Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma



Abstract
:Simple Summary
Abstract
1. Introduction
2. Interaction with Bone Marrow Stromal Cell
2.1. Cell Adhesion-Mediated Drug Resistance and Soluble Factor-Mediated Drug Resistance
2.2. Treatment for Interaction with CAM-DR and SFM-DR
3. Myeloma Cells in Hypoxia and Vascular Niche
3.1. Vascular Niche in Myeloma
Hypoxia in Myeloma
3.2. Treatment for Vascular Niche and Hypoxia
4. Endosteal Niche Contributes Survival of Myeloma Cells
4.1. Endosteal Niche is Constituted by Osteoclasts and Osteoblasts
4.2. Treatment for Endosteal Niche
5. Clonal Evolution
5.1. Two Types of Driver Mutation
5.2. Three Types of Clonal Evolution
5.3. Genetic Change during Treatment in PCL and EMD
5.4. HRCA is Associated with Clonal Evolution
5.5. Relation between Deep Response and Clonal Evolution
5.6. Nongenetic Clonal Diversity
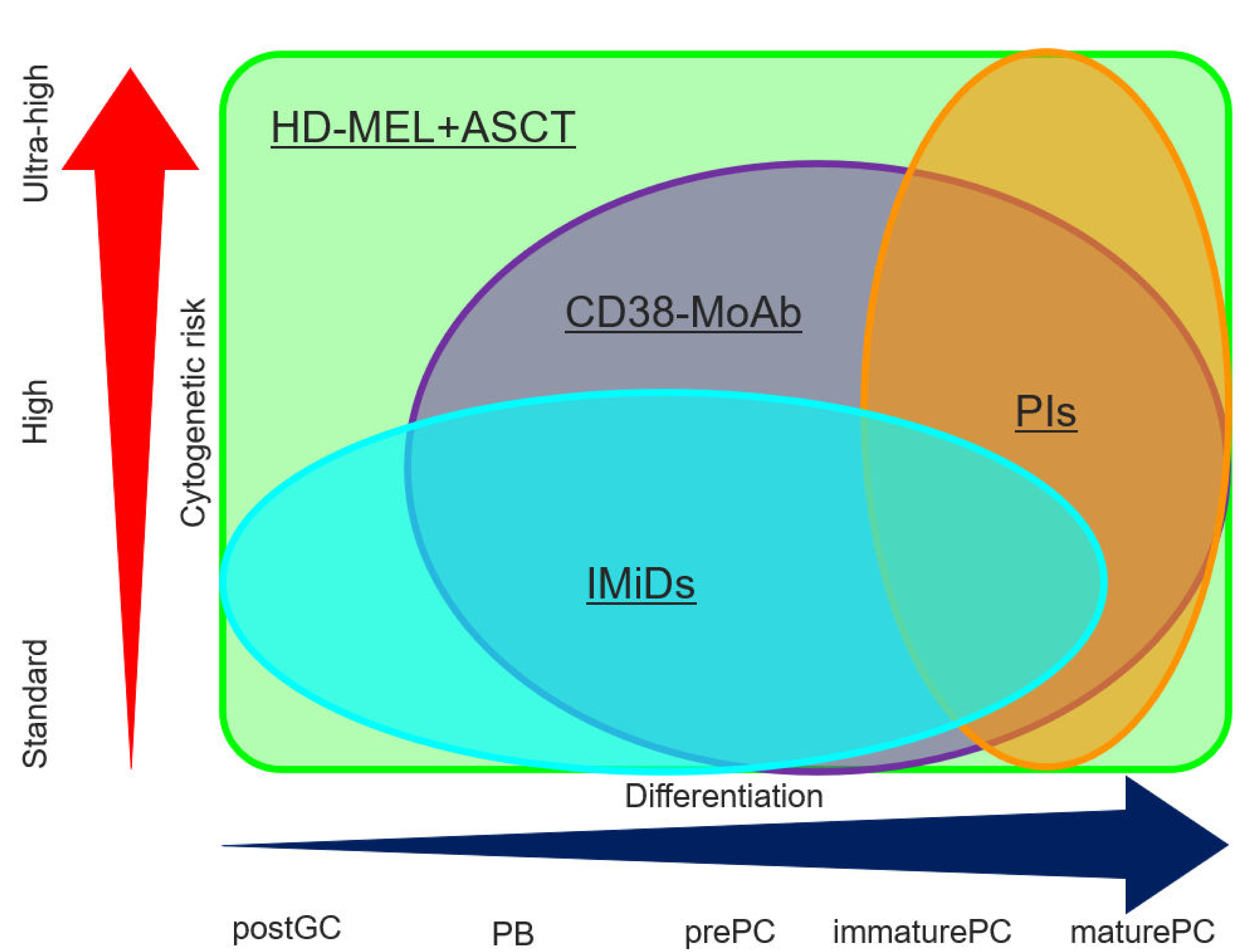
6. Overall Treatment Strategy Considering Microenvironment and Clonal Evolution
6.1. Concept of Treatment Concerning Microenvironment and Clonal Evolution
6.2. Autologous Stem Cell Transplantation for Microenvironment and Clonal Evolution
6.3. Clinical Significance of Immune Reconstitution
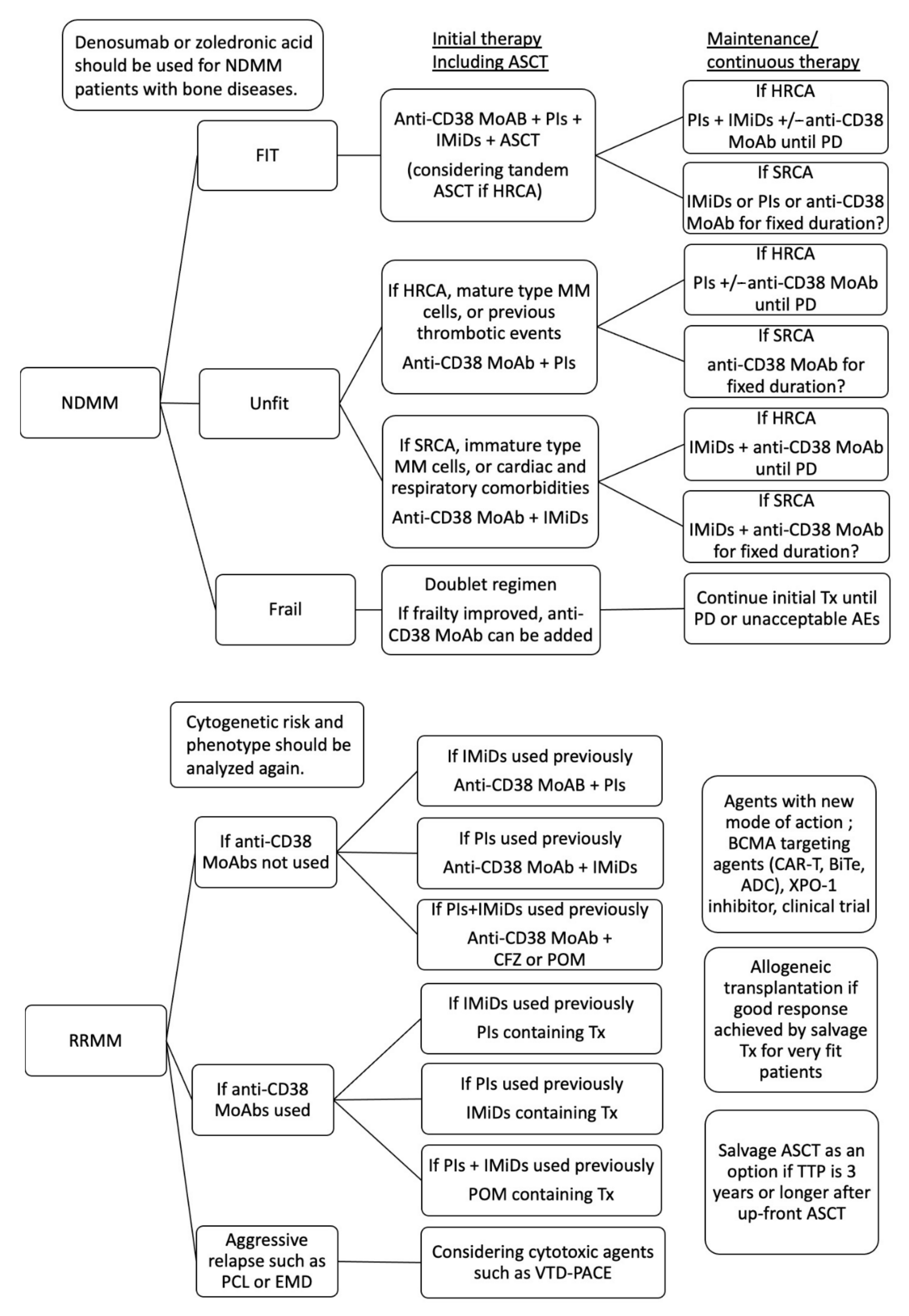
6.4. Treatment for Unfit and Frail NDMM, RRMM, and EMDs
6.5. Future Direction
7. Conclusions
Funding
Conflicts of Interest
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podar, K.; Tai, Y.T.; Lin, B.K.; Narsimhan, R.P.; Sattler, M.; Kijima, T.; Salgia, R.; Gupta, D.; Chauhan, D.; Anderson, K.C. Vascular endothelial growth factor-induced migration of multiple myeloma cells is associated with beta 1 integrin and phosphatidylinositol 3-kinase-dependent PKC alpha activation. J. Biol. Chem. 2002, 277, 7875–7881. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Vacca, A. Bone Marrow Stromal Cells-Induced Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 613. [Google Scholar] [CrossRef] [Green Version]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef] [Green Version]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Takamatsu, H. Clinical value of measurable residual disease testing for multiple myeloma and implementation in Japan. Int. J. Hematol. 2020, 111, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Liu, C.J.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 2019, 94, 1244–1253. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Manges, R.F.; Sonneveld, P.; Jagannath, S.; Somlo, G.; Krishnan, A.; Lentzsch, S.; Frank, R.C.; Zweegman, S.; Wijermans, P.W.; et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 161, 357–366. [Google Scholar] [CrossRef]
- Orlowski, R.Z.; Gercheva, L.; Williams, C.; Sutherland, H.; Robak, T.; Masszi, T.; Goranova-Marinova, V.; Dimopoulos, M.A.; Cavenagh, J.D.; Špička, I.; et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am. J. Hematol. 2015, 90, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Lacy, M.Q.; Alsina, M.; Fonseca, R.; Paccagnella, M.L.; Melvin, C.L.; Yin, D.; Sharma, A.; Enriquez Sarano, M.; Pollak, M.; Jagannath, S.; et al. Phase I, pharmacokinetic and pharmacodynamic study of the anti-insulinlike growth factor type 1 Receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. J. Clin. Oncol. 2008, 26, 3196–3203. [Google Scholar] [CrossRef]
- Somlo, G.; Lashkari, A.; Bellamy, W.; Zimmerman, T.M.; Tuscano, J.M.; O’Donnell, M.R.; Mohrbacher, A.F.; Forman, S.J.; Frankel, P.; Chen, H.X.; et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: A California Cancer Consortium trial. Br. J. Haematol. 2011, 154, 533–535. [Google Scholar] [CrossRef] [Green Version]
- White, D.; Kassim, A.; Bhaskar, B.; Yi, J.; Wamstad, K.; Paton, V.E. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer 2013, 119, 339–347. [Google Scholar] [CrossRef]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gütgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in patients with refractory or recurrent multiple myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef]
- Srkalovic, G.; Hussein, M.A.; Hoering, A.; Zonder, J.A.; Popplewell, L.L.; Trivedi, H.; Mazzoni, S.; Sexton, R.; Orlowski, R.Z.; Barlogie, B. A phase II trial of BAY 43-9006 (sorafenib) (NSC-724772) in patients with relapsing and resistant multiple myeloma: SWOG S0434. Cancer Med. 2014, 3, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.J.; Reece, D.E.; Marcellus, D.; Meyer, R.M.; Mathews, S.; Dong, R.P.; Eisenhauer, E. A phase II study of ZD6474 (Zactima, a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma—NCIC CTG IND.145. Investig. New Drugs 2006, 24, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; García-Sanz, R.; Durie, B.; Legieć, W.; Krejčí, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.S.; Moreau, P.; Terpos, E.; Benboubker, L.; Grząśko, N.; Holstein, S.A.; Oriol, A.; Huang, S.Y.; Beksac, M.; Kuliczkowski, K.; et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br. J. Haematol. 2017, 176, 783–795. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, J.; Koyama, D.; Wada, T.; Izumi, T.; Hofgaard, P.O.; Bogen, B.; Furukawa, Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Investig. 2015, 125, 4375–4390. [Google Scholar] [CrossRef]
