ADCC against MICA/B Is Mediated against Differentiated Oral and Pancreatic and Not Stem-Like/Poorly Differentiated Tumors by the NK Cells; Loss in Cancer Patients due to Down-Modulation of CD16 Receptor



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
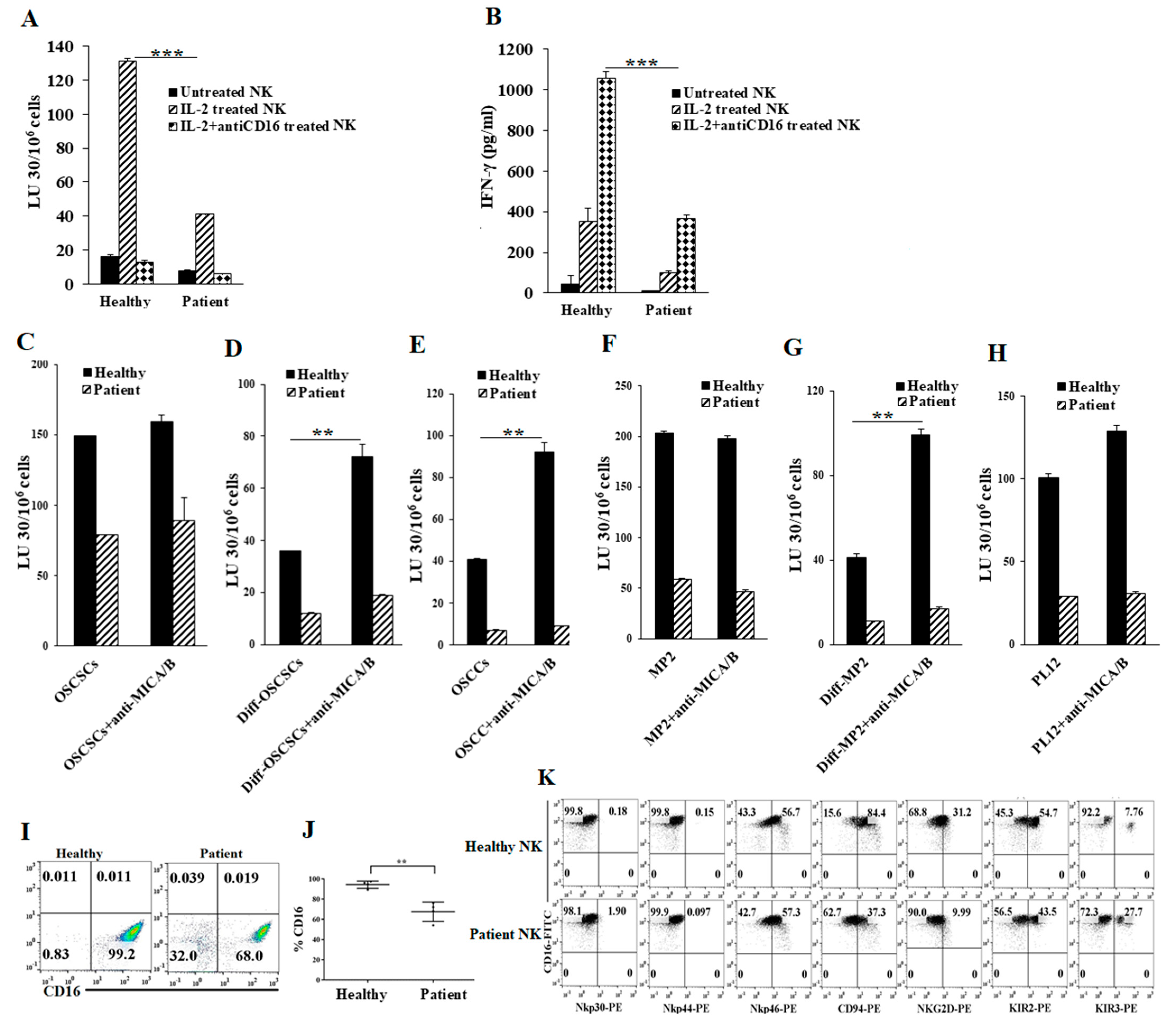
2.1. Cancer Patients’ NK Cells Exhibit Decreased Direct Killing and NK Cell-Mediated ADCC Compared to Healthy Individuals’ NK Cells
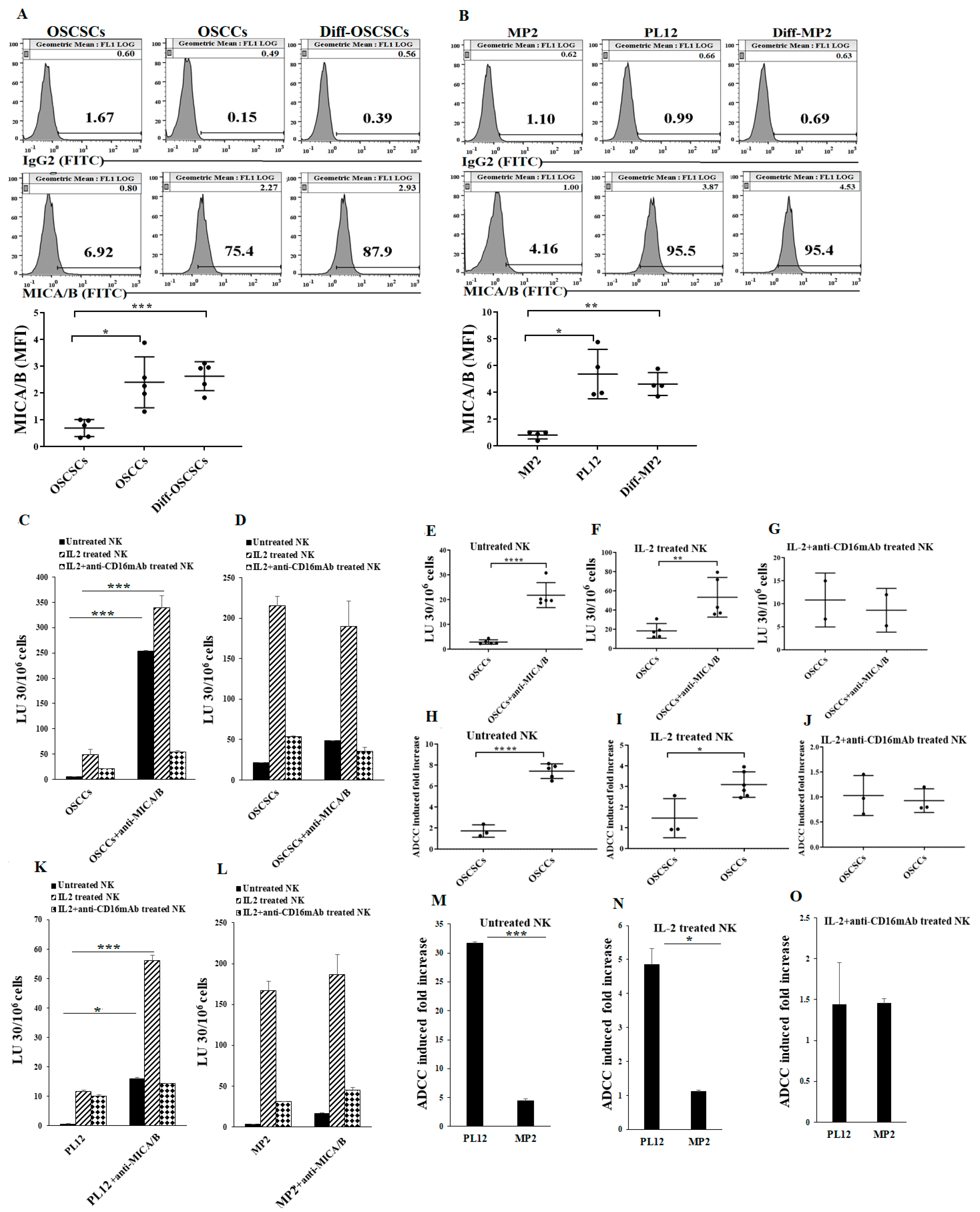
2.2. Differentiated Tumor Cells Expressed Higher Levels of Surface MICA/B and Were More Susceptible to NK Cell-Mediated ADCC in the Presence of Anti-MICA/B mAb Compared to Their Stem-Like/Poorly Differentiated Couterparts
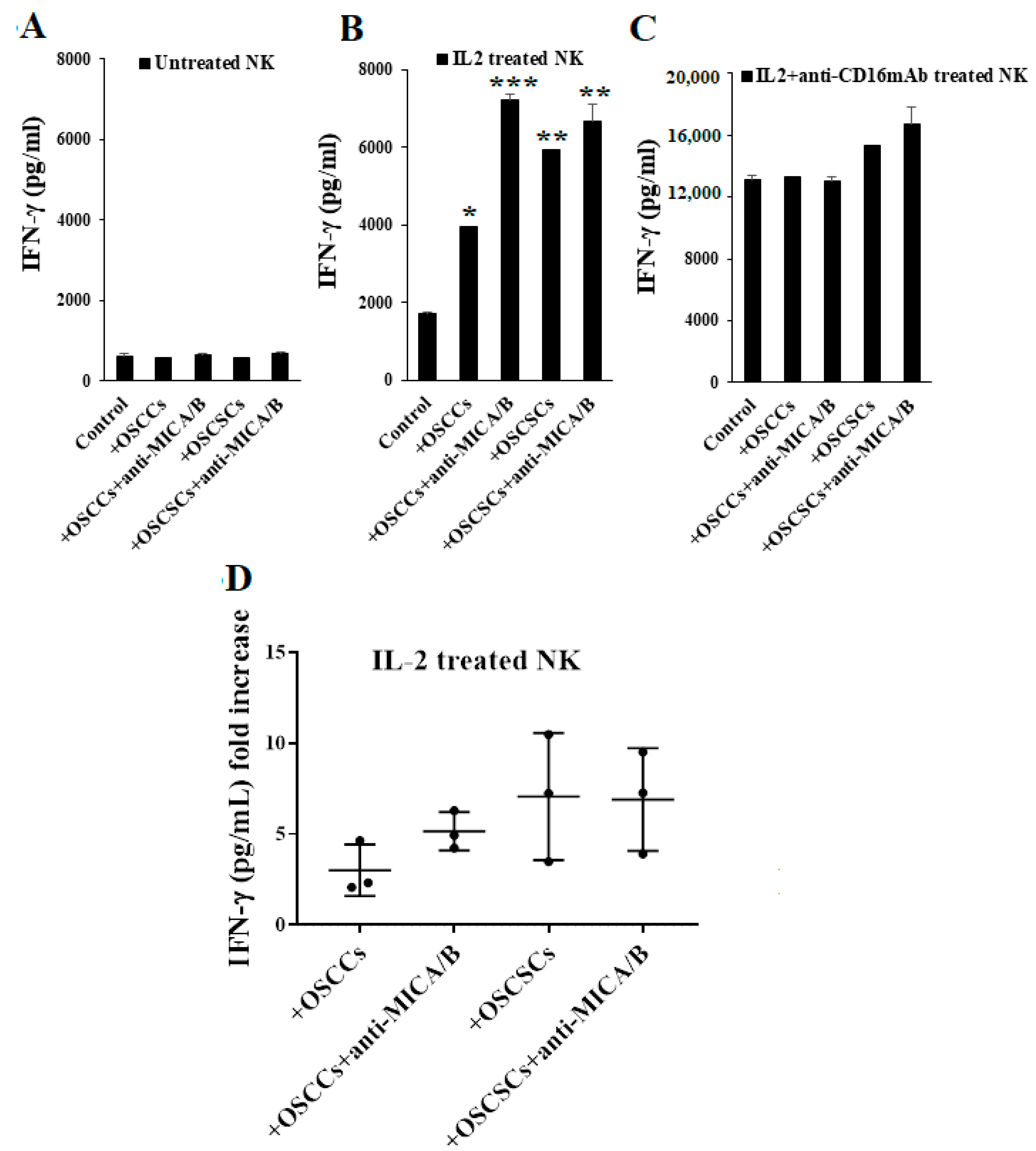
2.3. Differentiated Tumor Cells Treated with Anti-MICA/B mAb Triggered Increased IFN-γ Secretion by NK Cells
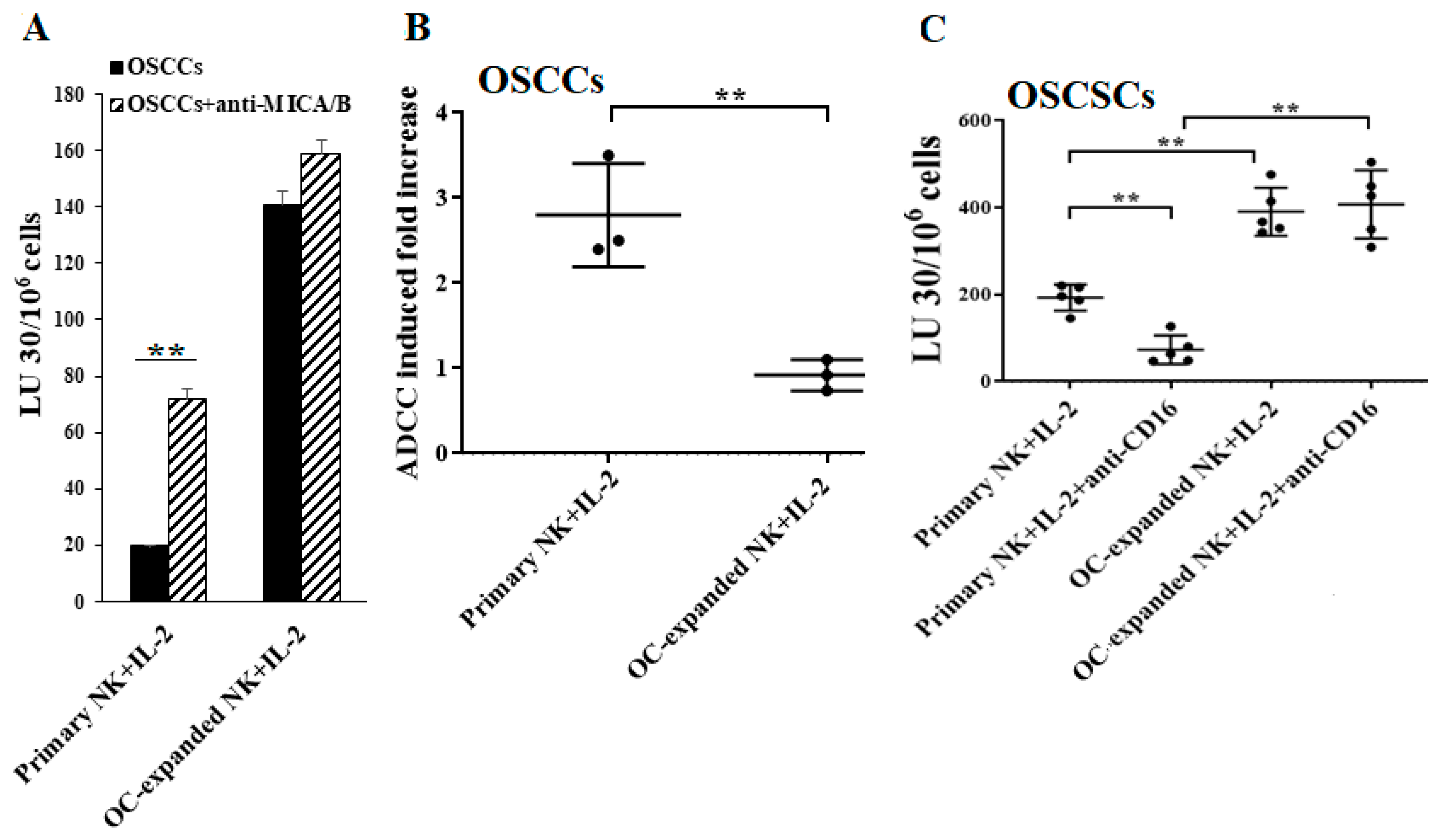
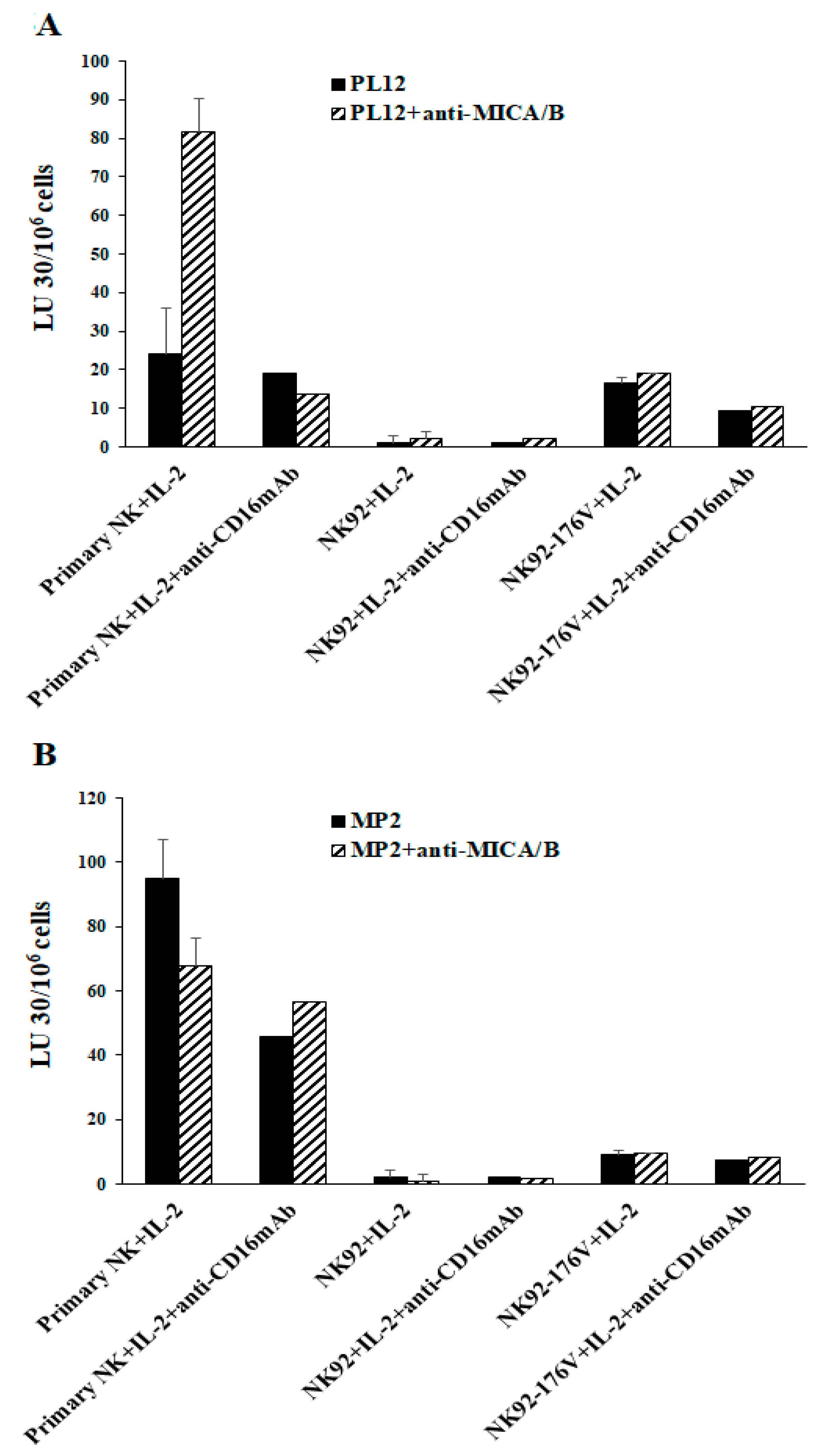
2.4. Higher Levels of NK Cell-Mediated ADCC Were Seen in Freshly Isolated Primary NK Cells When Compared to OC-Expanded NK Cells
2.5. Higher Levels of NK Cell-Mediated Direct Cytotoxicity and ADCC by Freshly Isolated NK Cells When Compared to Either Parental NK92 Tumors and Those Expressing CD16 Receptor
2.6. Differentiated Tumor Cells Expressed Higher Levels of Surface EGFR and PDL-1 and Were More Susceptible to NK Cell-Mediated ADCC in the Presence of Anti-EGFR and Anti-PDL1 mAbs Compared to Their Stem-Like/Poorly Differentiated Counterparts
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents, and Antibodies
4.2. Purification of Human NK Cells and Monocytes
4.3. NK Cells Induced Differentiation of OSCSCs and MP2 Tumors
4.4. Generation of Human OCs
4.5. Sonication of Probiotic Bacteria (AJ2)
4.6. Expansion of NK Cells
4.7. Enzyme-Linked Immunosorbent Assays (ELISAs)
4.8. 51Cr release Cytotoxicity Assay
4.9. Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) Measurements
4.10. Surface Staining Assay
4.11. Western Blot
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NK cells | Natural killer cells |
| MICA/B | Major histocompatibility complex-class I chain related proteins A and B |
| ADCC | Antibody-dependent cellular cytotoxicity (ADCC) |
| Hu-BLT | Humanized-bone marrow/liver/thymus |
| MP2 | Mia PaCa-2 pancreatic cancer stem cells |
| OSCSCs | Oral squamous cancer stem cells |
| OSCCs | Oral squamous carcinoma cells |
