | NA | Epigenetic transcriptional repression by recruiting HP1 (CBX1, CBX3 and/or CBX5) proteins to methylated histones | ||
| NTRK1 | Neurotrophic receptor tyrosine kinase 1 | 1q23.1 | Oncogene, involved in 1q gain | V | NA | Proliferative signaling; escaping programmed cell death; angiogenesis | ||
| FCRL4 | Fc receptor-like 4 | 1q23.1 | Oncogene, involved in 1q gain | V | NA | Escaping immunic response to cancer | ||
| FCGR2B | Fc fragment of IgG receptor IIb | 1q23.3 | Oncogene, involved in 1q gain | V | NA | Suppression of growth | Escaping programmed cell death | |
| DDR2 | Discoidin domain receptor tyrosine kinase 2 | 1q23.3 | Oncogene, involved in 1q gain | V | NA | Invasion and metastasis | ||
| PBX1 | PBX homeobox 1 | 1q23.3 | Oncogene, involved in 1q gain | V | NA | Angiogenesis; escaping programmed cell death; change of cellular energetics | ||
| ABL2 | ABL proto-oncogene 2, non-receptor tyrosine kinase | 1q25.2 | Oncogene, involved in 1q gain | V | NA | Proliferative signaling; invasion and metastasis; angiogenesis; genome instability and mutations; change of cellular energetics | Escaping programmed cell death | |
| MDM4 | MDM4 regulator of P53 | 1q32.1 | Oncogene, involved in 1q gain | V | NA | Proliferative signaling; invasion and metastasis; angiogenesis; escaping programmed cell death | Suppression of growth | |
| ELK4 | ETS transcription factor ELK4 | 1q32.1 | Oncogene, involved in 1q gain | V | NA | Proliferative signaling; escaping programmed cell death | ||
| RGS7 | Regulator of G protein signaling 7 | 1q43 | Oncogene, involved in 1q gain | V | NA | Change of cellular energetics | ||
| AKT3 | AKT serine/threonine Kinase 3 | 1q43-q44 | Oncogene, involved in 1q gain | V | NA | Proliferative signaling; suppression of growth; invasion and metastasis; angiogenesis; escaping programmed cell death; change of cellular energetics | Invasion and metastasis; angiogenesis; genome instability and mutations | |
| EPHA7 | EPH receptor A7 | 6q16.1 | Tumor suppressor gene, involved in 6q loss | V | NA | Escaping programmed cell death | ||
| CCNC | Cyclin C | 6q16.2 | Tumor suppressor gene, involved in 6q loss | V | NA | Proliferative signaling | ||
| PRDM1 | PR/SET domain 1 | 6q21 | Tumor suppressor gene, involved in 6q loss | V | NA | Suppression of growth | Escaping immunic response to cancer | |
| FOXO3 | Forkhead box O3 | 6q21 | Tumor suppressor gene, involved in 6q loss | V | NA | Change of cellular energetics | Escaping programmed cell death | |
| PTPRK | Protein tyrosine phosphatase receptor type K | 6q22.33 | Tumor suppressor gene, involved in 6q loss | V | NA | Escaping immunic response to cancer | Proliferative signaling | |
| BCLAF1 | BCL2-associated transcription factor 1 | 6q23.3 | Tumor suppressor gene, involved in 6q loss | V | NA | Escaping programmed cell death | ||
| TNFAIP3 | TNF alpha-induced protein 3 | 6q23.3 | Tumor suppressor gene, involved in 6q loss | V | NA | Escaping immunic response to cancer; tumor promoting inflammation | ||
| LATS1 | Large tumor suppressor kinase 1 | 6q25.1 | Tumor suppressor gene, involved in 6q loss | V | NA | Suppression of growth | Genome instability and mutations; escaping programmed cell death | |
| ESR1 | Estrogen receptor 1 | 6q25.1 | Tumor suppressor gene, involved in 6q loss | V | NA | Proliferative signaling; suppression of growth; escaping immunic response to cancer; invasion and metastasis | Invasion and metastasis | |
