JAK3 Is Expressed in the Nucleus of Malignant T Cells in Cutaneous T Cell Lymphoma (CTCL)

 , , , , , ,
, , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
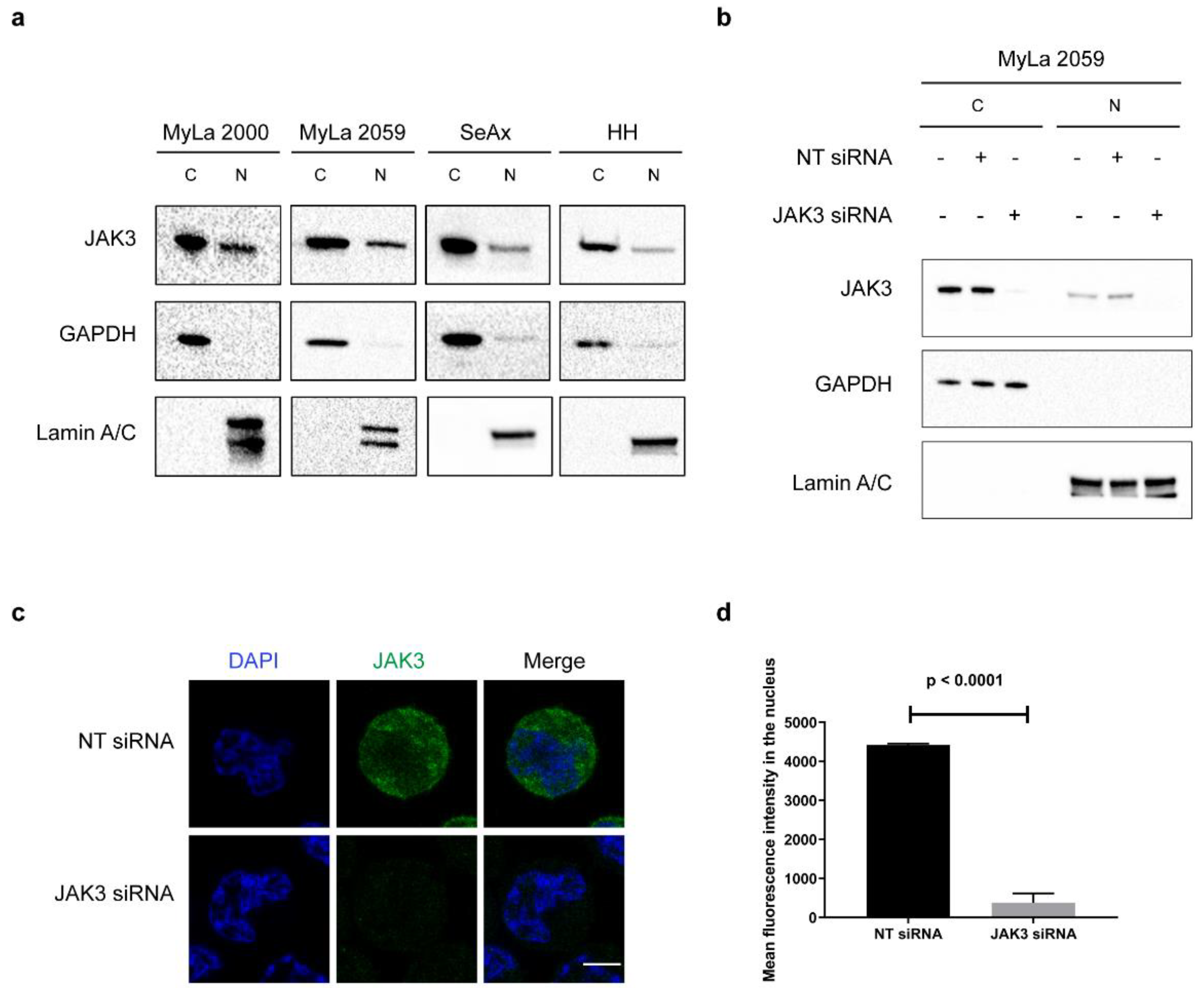
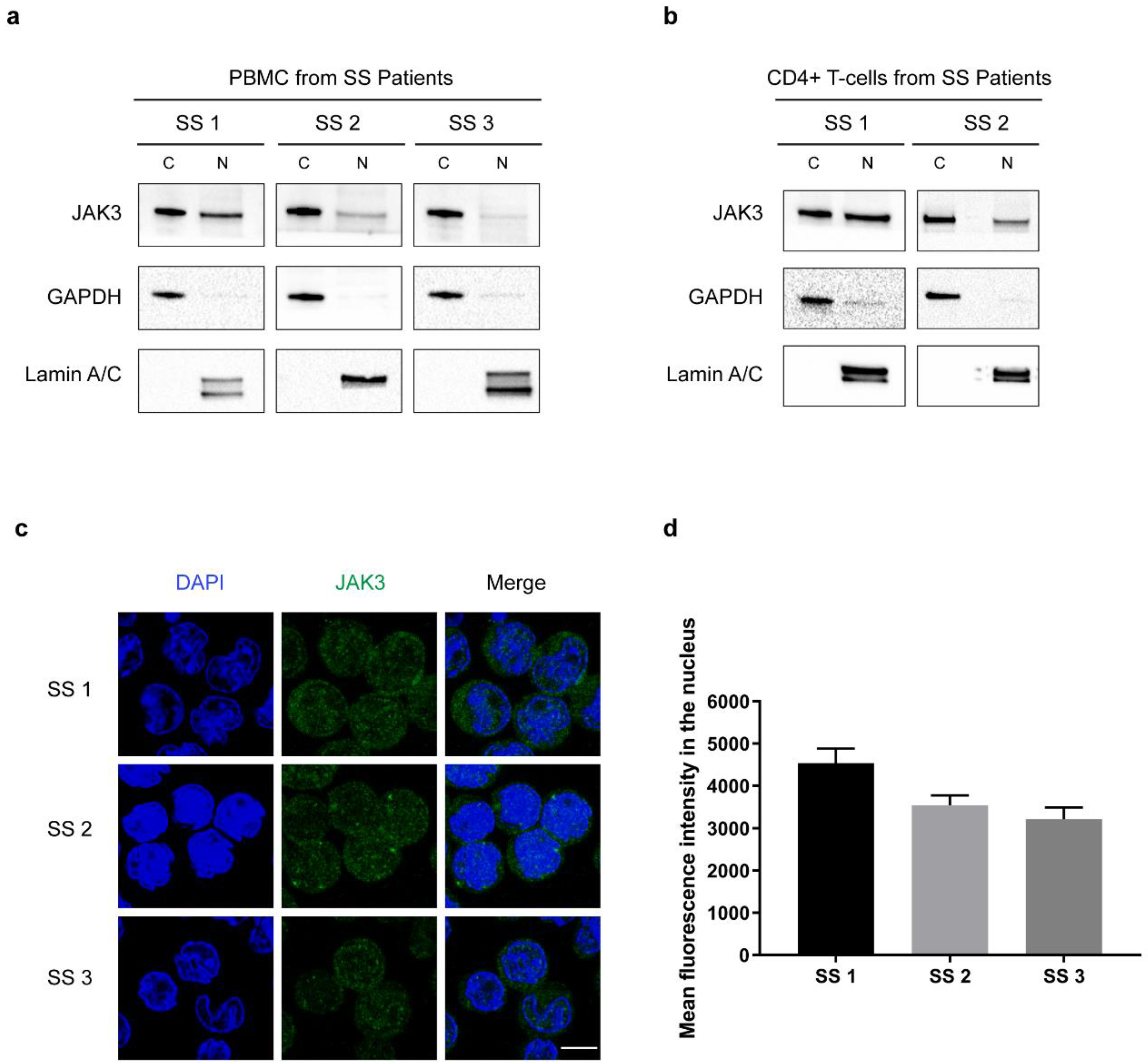
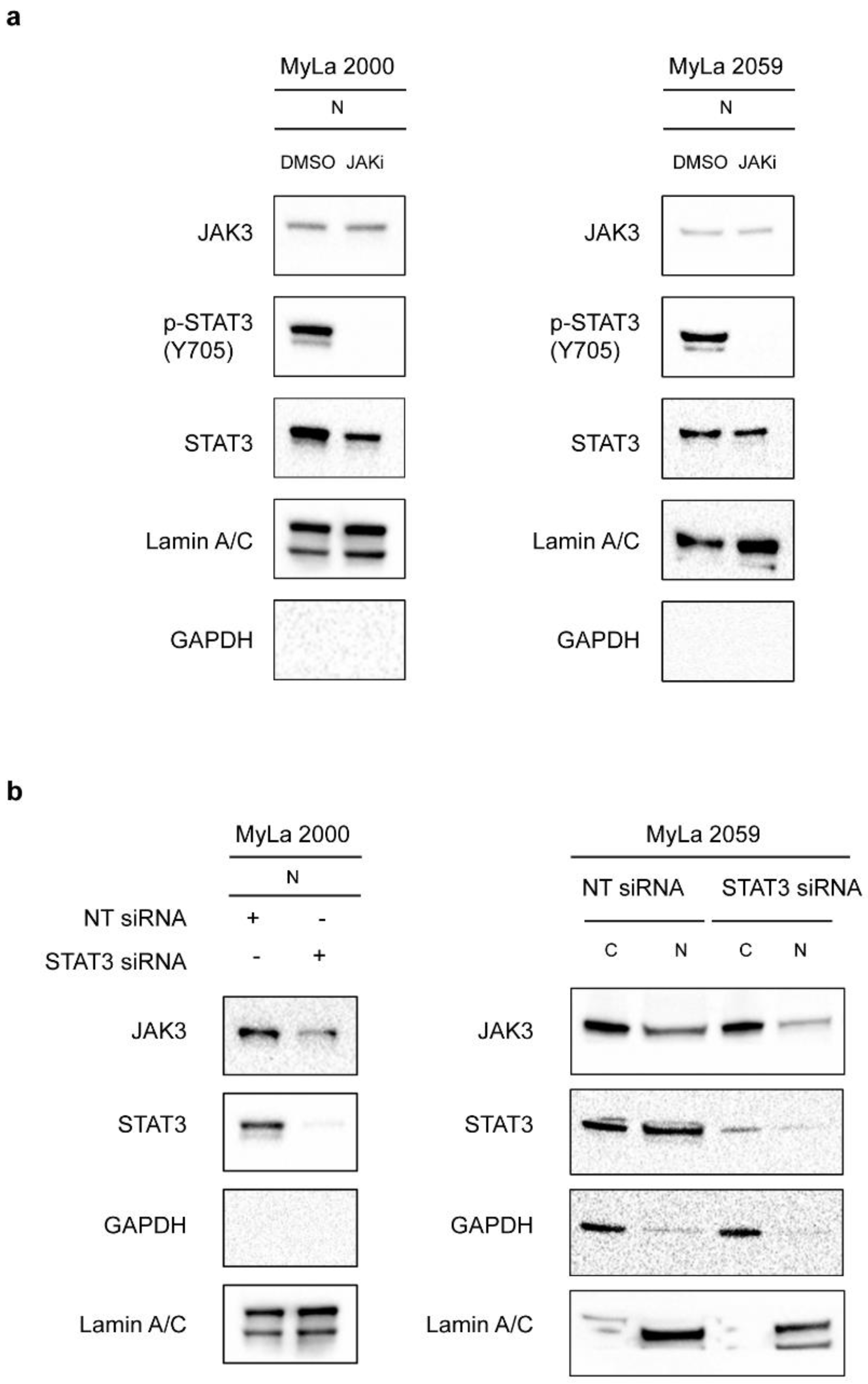
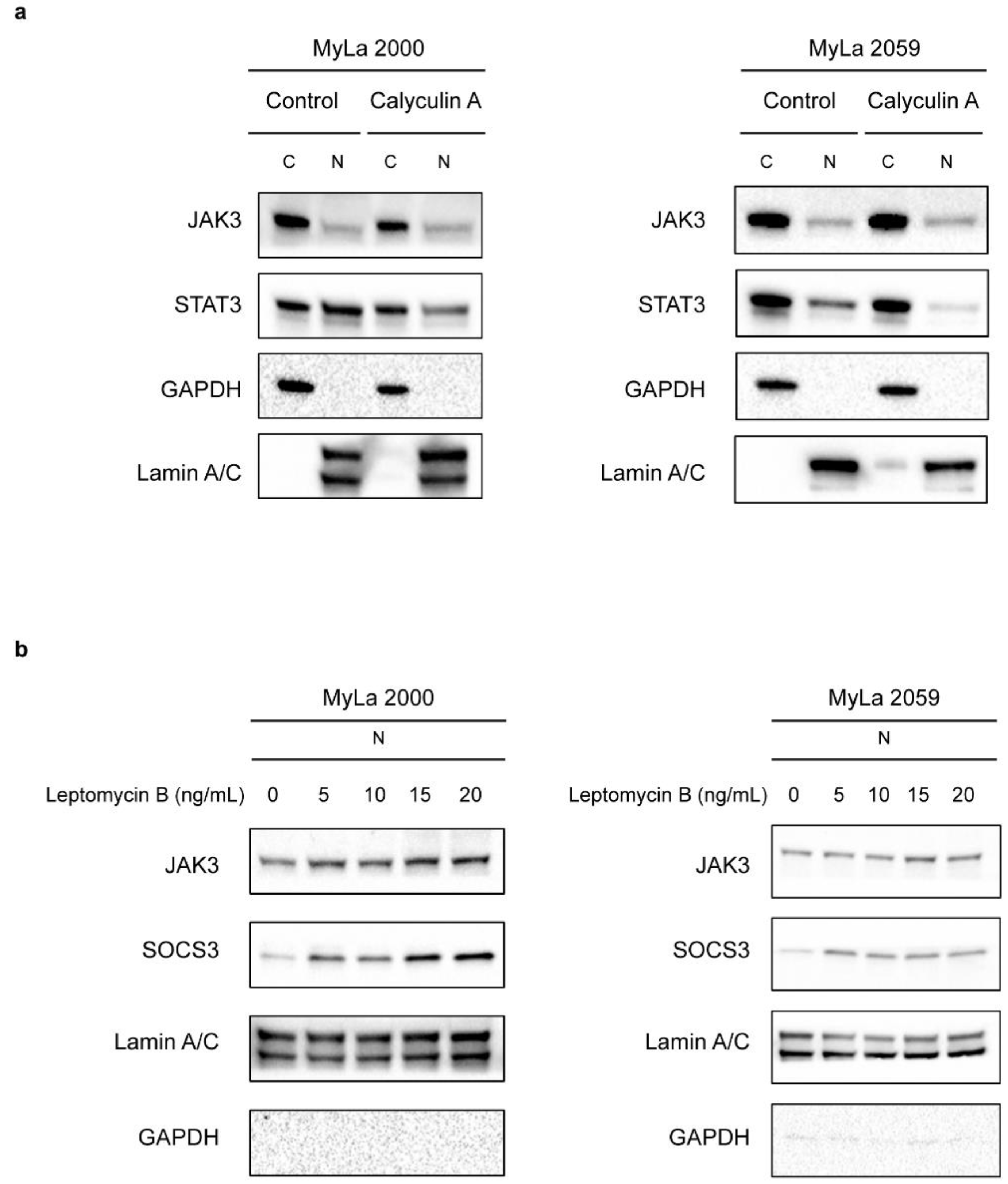
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Primary Cells and Cell Culture
4.2. Cell Fractionation and Western Blotting
4.3. Transient Transfection
4.4. Immunofluorescence
4.5. Tofacitinib Citrate Treatment
4.6. Calyculin A Treatment
4.7. Leptomycin B Treatment
4.8. Co-Immunoprecipitation
4.9. Radioactive In Vitro Kinase Assay
4.10. Antibodies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Hristov, A.C.; Tejasvi, T.; Wilcox, R.A. Mycosis fungoides and Sezary syndrome: 2019 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2019, 94, 1027–1041. [Google Scholar] [CrossRef] [PubMed]
- Girardi, M.; Heald, P.W.; Wilson, L.D. The Pathogenesis of Mycosis Fungoides. N. Engl. J. Med. 2004, 350, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Hess, S.; Richardson, S.K.; Newton, S.; Showe, L.C.; Benoit, B.M.; Ubriani, R.; Vittorio, C.C.; Junkins-Hopkins, J.M.; Wysocka, M.; et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J. Clin. Investig. 2005, 115, 798–812. [Google Scholar] [CrossRef]