| MHC-Class I | Major histocompatibility complex molecule class I |
| IFN-γ | Interferon-gamma |
| TNF-α | Tumor necrosis factor-α |
| CSCs | Cancer stem cells |
| rhIL-2 | Recombinant human IL-2 |
References
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Fildes, J.E.; Yonan, N.; Leonard, C.T. Natural killer cells and lung transplantation, roles in rejection, infection, and tolerance. Transpl. Immunol. 2008, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Caligiuri, M.A. Human natural killer cell development and biology. Blood Rev. 2006, 20, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef]
- Gogali, F.; Paterakis, G.; Rassidakis, G.Z.; Liakou, C.I.; Liapi, C. CD3(-)CD16(-)CD56(bright) immunoregulatory NK cells are increased in the tumor microenvironment and inversely correlate with advanced stages in patients with papillary thyroid cancer. Thyroid Off. J. Am. Thyroid Assoc. 2013, 23, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cobo, S.; Pieper, N.; Campos-Silva, C.; Garcia-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Vales-Gomez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology 2018, 7, e1392426. [Google Scholar] [CrossRef]
- Morel, P.A.; Ernst, L.K.; Metes, D. Functional CD32 molecules on human NK cells. Leuk. Lymphoma 1999, 35, 47–56. [Google Scholar] [CrossRef]
- Lanier, L.L.; Ruitenberg, J.J.; Phillips, J.H. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol. 1988, 141, 3478–3485. [Google Scholar]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Groh, V.; Spies, T.; Kimura, R.; Miyagi, T.; Mochizuki, K.; Sasaki, Y.; et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int. J. Cancer 2003, 104, 354–361. [Google Scholar] [CrossRef]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Moncayo, G.; Lin, D.; McCarthy, M.T.; Watson, A.A.; O’Callaghan, C.A. MICA Expression Is Regulated by Cell Adhesion and Contact in a FAK/Src-Dependent Manner. Front. Immunol. 2016, 7, 687. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.W.; Kim, A.B.; Li, P.J.; Wang, J.; Jackson, B.T.; Huang, K.T.H.; Zhang, L.; Raulet, D.H. Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, N.; Yu, Y.; Zhou, L.; Niu, C.; Liu, Y.; Tian, H.; Lv, Z.; Han, F.; Cui, J. Prognostic value of MICA/B in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 96384–96395. [Google Scholar] [CrossRef]
