
Drug-Induced Resistance and Phenotypic Switch in Triple-Negative Breast Cancer Can Be Controlled via Resolution and Targeting of Individualized Signaling Signatures

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Survival Assay
2.3. Cell Fixation and Permeabilization of Phosphoproteins for Flow Cytometry
2.4. Flow Cytometry Analysis
2.5. Animal Studies
2.6. Western Blot Analysis
2.7. PaSSS Analysis
2.8. Selection of TNBC Cell Lines for Experimental Validation of the PaSSS-Based Strategy
2.9. Computational Single Data Analysis for Calculation of Cell-Specific Signaling Signatures (CSSS)
3. Results
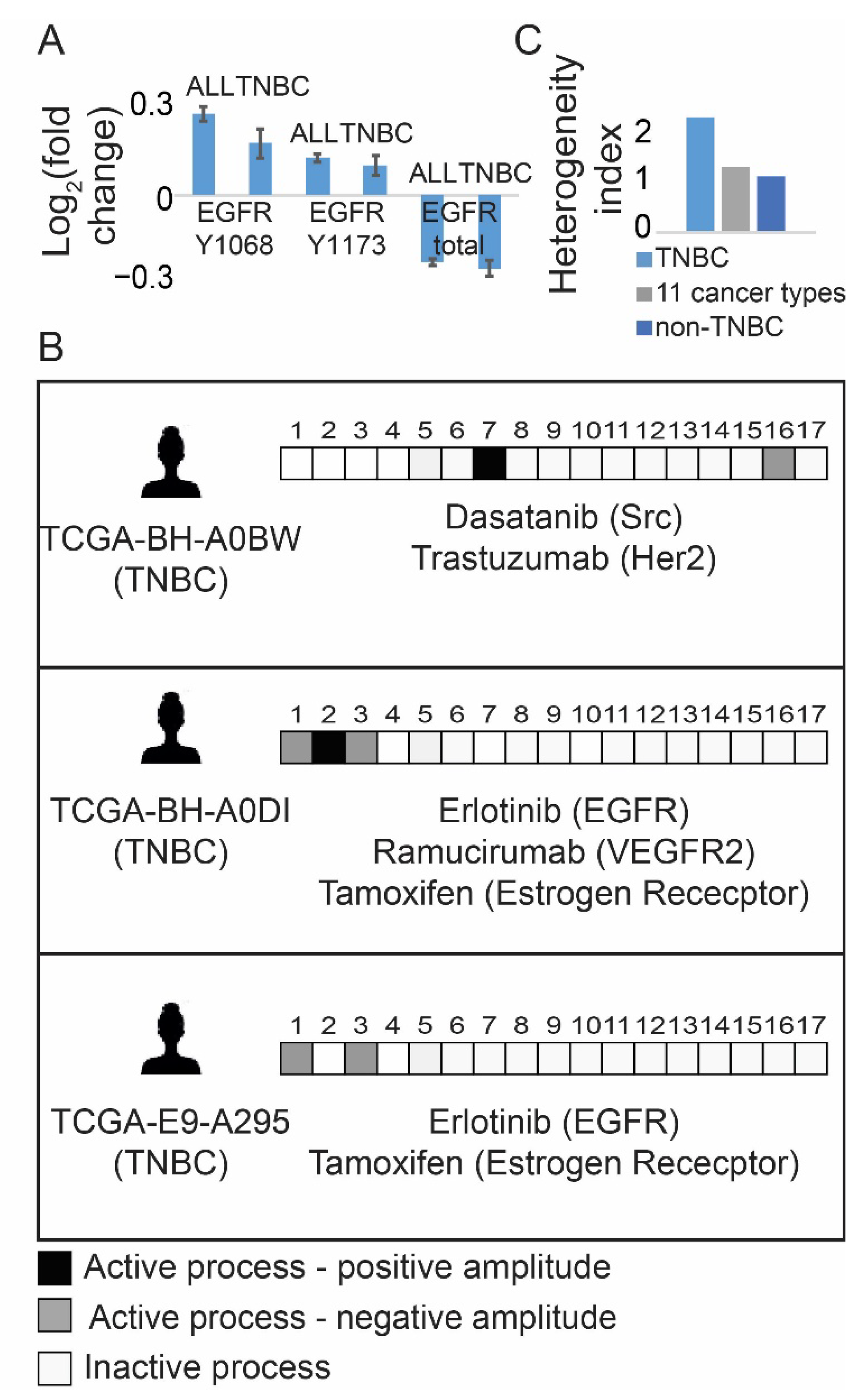
3.1. Resolution of the Signaling Structures in Breast Cancer Patients Suggests That Anti-EGFR Monotherapy Should Be Inefficient in TNBC in Contrast to Anti-ER Therapies
3.2. The PaSSS-Based Strategy Suggests How Anti-EGFR/Anti-ER Therapies Should Be Incorporated in Order to Reduce Efficiently the Individualized Signaling Fluxes and Cell Regrowth
3.3. Rationally Designed PaSSS-Based Drug Combinations Prevented the Development of Drug Resistance
3.4. Monotherapies and Wrong Drug Combinations Do Not Deplete the Signaling of the TNBC Cell Lines but Induce a Switch from One Signaling State to Another
3.5. The Predicted Drug Combinations Are Selective and Efficient in Preventing the Development of Tumor Regrowth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer - Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamandis, E.P. Present and Future of Cancer Biomarkers. Clin. Chem. Lab. Med. 2014, 52, 791–794. [Google Scholar] [CrossRef]
- Borrebaeck, C.A.K. Precision diagnostics: Moving towards protein biomarker signatures of clinical utility in cancer. Nat. Rev. Cancer 2017, 17, 199–204. [Google Scholar] [CrossRef]
- Zhao, L.; Lee, V.H.F.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief. Bioinform. 2018, 20, 572–584. [Google Scholar] [CrossRef]
- Costa, R.; Shah, A.N.; Santa-Maria, C.A.; Cruz, M.R.; Mahalingam, D.; Carneiro, B.A.; Chae, Y.K.; Cristofanilli, M.; Gradishar, W.J.; Giles, F.J. Targeting Epidermal Growth Factor Receptor in triple negative breast cancer: New discoveries and practical insights for drug development. Cancer Treat. Rev. 2017, 53, 111–119. [Google Scholar] [CrossRef]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular targeting therapy against egfr family in breast cancer: Progress and future potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Aichler, M.; Motschmann, M.; Jütting, U.; Luber, B.; Becker, K.; Ott, K.; Lordick, F.; Langer, R.; Feith, M.; Siewert, J.R.; et al. Epidermal growth factor receptor (EGFR) is an independent adverse prognostic factor in esophageal adenocarcinoma patients treated with cisplatin-based neoadjuvant chemotherapy. Oncotarget 2014, 5, 6620–6632. [Google Scholar] [CrossRef]