- Alzrigat, M.; Jernberg-Wiklund, H.; Licht, J.D. Targeting EZH2 in Multiple Myeloma-Multifaceted Anti-Tumor Activity. Epigenomes 2018, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Reagan, M.R.; Ghobrial, I.M. Multiple myeloma mesenchymal stem cells: Characterization, origin, and tumor-promoting effects. Clin. Cancer Res. 2012, 18, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di Marzo, L.; De Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2014, 28, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Paiva, B.; Corchete, L.A.; Vidriales, M.B.; Puig, N.; Maiso, P.; Rodriguez, I.; Alignani, D.; Burgos, L.; Sanchez, M.L.; Barcena, P.; et al. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: A new model to understand chemoresistance. Blood 2016, 127, 1896–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Abroun, S.; Ishikawa, H.; Tsuyama, N.; Liu, S.; Li, F.J.; Otsuyama, K.; Zheng, X.; Obata, M.; Kawano, M.M. Receptor synergy of interleukin-6 (IL-6) and insulin-like growth factor-I in myeloma cells that highly express IL-6 receptor alpha [corrected]. Blood 2004, 103, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, A.; Chauhan, D.; Teoh, G.; Treon, S.P.; Urashima, M.; Schlossman, R.L.; Anderson, K.C. IL-6 triggers cell growth via the Ras-dependent mitogen-activated protein kinase cascade. J. Immunol. 1997, 159, 2212–2221. [Google Scholar] [PubMed]
- Lauta, V.M. A review of the cytokine network in multiple myeloma: Diagnostic, prognostic, and therapeutic implications. Cancer 2003, 97, 2440–2452. [Google Scholar] [CrossRef]
- Jasrotia, S.; Gupta, R.; Sharma, A.; Halder, A.; Kumar, L. Cytokine profile in multiple myeloma. Cytokine 2020, 136, 155271. [Google Scholar] [CrossRef]
- Barlogie, B.; Tricot, G.; Rasmussen, E.; Anaissie, E.; van Rhee, F.; Zangari, M.; Fassas, A.; Hollmig, K.; Pineda-Roman, M.; Shaughnessy, J.; et al. Total therapy 2 without thalidomide in comparison with total therapy 1: Role of intensified induction and posttransplantation consolidation therapies. Blood 2006, 107, 2633–2638. [Google Scholar] [CrossRef] [Green Version]
- Sprynski, A.C.; Hose, D.; Caillot, L.; Réme, T.; Shaughnessy, J.D., Jr.; Barlogie, B.; Seckinger, A.; Moreaux, J.; Hundemer, M.; Jourdan, M.; et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009, 113, 4614–4626. [Google Scholar] [CrossRef]
- Vishwamitra, D.; George, S.K.; Shi, P.; Kaseb, A.O.; Amin, H.M. Type I insulin-like growth factor receptor signaling in hematological malignancies. Oncotarget 2017, 8, 1814–1844. [Google Scholar] [CrossRef] [Green Version]
- Chiron, D.; Maïga, S.; Surget, S.; Descamps, G.; Gomez-Bougie, P.; Traore, S.; Robillard, N.; Moreau, P.; Le Gouill, S.; Bataille, R.; et al. Autocrine insulin-like growth factor 1 and stem cell factor but not interleukin 6 support self-renewal of human myeloma cells. Blood Cancer J. 2013, 3, e120. [Google Scholar] [CrossRef] [Green Version]
- Georgii-Hemming, P.; Wiklund, H.J.; Ljunggren, O.; Nilsson, K. Insulin-like growth factor I is a growth and survival factor in human multiple myeloma cell lines. Blood 1996, 88, 2250–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Kirito, K.; Yoshida, K.; Mitsumori, T.; Nakajima, K.; Nozaki, Y.; Hamanaka, S.; Nagashima, T.; Kunitama, M.; Sakoe, K.; et al. Inhibition of hypoxia-inducible factor-1 function enhances the sensitivity of multiple myeloma cells to melphalan. Mol. Cancer Ther. 2009, 8, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standal, T.; Borset, M.; Lenhoff, S.; Wisloff, F.; Stordal, B.; Sundan, A.; Waage, A.; Seidel, C. Serum insulinlike growth factor is not elevated in patients with multiple myeloma but is still a prognostic factor. Blood 2002, 100, 3925–3929. [Google Scholar] [CrossRef] [PubMed]
- Tagoug, I.; Jordheim, L.P.; Herveau, S.; Matera, E.L.; Huber, A.L.; Chettab, K.; Manié, S.; Dumontet, C. Therapeutic enhancement of ER stress by insulin-like growth factor I sensitizes myeloma cells to proteasomal inhibitors. Clin. Cancer Res. 2013, 19, 3556–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, J.; Koyama, D.; Mukai, H.Y.; Furukawa, Y. Suitable drug combination with bortezomib for multiple myeloma under stroma-free conditions and in contact with fibronectin or bone marrow stromal cells. Int. J. Hematol. 2014, 99, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef]
- Kikuchi, J.; Kuroda, Y.; Koyama, D.; Osada, N.; Izumi, T.; Yasui, H.; Kawase, T.; Ichinohe, T.; Furukawa, Y. Myeloma Cells Are Activated in Bone Marrow Microenvironment by the CD180/MD-1 Complex, Which Senses Lipopolysaccharide. Cancer Res. 2018, 78, 1766–1778. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Anand, P.; Guha, S.; Aggarwal, B.B. Curcumin circumvents chemoresistance in vitro and potentiates the effect of thalidomide and bortezomib against human multiple myeloma in nude mice model. Mol. Cancer Ther. 2009, 8, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghose, J.; Viola, D.; Terrazas, C.; Caserta, E.; Troadec, E.; Khalife, J.; Gunes, E.G.; Sanchez, J.; McDonald, T.; Marcucci, G.; et al. Daratumumab induces CD38 internalization and impairs myeloma cell adhesion. Oncoimmunology 2018, 7, e1486948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int. Rev. Cell Mol. Biol. 2015, 314, 259–282. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P. Vascular niches for disseminated tumour cells in bone. J. Bone Oncol. 2016, 5, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberstein, L.E.; Lin, C.P. A new image of the hematopoietic stem cell vascular niche. Cell Stem. Cell 2013, 13, 514–516. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.K.; Diamond, P.; Gronthos, S.; Peet, D.J.; Zannettino, A.C. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia 2011, 25, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Vanderkerken, K. Understanding the hypoxic niche of multiple myeloma: Therapeutic implications and contributions of mouse models. Dis. Model Mech. 2012, 5, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Handisides, D.R.; Van Valckenborgh, E.; De Raeve, H.; Menu, E.; Vande Broek, I.; Liu, Q.; Sun, J.D.; Van Camp, B.; Hart, C.P.; et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood 2010, 116, 1524–1527. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef]