- Berger, C.L.; Warburton, D.; Raafat, J.; LoGerfo, P.; Edelson, R.L. Cutaneous T-cell lymphoma: Neoplasm of T cells with helper activity. Blood 1979, 53, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous T-cell lymphoma—a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Kim, Y.H.; Whittaker, S.J.; Wood, G.S.; Vermeer, M.H.; Prince, H.M.; Quaglino, P. Prognostic factors, prognostic indices and staging in mycosis fungoides and Sézary syndrome: Where are we now? Br. J. Dermatol. 2014, 170, 1226–1236. [Google Scholar] [CrossRef]
- Buus, T.B.; Willerslev-Olsen, A.; Fredholm, S.; Blümel, E.; Nastasi, C.; Gluud, M.; Hu, T.; Lindahl, L.M.; Iversen, L.; Fogh, H.; et al. Single-cell heterogeneity in Sézary syndrome. Blood Adv. 2018, 2, 2115–2126. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Geskin, L.J.; Bayan, C.A.; Conway, J.F.; Lafyatis, R.; Fuschiotti, P. Single-Cell Lymphocyte Heterogeneity in Advanced Cutaneous T-cell Lymphoma Skin Tumors. Clin. Cancer Res. 2019, 25, 4443–4454. [Google Scholar] [CrossRef]