- Ali, R.; Wendt, M.K. The paradoxical functions of EGFR during breast cancer progression. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Cowherd, S.; Miller, L.D.; Melin, S.A.; Akman, S.; Isom, S.; Cole, J.; Pullikuth, A.; Lawrence, J.A. A phase II clinical trial of weekly paclitaxel and carboplatin in combination with panitumumab in metastatic triple negative breast cancer. Cancer Biol. Ther. 2015, 16, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabholtz, J.M.; Abrial, C.; Mouret-Reynier, M.A.; Dauplat, M.M.; Weber, B.; Gligorov, J.; Forest, A.M.; Tredan, O.; Vanlemmens, L.; Petit, T.; et al. Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane based chemotherapy in operable triple negative breast cancer: Identification of biologically-defined signatures predicting treatment impact. Ann. Oncol. 2014, 25, 1570–1577. [Google Scholar] [CrossRef]
- Guerrab, A.; El Bamdad, M.; Bignon, Y.-J. Co-targeting EGFR and mTOR with gefitinib and everolimus in triple-negative breast cancer cells. Sci. Rep. 2020, 10, 6367. [Google Scholar] [CrossRef] [PubMed]
- You, K.S.; Weon Yi, Y.; Cho, J.; Seong, Y.S. Dual inhibition of akt and mek pathways potentiates the anti-cancer effect of gefitinib in triple-negative breast cancer cells. Cancers 2021, 13, 1205. [Google Scholar] [CrossRef]
- Jabbarzadeh Kaboli, P.; Salimian, F.; Aghapour, S.; Xiang, S.; Zhao, Q.; Li, M.; Wu, X.; Du, F.; Zhao, Y.; Shen, J.; et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer – A comprehensive review from chemotherapy to immunotherapy. Pharmacol. Res. 2020, 156, 104806. [Google Scholar] [CrossRef]
- Flashner-Abramson, E.; Vasudevan, S.; Adejumobi, I.A.; Sonnenblick, A.; Kravchenko-Balasha, N. Decoding cancer heterogeneity: Studying patient-specific signaling signatures towards personalized cancer therapy. Theranostics 2019, 9, 5149–5165. [Google Scholar] [CrossRef]
- Vasudevan, S.; Flashner-Abramson, E.; Alkhatib, H.; Roy Chowdhury, S.; Adejumobi, I.A.; Vilenski, D.; Stefansky, S.; Rubinstein, A.M.; Kravchenko-Balasha, N. Overcoming resistance to BRAFV600E inhibition in melanoma by deciphering and targeting personalized protein network alterations. NPJ Precis. Oncol. 2021, 5, 50. [Google Scholar] [CrossRef]
- Krutzik, P.O.; Nolan, G.P. Intracellular Phospho-protein Staining Techniques for Flow Cytometry: Monitoring Single Cell Signaling Events. Cytom. Part A 2003, 55, 61–70. [Google Scholar] [CrossRef]
- Jubran, M.R.; Rubinstein, A.M.; Cojocari, I.; Adejumobi, I.A.; Mogilevsky, M.; Tibi, S.; Sionov, R.V.; Verreault, M.; Idbaih, A.; Karni, R.; et al. Dissecting the role of crosstalk between glioblastoma subpopulations in tumor cell spreading. Oncogenesis 2020, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Flashner-Abramson, E.; Remacle, F.; Levine, R.D.; Kravchenko-Balasha, N. Personalized disease signatures through information-theoretic compaction of big cancer data. Proc. Natl. Acad. Sci. USA 2018, 115, 7694–7699. [Google Scholar] [CrossRef] [Green Version]
- Remacle, F.; Kravchenko-Balasha, N.; Levitzki, A.; Levine, R.D. Information-theoretic analysis of phenotype changes in early stages of carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 10324–10329. [Google Scholar] [CrossRef] [Green Version]