- Vacca, A.; Ria, R.; Semeraro, F.; Merchionne, F.; Coluccia, M.; Boccarelli, A.; Scavelli, C.; Nico, B.; Gernone, A.; Battelli, F.; et al. Endothelial cells in the bone marrow of patients with multiple myeloma. Blood 2003, 102, 3340–3348. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, A.; Ria, R.; Ribatti, D.; Semeraro, F.; Djonov, V.; Di Raimondo, F.; Dammacco, F. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica 2003, 88, 176–185. [Google Scholar] [PubMed]
- Adams, J.; Carder, P.J.; Downey, S.; Forbes, M.A.; MacLennan, K.; Allgar, V.; Kaufman, S.; Hallam, S.; Bicknell, R.; Walker, J.J.; et al. Vascular endothelial growth factor (VEGF) in breast cancer: Comparison of plasma, serum, and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res. 2000, 60, 2898–2905. [Google Scholar] [PubMed]
- Sujobert, P.; Bardet, V.; Cornillet-Lefebvre, P.; Hayflick, J.S.; Prie, N.; Verdier, F.; Vanhaesebroeck, B.; Muller, O.; Pesce, F.; Ifrah, N.; et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005, 106, 1063–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Kimlinger, T.K.; Greipp, P.R.; Rajkumar, S.V. Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: Evidence favoring loss of marrow angiogenesis inhibitory activity with disease progression. Blood 2004, 104, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V.; Mesa, R.A.; Fonseca, R.; Schroeder, G.; Plevak, M.F.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Witzig, T.E.; Gertz, M.A.; et al. Bone marrow angiogenesis in 400 patients with monoclonal gammopathy of undetermined significance, multiple myeloma, and primary amyloidosis. Clin. Cancer Res. 2002, 8, 2210–2216. [Google Scholar]
- Vacca, A.; Ribatti, D.; Roncali, L.; Ranieri, G.; Serio, G.; Silvestris, F.; Dammacco, F. Bone marrow angiogenesis and progression in multiple myeloma. Br. J. Haematol. 1994, 87, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Kikukawa, Y.; Fujiwara, S.; Wada, N.; Okuno, Y.; Mitsuya, H.; Hata, H. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int. J. Oncol. 2013, 43, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.S.; Rameshwar, P.; Chang, V.; Bandari, P. Oxygen saturation in the bone marrow of healthy volunteers. Blood 2002, 99, 394. [Google Scholar] [CrossRef]
- Asosingh, K.; De Raeve, H.; de Ridder, M.; Storme, G.A.; Willems, A.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Role of the hypoxic bone marrow microenvironment in 5T2MM murine myeloma tumor progression. Haematologica 2005, 90, 810–817. [Google Scholar]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlowski, R.Z. Why proteasome inhibitors cannot ERADicate multiple myeloma. Cancer Cell 2013, 24, 275–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Shi, J.; Tolomelli, G.; Xu, H.; Xia, J.; Wang, H.; Zhou, W.; Zhou, Y.; Das, S.; Gu, Z.; et al. RARα2 expression confers myeloma stem cell features. Blood 2013, 122, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kitadate, A.; Abe, F.; Saitoh, H.; Michishita, Y.; Hatano, Y.; Kawabata, Y.; Kitabayashi, A.; Teshima, K.; Kume, M.; et al. Hypoxia-inducible microRNA-210 regulates the DIMT1-IRF4 oncogenic axis in multiple myeloma. Cancer Sci. 2017, 108, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef] [Green Version]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal-Transition-like Transcriptional Activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef] [Green Version]
- Roccaro, A.M.; Sacco, A.; Purschke, W.G.; Moschetta, M.; Buchner, K.; Maasch, C.; Zboralski, D.; Zöllner, S.; Vonhoff, S.; Mishima, Y.; et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014, 9, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Bladé, J.; Fernández de Larrea, C.; Rosiñol, L.; Cibeira, M.T.; Jiménez, R.; Powles, R. Soft-tissue plasmacytomas in multiple myeloma: Incidence, mechanisms of extramedullary spread, and treatment approach. J. Clin. Oncol. 2011, 29, 3805–3812. [Google Scholar] [CrossRef]
- Bhutani, M.; Foureau, D.M.; Atrash, S.; Voorhees, P.M.; Usmani, S.Z. Extramedullary multiple myeloma. Leukemia 2020, 34, 1–20. [Google Scholar] [CrossRef]
- Galluzzo, M.; Pennacchietti, S.; Rosano, S.; Comoglio, P.M.; Michieli, P. Prevention of hypoxia by myoglobin expression in human tumor cells promotes differentiation and inhibits metastasis. J. Clin. Investig. 2009, 119, 865–875. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Sattler, M.; Tonon, G.; Grabher, C.; Lababidi, S.; Zimmerhackl, A.; Raab, M.S.; Vallet, S.; Zhou, Y.; Cartron, M.A.; et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 2009, 69, 5082–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Catacchio, I.; Berardi, S.; De Luisi, A.; Caivano, A.; Piccoli, C.; Ruggieri, V.; Frassanito, M.A.; Ribatti, D.; Nico, B.; et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin. Cancer Res. 2014, 20, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.L.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef] [Green Version]