- Hu, T.; Krejsgaard, T.; Nastasi, C.; Buus, T.B.; Nansen, A.; Hald, A.; Spee, P.; Nielsen, P.R.; Blümel, E.; Gluud, M.; et al. Expression of the Voltage-Gated Potassium Channel Kv1.3 in Lesional Skin from Patients with Cutaneous T-Cell Lymphoma and Benign Dermatitis. Dermatology 2020, 236, 123–132. [Google Scholar] [CrossRef]
- Iyer, A.; Hennessey, D.; O’Keefe, S.; Patterson, J.; Wang, W.; Wong, G.K.; Gniadecki, R. Branched evolution and genomic intratumor heterogeneity in the pathogenesis of cutaneous T-cell lymphoma. Blood Adv. 2020, 4, 2489–2500. [Google Scholar] [CrossRef]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic landscape of cutaneous T cell lymphoma. Nat. Genet. 2015, 47, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Ødum, N. Deregulated signalling and inflammation in cutaneous T-cell lymphoma. Br. J. Dermatol. 2020, 182, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Netchiporouk, E.; Litvinov, I.V.; Moreau, L.; Gilbert, M.; Sasseville, D.; Duvic, M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle 2014, 13, 3331–3335. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Queen, D.S.; Trager, M.H.; Akilov, O.E.; Geskin, L.; Fuschiotti, P. Genome-wide transcriptome analysis of the STAT6-regulated genes in advanced-stage cutaneous T-cell lymphoma. Blood 2020, 136, 1748–1759. [Google Scholar] [CrossRef]
- Geskin, L.J.; Viragova, S.; Stolz, D.B.; Fuschiotti, P. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 2015, 125, 2798–2805. [Google Scholar] [CrossRef]
- Seffens, A.; Herrera, A.; Tegla, C.; Buus, T.B.; Hymes, K.B.; Ødum, N.; Geskin, L.J.; Koralov, S.B. STAT3 Dysregulation in Mature T and NK Cell Lymphomas. Cancers 2019, 11, 1711. [Google Scholar] [CrossRef]
- Gluud, M.; Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Lindahl, L.M.; Buus, T.B.; Andersen, M.H.; Bonefeld, C.M.; Krejsgaard, T.; Litvinov, I.V.; Iversen, L.; et al. MicroRNAs in the Pathogenesis, Diagnosis, Prognosis and Targeted Treatment of Cutaneous T-Cell Lymphomas. Cancers 2020, 12, 1229. [Google Scholar] [CrossRef]
- Gluud, M.; Fredholm, S.; Blümel, E.; Willerslev-Olsen, A.; Buus, T.B.; Nastasi, C.; Krejsgaard, T.; Bonefeld, C.M.; Woetmann, A.; Iversen, L.; et al. MicroRNA-93 Targets p21 and Promotes Proliferation in Mycosis Fungoides T Cells. Dermatology 2020. [Google Scholar] [CrossRef]