- Poovathingal, S.K.; Kravchenko-Balasha, N.; Shin, Y.S.; Levine, R.D.; Heath, J.R. Critical Points in Tumorigenesis: A Carcinogen-Initiated Phase Transition Analyzed via Single-Cell Proteomics. Small 2016, 12, 1425–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravchenko-Balasha, N.; Levitzki, A.; Goldstein, A.; Rotter, V.; Gross, A.; Remacle, F.; Levine, R.D. On a fundamental structure of gene networks in living cells. Proc. Natl. Acad. Sci. USA 2012, 109, 4702–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhatib, H.; Rubinstein, A.M.; Vasudevan, S.; Flashner-Abramson, E.; Oguche, S.; Peretz-Yablonsky, T.; Granit, A.; Granot, Z.; Sheva, K.; Meirovitz, A.; et al. Mapping cellular subpopulations within triple negative breast cancer tumors provides a tool for cancer sensitization to radiotherapy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kravchenko-Balasha, N. Translating Cancer Molecular Variability into Personalized Information Using Bulk and Single Cell Approaches. Proteomics 2020, 20, e1900227. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.Y.; Broom, B.M.; W Verhaak, R.G.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, A.A.; Naz, S.; Hashmi, S.K.; Irfan, M.; Hussain, Z.F.; Khan, E.Y.; Asif, H.; Faridi, N. Epidermal growth factor receptor (EGFR) overexpression in triple-negative breast cancer: Association with clinicopathologic features and prognostic parameters. Surg. Exp. Pathol. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Ismail-Khan, R.; Bui, M.M. A review of triple-negative breast cancer. Cancer Control 2010, 17, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Volinsky, N.; Kholodenko, B.N. Complexity of receptor tyrosine kinase signal processing. Cold Spring Harb. Perspect. Biol. 2013, 5, a009043. [Google Scholar] [CrossRef] [Green Version]
- Lan, S.-H.; Wu, S.-Y.; Raghavaraju, G.; Chow, N.-H.; Liu, H.-S. The Crosstalk of c-MET with Related Receptor Tyrosine Kinases in Urothelial Bladder Cancer. In Advances in the Scientific Evaluation of Bladder Cancer and Molecular Basis for Diagnosis and Treatment; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Pazarentzos, E.; Bivona, T.G. Adaptive stress signaling in targeted cancer therapy resistance. Oncogene 2015, 34, 5599–5606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.J.M.; Li, J.; Wang, Y.; Akbani, R.; Lu, Y.; Mills, G.B.; Liang, H. TCPA v3.0: An integrative platform to explore the pan-cancer analysis of functional proteomic data. Mol. Cell. Proteom. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasudevan, S.; Adejumobi, I.A.; Alkhatib, H.; Roy Chowdhury, S.; Stefansky, S.; Rubinstein, A.M.; Kravchenko-Balasha, N. Drug-Induced Resistance and Phenotypic Switch in Triple-Negative Breast Cancer Can Be Controlled via Resolution and Targeting of Individualized Signaling Signatures. Cancers 2021, 13, 5009. https://doi.org/10.3390/cancers13195009
Vasudevan S, Adejumobi IA, Alkhatib H, Roy Chowdhury S, Stefansky S, Rubinstein AM, Kravchenko-Balasha N. Drug-Induced Resistance and Phenotypic Switch in Triple-Negative Breast Cancer Can Be Controlled via Resolution and Targeting of Individualized Signaling Signatures. Cancers. 2021; 13(19):5009. https://doi.org/10.3390/cancers13195009
Chicago/Turabian StyleVasudevan, Swetha, Ibukun A. Adejumobi, Heba Alkhatib, Sangita Roy Chowdhury, Shira Stefansky, Ariel M. Rubinstein, and Nataly Kravchenko-Balasha. 2021. "Drug-Induced Resistance and Phenotypic Switch in Triple-Negative Breast Cancer Can Be Controlled via Resolution and Targeting of Individualized Signaling Signatures" Cancers 13, no. 19: 5009. https://doi.org/10.3390/cancers13195009