- Roccaro, A.M.; Hideshima, T.; Raje, N.; Kumar, S.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Ribatti, D.; Nico, B.; Vacca, A.; et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006, 66, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Dredge, K.; Marriott, J.B.; Macdonald, C.D.; Man, H.W.; Chen, R.; Muller, G.W.; Stirling, D.; Dalgleish, A.G. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br. J. Cancer 2002, 87, 1166–1172. [Google Scholar] [CrossRef]
- De Luisi, A.; Ferrucci, A.; Coluccia, A.M.; Ria, R.; Moschetta, M.; de Luca, E.; Pieroni, L.; Maffia, M.; Urbani, A.; Di Pietro, G.; et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin. Cancer Res. 2011, 17, 1935–1946. [Google Scholar] [CrossRef] [Green Version]
- Breitkreutz, I.; Raab, M.S.; Vallet, S.; Hideshima, T.; Raje, N.; Mitsiades, C.; Chauhan, D.; Okawa, Y.; Munshi, N.C.; Richardson, P.G.; et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia 2008, 22, 1925–1932. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Payvandi, F.; Wu, L.; Zhang, L.H.; Hariri, R.J.; Man, H.W.; Chen, R.S.; Muller, G.W.; Hughes, C.C.; Stirling, D.I.; et al. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc. Res. 2009, 77, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Chen, J.; Gao, H.; Kong, S.N.; Deivasigamani, A.; Shi, M.; Xie, T.; Hui, K.M. Hypoxia-induced modulation of glucose transporter expression impacts 18 F-fluorodeoxyglucose PET-CT imaging in hepatocellular carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Rosiñol, L.; Cibeira, M.T.; Bladé, J.; Esteve, J.; Aymerich, M.; Rozman, M.; Segarra, M.; Cid, M.C.; Filella, X.; Montserrat, E. Extramedullary multiple myeloma escapes the effect of thalidomide. Haematologica 2004, 89, 832–836. [Google Scholar] [PubMed]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Moreau, P.; Zweegman, S.; Perrot, A.; Hulin, C.; Caillot, D.; Facon, T.; Leleu, X.; Belhadj, K.; Karlin, L.; Benboubker, L.; et al. Evaluation of the Prognostic Value of Positron Emission Tomography-Computed Tomography (PET-CT) at Diagnosis and Follow-up in Transplant-Eligible Newly Diagnosed Multiple Myeloma (TE NDMM) Patients Treated in the Phase 3 Cassiopeia Study: Results of the Cassiopet Companion Study. Blood 2019, 134 (Suppl. 1), 692. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Caillot, D.; Macro, M.; Karlin, L.; Garderet, L.; Facon, T.; Benboubker, L.; Escoffre-Barbe, M.; Stoppa, A.M.; et al. Prospective Evaluation of Magnetic Resonance Imaging and [ 18 F]Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography at Diagnosis and Before Maintenance Therapy in Symptomatic Patients With Multiple Myeloma Included in the IFM/DFCI 2009 Trial: Results of the IMAJEM Study. J. Clin. Oncol. 2017, 35, 2911–2918. [Google Scholar] [CrossRef]
- Wu, C.; Yang, T.; Liu, Y.; Lu, Y.; Yang, Y.; Liu, X.; Liu, X.; Ye, L.; Sun, Y.; Wang, X.; et al. ARNT/HIF-1β links high-risk 1q21 gain and microenvironmental hypoxia to drug resistance and poor prognosis in multiple myeloma. Cancer Med. 2018, 7, 3899–3911. [Google Scholar] [CrossRef]
- Lakshman, A.; Singh, P.P.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Dingli, D.; Hwa, Y.L.; Fonder, A.L.; et al. Efficacy of VDT PACE-like regimens in treatment of relapsed/refractory multiple myeloma. Am. J. Hematol. 2018, 93, 179–186. [Google Scholar] [CrossRef]
- Capp, J.P.; Bataille, R. Multiple Myeloma as a Bone Disease? The Tissue Disruption-Induced Cell Stochasticity (TiDiS) Theory. Cancers 2020, 12, 2158. [Google Scholar] [CrossRef]
- Terpos, E.; Szydlo, R.; Apperley, J.F.; Hatjiharissi, E.; Politou, M.; Meletis, J.; Viniou, N.; Yataganas, X.; Goldman, J.M.; Rahemtulla, A. Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: Proposal for a novel prognostic index. Blood 2003, 102, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Kido, S.; Harada, T.; Tanaka, O.; Miki, H.; Nakamura, S.; et al. Tgf-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLoS ONE 2010, 5, e9870. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.W.; Shaughnessy, J.D., Jr.; Yaccoby, S. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood 2008, 112, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Cremer, F.W.; Reme, T.; Raab, M.; Mahtouk, K.; Kaukel, P.; Pantesco, V.; De Vos, J.; Jourdan, E.; Jauch, A.; et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 2005, 106, 1021–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, M.; Kido, S.; Hiasa, M.; Nakano, A.; Oda, A.; Amou, H.; Matsumoto, T. BAFF and APRIL as osteoclast-derived survival factors for myeloma cells: A rationale for TACI-Fc treatment in patients with multiple myeloma. Leukemia 2006, 20, 1313–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, D.A., 2nd; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Bellido, T.; Roodman, G.D. Role of osteocytes in multiple myeloma bone disease. Curr. Opin. Support. Palliat. Care 2014, 8, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Moreaux, J.; Hose, D.; Kassambara, A.; Reme, T.; Moine, P.; Requirand, G.; Goldschmidt, H.; Klein, B. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood 2011, 117, 1280–1290. [Google Scholar] [CrossRef] [Green Version]