- Lin, S.R.; Wen, Y.C.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Mokgautsi, N.; Huang, J.; Chen, W.Y.; Liu, Y.N. EGFR-upregulated LIFR promotes SUCLG2-dependent castration resistance and neuroendocrine differentiation of prostate cancer. Oncogene 2020, 39, 6757–6775. [Google Scholar] [CrossRef]
- Eriksen, J.G.; Steiniche, T.; Askaa, J.; Alsner, J.; Overgaard, J. The prognostic value of epidermal growth factor receptor is related to tumor differentiation and the overall treatment time of radiotherapy in squamous cell carcinomas of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 561–566. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Tseng, H.C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-γ after differentiation of glioblastoma by human natural killer cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef]
- Bui, V.T.; Tseng, H.C.; Kozlowska, A.; Maung, P.O.; Kaur, K.; Topchyan, P.; Jewett, A. Augmented IFN-γ and TNF-α Induced by Probiotic Bacteria in NK Cells Mediate Differentiation of Stem-Like Tumors Leading to Inhibition of Tumor Growth and Reduction in Inflammatory Cytokine Release; Regulation by IL-10. Front. Immunol. 2015, 6, 576. [Google Scholar] [CrossRef]
- Alderson, K.L.; Sondel, P.M. Clinical Cancer Therapy by NK Cells via Antibody-Dependent Cell-Mediated Cytotoxicity. J. Biomed. Biotechnol. 2011, 2011, 379123. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed]
- Yeap, W.H.; Wong, K.L.; Shimasaki, N.; Teo, E.C.Y.; Quek, J.K.S.; Yong, H.X.; Diong, C.P.; Bertoletti, A.; Linn, Y.C.; Wong, S.C. CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci. Rep. 2016, 6, 34310. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, N.; Ahmad, F.; Hong, H.S.; Eberhard, J.; Lu, I.N.; Ballmaier, M.; Schmidt, R.E.; Jacobs, R.; Meyer-Olson, D. FcgammaRIII (CD16)-mediated ADCC by NK cells is regulated by monocytes and FcgammaRII (CD32). Eur. J. Immunol. 2014, 44, 3368–3379. [Google Scholar] [CrossRef] [PubMed]
- Oboshi, W.; Watanabe, T.; Matsuyama, Y.; Kobara, A.; Yukimasa, N.; Ueno, I.; Aki, K.; Tada, T.; Hosoi, E. The influence of NK cell-mediated ADCC: Structure and expression of the CD16 molecule differ among FcgammaRIIIa-V158F genotypes in healthy Japanese subjects. Hum. Immunol. 2016, 77, 165–171. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.S.; Richard, J.; Lee, W.S.; Vanderven, H.; Grant, M.D.; Finzi, A.; Kent, S.J. NKG2D Acts as a Co-Receptor for Natural Killer Cell-Mediated Anti-HIV-1 Antibody-Dependent Cellular Cytotoxicity. Aids Res. Hum. Retrovir. 2016, 32, 1089–1096. [Google Scholar] [CrossRef]