| ARID1B | AT-rich interaction domain 1B | 6q25.3 | Tumor suppressor gene, involved in 6q loss | V | NA | Suppression of growth; cell replicative immortality | Cell replicative immortality; invasion and metastasis; genome instability and mutations; escaping programmed cell death | |
| QKI | QKI, KH domain-containing RNA binding | 6q26 | Tumor suppressor gene, involved in 6q loss | V | NA | Suppression of growth; escaping programmed cell death | Escaping programmed cell death | |
| PF-EPN-B | LATS2 | Large tumor suppressor kinase 2 | 13q12.11 | Tumor suppressor gene, involved in 13 q loss | V | NA | Suppression of growth; invasion and metastasis | Invasion and metastasis; genome instability and mutations; escaping programmed cell death |
| CDX2 | Caudal type homeobox 2 | 13q12.2 | Tumor suppressor gene, involved in 13 q loss | V | NA | Proliferative signaling | ||
| BRCA2 | BRCA2 DNA repair associated | 13q13.1 | Tumor suppressor gene, involved in 13 q loss | V | NA | Genome instability and mutations; escaping programmed cell death | ||
| RB1 | RB transcriptional corepressor 1 | 13q14.2 | Tumor suppressor gene, involved in 13 q loss | V | NA | Suppression of growth; escaping programmed cell death; change of cellular energetics | Escaping immunic response to cancer; invasion and metastasis; genome instability and mutations; escaping programmed cell death | |
| GPC5 | Glypican 5 | 13q31.3 | Tumor suppressor gene, involved in 13 q loss | V | NA | Suppression of growth; invasion and metastasis | ||
| SOX21 | SRY-box transcription factor 21 | 13q32.1 | Tumor suppressor gene, involved in 13 q loss | V | NA | Suppression of growth | Proliferative signaling | |
| ERCC5 | ERCC excision repair 5, endonuclease | 13q33.1 | Tumor suppressor gene, involved in 13 q loss | V | NA | Genome instability and mutations; escaping programmed cell death | Genome instability and mutations | |
| SP-MPE | HOXB13 | Homeobox B13 | 17q21.32 | Amplification | III | Tier II, level C | Change of cellular energetics | Escaping programmed cell death |
| SP-EPN-MYCN | MYCN | MYCN proto-oncogene, BHLH transcription factor | 2p24.3 | Amplification | II | Tier I, level A | Proliferative signaling; escaping immunic response to cancer; angiogenesis; genome instability and mutations; change of cellular energetics | Cell replicative immortality; invasion and metastasis; escaping programmed cell death |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaytseva, M.; Papusha, L.; Novichkova, G.; Druy, A. Molecular Stratification of Childhood Ependymomas as a Basis for Personalized Diagnostics and Treatment. Cancers 2021, 13, 4954. https://doi.org/10.3390/cancers13194954
Zaytseva M, Papusha L, Novichkova G, Druy A. Molecular Stratification of Childhood Ependymomas as a Basis for Personalized Diagnostics and Treatment. Cancers. 2021; 13(19):4954. https://doi.org/10.3390/cancers13194954
Chicago/Turabian StyleZaytseva, Margarita, Ludmila Papusha, Galina Novichkova, and Alexander Druy. 2021. "Molecular Stratification of Childhood Ependymomas as a Basis for Personalized Diagnostics and Treatment" Cancers 13, no. 19: 4954. https://doi.org/10.3390/cancers13194954
APA StyleZaytseva, M., Papusha, L., Novichkova, G., & Druy, A. (2021). Molecular Stratification of Childhood Ependymomas as a Basis for Personalized Diagnostics and Treatment. Cancers, 13(19), 4954. https://doi.org/10.3390/cancers13194954