- Politou, M.C.; Heath, D.J.; Rahemtulla, A.; Szydlo, R.; Anagnostopoulos, A.; Dimopoulos, M.A.; Croucher, P.I.; Terpos, E. Serum concentrations of Dickkopf-1 protein are increased in patients with multiple myeloma and reduced after autologous stem cell transplantation. Int. J. Cancer 2006, 119, 1728–1731. [Google Scholar] [CrossRef]
- Heider, U.; Kaiser, M.; Mieth, M.; Lamottke, B.; Rademacher, J.; Jakob, C.; Braendle, E.; Stover, D.; Sezer, O. Serum concentrations of DKK-1 decrease in patients with multiple myeloma responding to anti-myeloma treatment. Eur. J. Haematol. 2009, 82, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A.; et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 2007, 110, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangari, M.; Yaccoby, S.; Pappas, L.; Cavallo, F.; Kumar, N.S.; Ranganathan, S.; Suva, L.J.; Gruenwald, J.M.; Kern, S.; Zhan, F.; et al. A prospective evaluation of the biochemical, metabolic, hormonal and structural bone changes associated with bortezomib response in multiple myeloma patients. Haematologica 2011, 96, 333–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Raje, N.; Schoonmaker, J.A.; Liu, J.C.; Hideshima, T.; Wein, M.N.; Jones, D.C.; Vallet, S.; Bouxsein, M.L.; Pozzi, S.; et al. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J. Clin. Investig. 2008, 118, 491–504. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Hu, B.; Chen, Y.; Zhong, Y.; Shi, B.; Barlogie, B.; Shaughnessy, J.D., Jr. Bortezomib induces osteoblast differentiation via Wnt-independent activation of beta-catenin/TCF signaling. Blood 2009, 113, 4319–4330. [Google Scholar] [CrossRef] [Green Version]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef] [Green Version]
- Caraux, A.; Vincent, L.; Bouhya, S.; Quittet, P.; Moreaux, J.; Requirand, G.; Veyrune, J.L.; Olivier, G.; Cartron, G.; Rossi, J.F.; et al. Residual malignant and normal plasma cells shortly after high dose melphalan and stem cell transplantation. Highlight of a putative therapeutic window in Multiple Myeloma? Oncotarget 2012, 3, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.; Toscani, D.; Chillemi, A.; Quarona, V.; Bolzoni, M.; Marchica, V.; Vescovini, R.; Mancini, C.; Martella, E.; Campanini, N.; et al. Expression of CD38 in myeloma bone niche: A rational basis for the use of anti-CD38 immunotherapy to inhibit osteoclast formation. Oncotarget 2017, 8, 56598–56611. [Google Scholar] [CrossRef] [Green Version]
- Boutsikas, G.; Terpos, E.; Papatheodorou, A.; Tsirkinidis, P.; Tsirigotis, P.; Meletiou, A.; Lalou, E.; Telonis, V.; Zannou, A.; Kanellopoulos, A.; et al. Study of bone metabolism and angiogenesis in patients undergoing high-dose chemotherapy/autologous hematopoietic stem cell transplantation. Eur. J. Haematol. 2018, 100, 131–139. [Google Scholar] [CrossRef]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Molecular basis of clonal evolution in multiple myeloma. Int. J. Hematol. 2020, 111, 496–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.B.; Shi, C.X.; Tembe, W.; Christoforides, A.; Kurdoglu, A.; Sinari, S.; Middha, S.; Asmann, Y.; Schmidt, J.; Braggio, E.; et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012, 120, 1060–1066. [Google Scholar] [CrossRef]
- Todoerti, K.; Agnelli, L.; Fabris, S.; Lionetti, M.; Tuana, G.; Mosca, L.; Lombardi, L.; Grieco, V.; Bianchino, G.; D’Auria, F.; et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin. Cancer Res. 2013, 19, 3247–3258. [Google Scholar] [CrossRef] [Green Version]
- Cifola, I.; Lionetti, M.; Pinatel, E.; Todoerti, K.; Mangano, E.; Pietrelli, A.; Fabris, S.; Mosca, L.; Simeon, V.; Petrucci, M.T.; et al. Whole-exome sequencing of primary plasma cell leukemia discloses heterogeneous mutational patterns. Oncotarget 2015, 6, 17543–17558. [Google Scholar] [CrossRef]
- Ohta, T.; Kimura, M. A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population*. Genet. Res. 2007, 89, 367–370. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Molecular pathogenesis of multiple myeloma. Int. J. Clin. Oncol. 2015, 20, 413–422. [Google Scholar] [CrossRef]
- Johnson, D.C.; Lenive, O.; Mitchell, J.; Jackson, G.; Owen, R.; Drayson, M.; Cook, G.; Jones, J.R.; Pawlyn, C.; Davies, F.E.; et al. Neutral tumor evolution in myeloma is associated with poor prognosis. Blood 2017, 130, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Heuck, C.; Mitchell, A.; Szymonifka, J.; Nair, B.; Hoering, A.; Alsayed, Y.; Waheed, S.; Haider, S.; Restrepo, A.; et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012, 97, 1761–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrábel, D.; Pour, L.; Ševčíková, S. The impact of NF-κB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. 2019, 34, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van de Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and Targeting of TP53 Pathway in Multiple Myeloma. Front. Oncol. 2019, 8, 665. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Tian, E.; Qu, P.; Mathur, P.; Schinke, C.; van Rhee, F.; Zangari, M.; Rasche, L.; Weinhold, N.; Alapat, D.; et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica 2017, 102, e364–e367. [Google Scholar] [CrossRef] [Green Version]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Chin, M.; Sive, J.I.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. 2017, 7, e610. [Google Scholar] [CrossRef] [Green Version]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshman, A.; Painuly, U.; Rajkumar, S.V.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; et al. Impact of acquired del(17p) in multiple myeloma. Blood Adv. 2019, 3, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Lancman, G.; Tremblay, D.; Barley, K.; Barlogie, B.; Cho, H.J.; Jagannath, S.; Madduri, D.; Moshier, E.; Parekh, S.; Chari, A. The effect of novel therapies in high-molecular-risk multiple myeloma. Clin. Adv. Hematol. Oncol. 2017, 15, 870–879. [Google Scholar] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Zhou, X.; Dierks, A.; Kertels, O.; Samnick, S.; Kircher, M.; Buck, A.K.; Haertle, L.; Knorz, S.; Böckle, D.; Scheller, L.; et al. The Link between Cytogenetics/Genomics and Imaging Patterns of Relapse and Progression in Patients with Relapsed/Refractory Multiple Myeloma: A Pilot Study Utilizing 18F-FDG PET/CT. Cancers 2020, 12, E2399. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef]
- Pawlyn, C.; Loehr, A.; Ashby, C.; Tytarenko, R.; Deshpande, S.; Sun, J.; Fedorchak, K.; Mughal, T.; Davies, F.E.; Walker, B.A.; et al. Loss of heterozygosity as a marker of homologous repair deficiency in multiple myeloma: A role for PARP inhibition? Leukemia 2018, 32, 1561–1566. [Google Scholar] [CrossRef] [Green Version]
- Weißbach, S.; Langer, C.; Puppe, B.; Nedeva, T.; Bach, E.; Kull, M.; Bargou, R.; Einsele, H.; Rosenwald, A.; Knop, S.; et al. The molecular spectrum and clinical impact of DIS3 mutations in multiple myeloma. Br. J. Haematol. 2015, 169, 57–70. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- An, G.; Yan, Y.; Xu, Y.; Mao, X.; Liu, J.; Fan, H.; Wang, Q.; Du, C.; Li, Z.; Yi, S.; et al. Monitoring the cytogenetic architecture of minimal residual plasma cells indicates therapy-induced clonal selection in multiple myeloma. Leukemia 2020, 34, 578–588. [Google Scholar] [CrossRef]
- Jones, J.R.; Weinhold, N.; Ashby, C.; Walker, B.A.; Wardell, C.; Pawlyn, C.; Rasche, L.; Melchor, L.; Cairns, D.A.; Gregory, W.M.; et al. Clonal evolution in myeloma: The impact of maintenance lenalidomide and depth of response on the genetics and sub-clonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica 2019, 104, 1440–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, L.; Alapat, D.; Kumar, M.; Gershner, G.; McDonald, J.; Wardell, C.P.; Samant, R.; Van Hemert, R.; Epstein, J.; Williams, A.F.; et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia 2019, 33, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Barnes, C.P.; Cowan, G.; May, P.C.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Pourabdollah, M.; Bahmanyar, M.; Atenafu, E.G.; Reece, D.; Hou, J.; Chang, H. High IKZF1/3 protein expression is a favorable prognostic factor for survival of relapsed/refractory multiple myeloma patients treated with lenalidomide. J. Hematol. Oncol. 2016, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, M.; Cren, M.; Schafer, P.; Robert, N.; Duperray, C.; Vincent, L.; Ceballos, P.; Cartron, G.; Rossi, J.F.; Moreaux, J.; et al. Differential effects of lenalidomide during plasma cell differentiation. Oncotarget. 2016, 7, 28096–28111. [Google Scholar] [CrossRef]
- Hosen, N.; Matsuoka, Y.; Kishida, S.; Nakata, J.; Mizutani, Y.; Hasegawa, K.; Mugitani, A.; Ichihara, H.; Aoyama, Y.; Nishida, S.; et al. CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia. 2012, 26, 2135–2141. [Google Scholar] [CrossRef]
- Jakubikova, J.; Adamia, S.; Kost-Alimova, M.; Klippel, S.; Cervi, D.; Daley, J.F.; Cholujova, D.; Kong, S.Y.; Leiba, M.; Blotta, S.; et al. Lenalidomide targets clonogenic side population in multiple myeloma: Pathophysiologic and clinical implications. Blood 2011, 117, 4409–4419. [Google Scholar] [CrossRef]
- Binder, M.; Rajkumar, S.V.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Dispenzieri, A.; Hwa, Y.L.; Fonder, A.; Hobbs, M.; Hayman, S.R.; et al. Peripheral blood biomarkers of early immune reconstitution in newly diagnosed multiple myeloma. Am. J. Hematol. 2019, 94, 306–311. [Google Scholar] [CrossRef]
- Jimenez-Zepeda, V.H.; Reece, D.E.; Trudel, S.; Chen, C.; Franke, N.; Winter, A.; Tiedemann, R.; Kukreti, V. Absolute lymphocyte count as predictor of overall survival for patients with multiple myeloma treated with single autologous stem cell transplant. Leuk. Lymphoma 2015, 56, 2668–2673. [Google Scholar] [CrossRef]
- Arteche-López, A.; Kreutzman, A.; Alegre, A.; Sanz Martín, P.; Aguado, B.; González-Pardo, M.; Espiño, M.; Villar, L.M.; García Belmonte, D.; de la Cámara, R.; et al. Multiple myeloma patients in long-term complete response after autologous stem cell transplantation express a particular immune signature with potential prognostic implication. Bone Marrow Transplant. 2017, 52, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Bryant, C.; Suen, H.; Brown, R.; Yang, S.; Favaloro, J.; Aklilu, E.; Gibson, J.; Ho, P.J.; Iland, H.; Fromm, P.; et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013, 3, e148. [Google Scholar] [CrossRef]