- Di Modica, M.; Sfondrini, L.; Regondi, V.; Varchetta, S.; Oliviero, B.; Mariani, G.; Bianchi, G.V.; Generali, D.; Balsari, A.; Triulzi, T.; et al. Taxanes enhance trastuzumab-mediated ADCC on tumor cells through NKG2D-mediated NK cell recognition. Oncotarget 2016, 7, 255–265. [Google Scholar] [CrossRef]
- Märklin, M.; Hagelstein, I.; Koerner, S.P.; Rothfelder, K.; Pfluegler, M.S.; Schumacher, A.; Grosse-Hovest, L.; Jung, G.; Salih, H.R. Bispecific NKG2D-CD3 and NKG2D-CD16 fusion proteins for induction of NK and T cell reactivity against acute myeloid leukemia. J. Immunother. Cancer 2019, 7, 143. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Li, P.; Morris, D.L.; Willcox, B.E.; Steinle, A.; Spies, T.; Strong, R.K. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat. Immunol. 2001, 2, 443–451. [Google Scholar] [CrossRef]
- Cerwenka, A.; Baron, J.L.; Lanier, L.L. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 11521–11526. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.G.; Kiessling, R.; Malmberg, K.J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Takahashi, A.; Ichihara, F.; Sugai, H.; Fujii, H.; Matsumoto, Y. Impaired antibody-dependent cellular cytotoxicity mediated by herceptin in patients with gastric cancer. Cancer Res. 2002, 62, 5813–5817. [Google Scholar] [PubMed]
- Kawaguchi, Y.; Kono, K.; Mimura, K.; Sugai, H.; Akaike, H.; Fujii, H. Cetuximab induce antibody-dependent cellular cytotoxicity against EGFR-expressing esophageal squamous cell carcinoma. Int. J. Cancer 2007, 120, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kono, K.; Kawaguchi, Y.; Mizukami, Y.; Mimura, K.; Maruyama, T.; Izawa, S.; Fujii, H. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis. Esophagus 2010, 23, 675–681. [Google Scholar] [CrossRef]
- Nakajima, T.; Okayama, H.; Ashizawa, M.; Noda, M.; Aoto, K.; Saito, M.; Monma, T.; Ohki, S.; Shibata, M.; Takenoshita, S.; et al. Augmentation of antibody-dependent cellular cytotoxicity with defucosylated monoclonal antibodies in patients with GI-tract cancer. Oncol. Lett. 2018, 15, 2604–2610. [Google Scholar] [CrossRef]
- Tseng, H.C.; Cacalano, N.; Jewett, A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget 2015, 6, 8947–8959. [Google Scholar] [CrossRef]