- Paiva, B.; Cedena, M.T.; Puig, N.; Arana, P.; Vidriales, M.B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.L.; Martinez-Lopez, J.; et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood 2016, 127, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- De Larrea, C.F.; Cibeira, M.T.; Elena, M.; Arostegui, J.I.; Rosiñol, L.; Rovira, M.; Filella, X.; Yagüe, J.; Bladé, J. Abnormal serum free light chain ratio in patients with multiple myeloma in complete remission has strong association with the presence of oligoclonal bands: Implications for stringent complete remission definition. Blood 2009, 114, 4954–4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zent, C.S.; Wilson, C.S.; Tricot, G.; Jagannath, S.; Siegel, D.; Desikan, K.R.; Munshi, N.; Bracy, D.; Barlogie, B.; Butch, A.W. Oligoclonal protein bands and Ig isotype switching in multiple myeloma treated with high-dose therapy and hematopoietic cell transplantation. Blood 1998, 91, 3518–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef]
- Giri, S.; Grimshaw, A.; Bal, S.; Godby, K.; Kharel, P.; Djulbegovic, B.; Dimopoulos, M.A.; Facon, T.; Usmani, S.Z.; Mateos, M.V.; et al. Evaluation of Daratumumab for the Treatment of Multiple Myeloma in Patients With High-risk Cytogenetic Factors: A Systematic Review and Meta-analysis. JAMA Oncol. 2020, e204338. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omedè, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M.; et al. High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica 2014, 99, e14–e16. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Lau, E.K.; Al-Shabeeb, A.; Nikolic, A.; Catalano, A.; Iland, H.; Horvath, N.; Ho, P.J.; Harrison, S.; Fleming, S.; et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 2012, 97, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.D., Jr.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef]
- Hayashi, T.; Hideshima, T.; Akiyama, M.; Podar, K.; Yasui, H.; Raje, N.; Kumar, S.; Chauhan, D.; Treon, S.P.; Richardson, P.G.; et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: Clinical application. Br. J. Haematol. 2005, 128, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.; Janji, B.; Berchem, G. Activation of NK cells and disruption of PD-L1/PD-1 axis: Two different ways for lenalidomide to block myeloma progression. Oncotarget 2017, 8, 24031–24044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Krönke, J.; Kuchenbauer, F.; Kull, M.; Teleanu, V.; Bullinger, L.; Bunjes, D.; Greiner, A.; Kolmus, S.; Köpff, S.; Schreder, M.; et al. IKZF1 expression is a prognostic marker in newly diagnosed standard-risk multiple myeloma treated with lenalidomide and intensive chemotherapy: A study of the German Myeloma Study Group (DSMM). Leukemia 2017, 31, 1363–1367. [Google Scholar] [CrossRef]
- Seckinger, A.; Hillengass, J.; Emde, M.; Beck, S.; Kimmich, C.; Dittrich, T.; Hundemer, M.; Jauch, A.; Hegenbart, U.; Raab, M.S.; et al. CD38 as Immunotherapeutic Target in Light Chain Amyloidosis and Multiple Myeloma-Association With Molecular Entities, Risk, Survival, and Mechanisms of Upfront Resistance. Front. Immunol. 2018, 9, 1676. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of myeloma [corrected] and their uses in immunotherapies. Blood Cancer J. 2015, 5, e306. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; San-Miguel, J.; Belch, A.; White, D.; Benboubker, L.; Cook, G.; Leiba, M.; Morton, J.; Ho, P.J.; Kim, K.; et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of POLLUX. Haematologica 2018, 103, 2088–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calandra, G.; McCarty, J.; McGuirk, J.; Tricot, G.; Crocker, S.A.; Badel, K.; Grove, B.; Dye, A.; Bridger, G. AMD3100 plus G-CSF can successfully mobilize CD34+ cells from non-Hodgkin’s lymphoma, Hodgkin’s disease and multiple myeloma patients previously failing mobilization with chemotherapy and/or cytokine treatment: Compassionate use data. Bone Marrow Transplant. 2008, 41, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, F.; Cerrato, C.; Scalabrini, D.R.; Galli, M.; Belotti, A.; Zamagni, E.; Ledda, A.; Grasso, M.; Emanuele Angelucci, E.; Liberati, A.M.; et al. Carfilzomib-Lenalidomide-Dexamethasone (KRd) Induction-Autologous Transplant (ASCT)-Krd Consolidation Vs KRd 12 Cycles Vs Carfilzomib-Cyclophosphamide-Dexamethasone (KCd) Induction-ASCT-KCd Consolidation: Analysis of the Randomized Forte Trial in Newly Diagnosed Multiple Myeloma (NDMM). Blood 2018, 132, 121. [Google Scholar]
- Oliva, S.; Genuardi, E.; Belotti, A.; Frascione, P.M.M.; Galli, M.; Capra, A.; Offidani, M.; Vozella, F.; Zambello, R.; Auclair, D.; et al. Multiparameter flow cytometry (MFC) and next generation sequencing (NGS) for minimal residual disease (MRD) evaluation: Results of the FORTE trial in newly diagnosed multiple myeloma (MM). J. Clin. Oncol. 2020, 38, 8533. [Google Scholar] [CrossRef]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dörner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef]
- Paiva, B.; Pérez-Andrés, M.; Vídriales, M.B.; Almeida, J.; de las Heras, N.; Mateos, M.V.; López-Corral, L.; Gutiérrez, N.C.; Blanco, J.; Oriol, A.; et al. Competition between clonal plasma cells and normal cells for potentially overlapping bone marrow niches is associated with a progressively altered cellular distribution in MGUS vs myeloma. Leukemia 2011, 25, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, L.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef]