- Jewett, A.; Teruel, A.; Romero, M.; Head, C.; Cacalano, N. Rapid and potent induction of cell death and loss of NK cell cytotoxicity against oral tumors by F(ab’)2 fragment of anti-CD16 antibody. Cancer Immunol. Immunother. 2008, 57, 1053–1066. [Google Scholar] [CrossRef]
- Bonavida, B.; Lebow, L.T.; Jewett, A. Natural killer cell subsets: Maturation, differentiation and regulation. Nat. Immun. 1993, 12, 194–208. [Google Scholar]
- Kaur, K.; Nanut, M.P.; Ko, M.W.; Safaie, T.; Kos, J.; Jewett, A. Natural killer cells target and differentiate cancer stem-like cells/undifferentiated tumors: Strategies to optimize their growth and expansion for effective cancer immunotherapy. Curr. Opin. Immunol. 2018, 51, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Vela-Ojeda, J.; Esparza, M.A.G.; Majluf-Cruz, A.; Garcia-Chavez, J.; Montiel-Cervantes, L.A.; Reyes-Maldonado, E.; Hernandez-Caballero, A.; Rodriguez-Gonzalez, M.G. Post-treatment improvement of NK cell numbers predicts better survival in myeloma patients treated with thalidomide-based regimens. Int. J. Hematol. 2019, 110, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-P.; Xie, M.-Z.; Li, K.-Z.; Li, J.-L.; Cai, Z.-M.; Hu, B.-L. Prognostic value of peripheral blood natural killer cells in colorectal cancer. BMC Gastroenterol. 2020, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.M.; Roberti, M.P.; Mordoh, J. Natural killer cells in human cancer: From biological functions to clinical applications. J. Biomed. Biotechnol. 2011, 2011, 676198. [Google Scholar] [CrossRef]
- Kuss, I.; Hathaway, B.; Ferris, R.L.; Gooding, W.; Whiteside, T.L. Decreased absolute counts of T lymphocyte subsets and their relation to disease in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2004, 10, 3755–3762. [Google Scholar] [CrossRef]
- Kim, J.W.; Tsukishiro, T.; Johnson, J.T.; Whiteside, T.L. Expression of pro- and antiapoptotic proteins in circulating CD8+ T cells of patients with squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2004, 10, 5101–5110. [Google Scholar] [CrossRef]
- Burke, S.; Lakshmikanth, T.; Colucci, F.; Carbone, E. New views on natural killer cell-based immunotherapy for melanoma treatment. Trends Immunol. 2010, 31, 339–345. [Google Scholar] [CrossRef]
- Larsen, S.K.; Gao, Y.; Basse, P.H. NK cells in the tumor microenvironment. Crit. Rev. Oncog. 2014, 19, 91–105. [Google Scholar] [CrossRef]
- Nolibe, D.; Poupon, M.F. Enhancement of pulmonary metastases induced by decreased lung natural killer cell activity. J. Natl. Cancer Inst. 1986, 77, 99–103. [Google Scholar]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Harning, R.; Koo, G.C.; Szalay, J. Regulation of the metastasis of murine ocular melanoma by natural killer cells. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1909–1915. [Google Scholar]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D.M. A think tank of TINK/TANKs: Tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J. Natl. Cancer Inst. 2014, 106, dju200. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Sunwoo, J.B.; Bui, J.D. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. 2013, 19, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Mirjacic Martinovic, K.M.; Babovic, N.; Dzodic, R.R.; Jurisic, V.B.; Tanic, N.T.; Konjevic, G.M. Decreased expression of NKG2D, NKp46, DNAM-1 receptors, and intracellular perforin and STAT-1 effector molecules in NK cells and their dim and bright subsets in metastatic melanoma patients. Melanoma Res. 2014, 24, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, J.A.; Felder, M.; Horibata, S.; Belisle, J.A.; Kapur, A.; Holden, H.; Petrie, S.; Migneault, M.; Rancourt, C.; Connor, J.P.; et al. MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol. Cancer 2010, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef]
- Krockenberger, M.; Dombrowski, Y.; Weidler, C.; Ossadnik, M.; Honig, A.; Hausler, S.; Voigt, H.; Becker, J.C.; Leng, L.; Steinle, A.; et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J. Immunol. 2008, 180, 7338–7348. [Google Scholar] [CrossRef]
- Kaur, K.; Cook, J.; Park, S.H.; Topchyan, P.; Kozlowska, A.; Ohanian, N.; Fang, C.; Nishimura, I.; Jewett, A. Novel Strategy to Expand Super-Charged NK Cells with Significant Potential to Lyse and Differentiate Cancer Stem Cells: Differences in NK Expansion and Function between Healthy and Cancer Patients. Front. Immunol. 2017, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Kozlowska, A.K.; Topchyan, P.; Ko, M.W.; Ohanian, N.; Chiang, J.; Cook, J.; Maung, P.O.; Park, S.H.; Cacalano, N.; et al. Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice. Cancers 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Topchyan, P.; Kozlowska, A.K.; Ohanian, N.; Chiang, J.; Maung, P.O.; Park, S.H.; Ko, M.W.; Fang, C.; Nishimura, I.; et al. Super-charged NK cells inhibit growth and progression of stem-like/poorly differentiated oral tumors in vivo in humanized BLT mice; effect on tumor differentiation and response to chemotherapeutic drugs. Oncoimmunology 2018, 7, e1426518. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Bui, V.; Man, Y.G.; Cacalano, N.; Jewett, A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Front. Immunol. 2014, 5, 269. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Man, Y.-G.; Tseng, H.-C. Dual Functions of Natural Killer Cells in Selection and Differentiation of Stem Cells; Role in Regulation of Inflammation and Regeneration of Tissues. J. Cancer 2013, 4, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-C.; Arasteh, A.; Paranjpe, A.; Teruel, A.; Yang, W.; Behel, A.; Alva, J.A.; Walter, G.; Head, C.; Ishikawa, T.-O.; et al. Increased Lysis of Stem Cells but Not Their Differentiated Cells by Natural Killer Cells; De-Differentiation or Reprogramming Activates NK Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef]
- Jing, Y.; Ni, Z.; Wu, J.; Higgins, L.; Markowski, T.W.; Kaufman, D.S.; Walcheck, B. Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS ONE 2015, 10, e0121788. [Google Scholar] [CrossRef]
- An, B.C.; Hong, S.; Park, H.J.; Kim, B.K.; Ahn, J.Y.; Ryu, Y.; An, J.H.; Chung, M.J. Anti-Colorectal Cancer Effects of Probiotic-Derived p8 Protein. Genes 2019, 10, 624. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Topchyan, P.; Kaur, K.; Tseng, H.C.; Teruel, A.; Hiraga, T.; Jewett, A. Differentiation by NK cells is a prerequisite for effective targeting of cancer stem cells/poorly differentiated tumors by chemopreventive and chemotherapeutic drugs. J. Cancer 2017, 8, 537–554. [Google Scholar] [CrossRef]
- Magister, Š.; Tseng, H.C.; Bui, V.T.; Kos, J.; Jewett, A. Regulation of split anergy in natural killer cells by inhibition of cathepsins C and H and cystatin F. Oncotarget 2015, 6, 22310–22327. [Google Scholar] [CrossRef] [PubMed]