- Ross, F.M.; Ibrahim, A.H.; Vilain-Holmes, A.; Winfield, M.O.; Chiecchio, L.; Protheroe, R.K.; Strike, P.; Gunasekera, J.L.; Jones, A.; Harrison, C.J.; et al. Age has a profound effect on the incidence and significance of chromosome abnormalities in myeloma. Leukemia 2005, 19, 1634–1642. [Google Scholar] [CrossRef] [Green Version]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and immunotherapy in multiple myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef]
- San Miguel, J.F.; González, M.; Gascón, A.; Moro, M.J.; Hernández, J.M.; Ortega, F.; Jiménez, R.; Guerras, L.; Romero, M.; Casanova, F.; et al. Lymphoid subsets and prognostic factors in multiple myeloma. Cooperative Group for the Study of Monoclonal Gammopathies. Br. J. Haematol. 1992, 80, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.A.; Sive, J.; Ambrose, J.; Roddie, C.; Counsell, N.; Lach, A.; Abbasian, M.; Popat, R.; Cavenagh, J.D.; Oakervee, H.; et al. RNA-seq of newly diagnosed patients in the PADIMAC study leads to a bortezomib/lenalidomide decision signature. Blood 2018, 132, 2154–2165. [Google Scholar] [CrossRef]
- Ubels, J.; Sonneveld, P.; van Beers, E.H.; Broijl, A.; van Vliet, M.H.; de Ridder, J. Predicting treatment benefit in multiple myeloma through simulation of alternative treatment effects. Nat. Commun. 2018, 9, 2943. [Google Scholar] [CrossRef] [Green Version]
- Korst, C.L.B.M.; van de Donk, N.W.C.J. Should all newly diagnosed MM patients receive CD38 antibody–based treatment? Hematol. Am. Soc. Hematol. Educ. Program. 2020, 259–263. [Google Scholar] [CrossRef]
- Liwing, J.; Uttervall, K.; Lund, J.; Aldrin, A.; Blimark, C.; Carlson, K.; Enestig, J.; Flogegård, M.; Forsberg, K.; Gruber, A.; et al. Improved survival in myeloma patients: Starting to close in on the gap between elderly patients and a matched normal population. Br. J. Haematol. 2014, 164, 684–693. [Google Scholar] [CrossRef]
- Beksac, M.; Seval, G.C.; Kanellias, N.; Coriu, D.; Rosiñol, L.; Ozet, G.; Goranova-Marinova, V.; Unal, A.; Bila, J.; Ozsan, H.; et al. A real world multicenter retrospective study on extramedullary disease from Balkan Myeloma Study Group and Barcelona University: Analysis of parameters that improve outcome. Haematologica 2020, 105, 201–208. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.J.; Jung, C.W.; Jang, J.H.; Kim, S.J.; Kim, W.S.; Kim, K. DCEP for relapsed or refractory multiple myeloma after therapy with novel agents. Ann. Hematol. 2014, 93, 99–105. [Google Scholar] [CrossRef]
- Abdallah, A.O.; Sigle, M.; Mohyuddin, G.R.; Coggins, E.; Remker, C.; Shune, L.; Mahmoudjafari, Z.; McGuirk, J.; Ganguly, S. Outcomes of VD-PACE With Immunomodulatory Agent as a Salvage Therapy for Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2020. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 38, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Bringhen, S.; Voorhees, P.; Plesner, T.; Mellqvist, U.H.; Reeves, B.; Paba-Prada, C.; Zubair, H.; Byrne, C.; Chauhan, D.; et al. Melflufen plus dexamethasone in relapsed and refractory multiple myeloma (O-12-M1): A multicentre, international, open-label, phase 1-2 study. Lancet Haematol. 2020, 7, e395–e407. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Target | Experimental Arm | Control Arm | Disease Status | Phase | Endopoint | Outcome | References |
|---|---|---|---|---|---|---|---|
| Stromal cell and adhesion | |||||||
| CXCR4 | Plerixafor + BOR | – | RRMM prior BOR | 2 | ORR | 45% (CBR = 60.6%) | [14] |
| IL-6 | Siltuximab + BOR | – | RRMM | 2 | ORR | 0% (siltuximab only), 8% (plus DEX) | [15] |
| IL-6 | Siltuximab ± DEX | – | RRMM | 2 | PFS | 8.0 vs. 7.6 mo (p = 0.345) | [16] |
| IGF-1R | Figitumumab ± DEX | – | RRMM | 1 | ORR | 33% | [17] |
| Vascular niche | |||||||
| VEGF-A | Bevacizumab + THAL | – | RRMM | 2 | ORR | 33%, EFS = 37−369 days | [18] |
| VEGF-A | Bevacizumab + BOR | BOR | RRMM | 2 | PFS | 6.2 vs. 5.1 mo (p = 0.28) | [19] |
| VEGFR | Sorafenib | – | RRMM | 2 | ORR | 9% (CBR = 18%) | [20] |
| VEGFR | Sorafenib | – | RRMM | 2 | ORR | 0% | [21] |
| VEGFR-2 | Vandetanib | – | RRMM | 2 | ORR | 0% | [22] |
| Endosteal niche | |||||||
| RANKL | Denosumab | Zoledronic acid | NDMM | 3 | Time to skeletal events, PFS | 22.8 vs. 24.0% (p = 0.01, non-inferior), PFS = 46.1 vs. 35.4 mo (p = 0.036) | [23] |
| DKK-1 | BHQ880 | – | RRMM | 1b | ORR | 15%, CBR = 23% (10 mg/kg) | [24] |
| BAFF | Tabalumab + BOR + DEX | BOR + DEX | RRMM | 2 | PFS | 6.6 (100 mg) vs. 7.5 (300 mg) vs. 7.6 mo (PCB) (p = NS) | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, K.; Nishiwaki, K.; Yano, S. Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma. Cancers 2021, 13, 215. https://doi.org/10.3390/cancers13020215
Suzuki K, Nishiwaki K, Yano S. Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma. Cancers. 2021; 13(2):215. https://doi.org/10.3390/cancers13020215
Chicago/Turabian StyleSuzuki, Kazuhito, Kaichi Nishiwaki, and Shingo Yano. 2021. "Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma" Cancers 13, no. 2: 215. https://doi.org/10.3390/cancers13020215
APA StyleSuzuki, K., Nishiwaki, K., & Yano, S. (2021). Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma. Cancers, 13(2), 215. https://doi.org/10.3390/cancers13020215