- Pazina, T.; James, A.M.; MacFarlane, A.W.t.; Bezman, N.A.; Henning, K.A.; Bee, C.; Graziano, R.F.; Robbins, M.D.; Cohen, A.D.; Campbell, K.S. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology 2017, 6, e1339853. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, J.; Azzoni, L.; Rao, A.; Perussia, B. Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: Regulation upon CD16 ligand binding. J. Exp. Med. 1995, 182, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Tamm, A.; Schmidt, R.E. The binding epitopes of human CD16 (Fc gamma RIII) monoclonal antibodies. Implications for ligand binding. J. Immunol. 1996, 157, 1576–1581. [Google Scholar] [PubMed]
- Tseng, H.C.; Inagaki, A.; Bui, V.T.; Cacalano, N.; Kasahara, N.; Man, Y.G.; Jewett, A. Differential Targeting of Stem Cells and Differentiated Glioblastomas by NK Cells. J. Cancer 2015, 6, 866–876. [Google Scholar] [CrossRef]
- Jewett, A.; Bonavida, B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J. Immunol. 1996, 156, 907–915. [Google Scholar] [PubMed]
- Jewett, A.; Wang, M.Y.; Teruel, A.; Poupak, Z.; Bostanian, Z.; Park, N.H. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: Effect on the function of natural killer cells. Hum. Immunol. 2003, 64, 505–520. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, K.; Safaie, T.; Ko, M.-W.; Wang, Y.; Jewett, A. ADCC against MICA/B Is Mediated against Differentiated Oral and Pancreatic and Not Stem-Like/Poorly Differentiated Tumors by the NK Cells; Loss in Cancer Patients due to Down-Modulation of CD16 Receptor. Cancers 2021, 13, 239. https://doi.org/10.3390/cancers13020239
Kaur K, Safaie T, Ko M-W, Wang Y, Jewett A. ADCC against MICA/B Is Mediated against Differentiated Oral and Pancreatic and Not Stem-Like/Poorly Differentiated Tumors by the NK Cells; Loss in Cancer Patients due to Down-Modulation of CD16 Receptor. Cancers. 2021; 13(2):239. https://doi.org/10.3390/cancers13020239
Chicago/Turabian StyleKaur, Kawaljit, Tahmineh Safaie, Meng-Wei Ko, Yuhao Wang, and Anahid Jewett. 2021. "ADCC against MICA/B Is Mediated against Differentiated Oral and Pancreatic and Not Stem-Like/Poorly Differentiated Tumors by the NK Cells; Loss in Cancer Patients due to Down-Modulation of CD16 Receptor" Cancers 13, no. 2: 239. https://doi.org/10.3390/cancers13020239
APA StyleKaur, K., Safaie, T., Ko, M.-W., Wang, Y., & Jewett, A. (2021). ADCC against MICA/B Is Mediated against Differentiated Oral and Pancreatic and Not Stem-Like/Poorly Differentiated Tumors by the NK Cells; Loss in Cancer Patients due to Down-Modulation of CD16 Receptor. Cancers, 13(2), 239. https://doi.org/10.3390/cancers13020239