- van der Fits, L.; van Kester, M.S.; Qin, Y.; Out-Luiting, J.J.; Smit, F.; Zoutman, W.H.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. MicroRNA-21 expression in CD4+ T cells is regulated by STAT3 and is pathologically involved in Sézary syndrome. J. Investig. Dermatol. 2011, 131, 762–768. [Google Scholar] [CrossRef]
- Fredholm, S.; Willerslev-Olsen, A.; Met, Ö.; Kubat, L.; Gluud, M.; Mathiasen, S.L.; Friese, C.; Blümel, E.; Petersen, D.L.; Hu, T.; et al. SATB1 in Malignant T Cells. J. Investig. Dermatol. 2018, 138, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.; Fredholm, S.; Cheng, A.; Mimitou, E.P.; Seffens, A.; Bar-Natan, M.; Sun, A.; Latkowski, J.A.; Willerslew-Olsen, A.; Buus, T.B.; et al. Low SATB1 Expression Promotes IL-5 and IL-9 Expression in Sézary Syndrome. J. Investig. Dermatol. 2020, 140, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.M.; Velusamy, T.; Chung, F.; Schaller, M.; Bailey, N.G.; Betz, B.L.; Miranda, R.N.; Porcu, P.; et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sézary syndrome. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bastidas Torres, A.N.; Cats, D.; Mei, H.; Szuhai, K.; Willemze, R.; Vermeer, M.H.; Tensen, C.P. Genomic analysis reveals recurrent deletion of JAK-STAT signaling inhibitors HNRNPK and SOCS1 in mycosis fungoides. Genes Chromosomes Cancer 2018, 57, 653–664. [Google Scholar] [CrossRef] [PubMed]
- McGirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Willerslev-Olsen, A.; Krejsgaard, T.; Lindahl, L.M.; Litvinov, I.V.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Gniadecki, R.; Mongan, N.P.; Sasseville, D.; et al. Staphylococcal enterotoxin A (SEA) stimulates STAT3 activation and IL-17 expression in cutaneous T-cell lymphoma. Blood 2016, 127, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Willerslev-Olsen, A.; Buus, T.B.; Nastasi, C.; Blümel, E.; Gluud, M.; Bonefeld, C.M.; Geisler, C.; Lindahl, L.M.; Vermeer, M.; Wasik, M.A.; et al. Staphylococcus aureus enterotoxins induce FOXP3 in neoplastic T cells in Sézary syndrome. Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Lindahl, L.M.; Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Nielsen, P.R.; Blümel, E.; Rittig, A.H.; Celis, P.; Herpers, B.; Becker, J.C.; Stausbøl-Grøn, B.; et al. Antibiotics inhibit tumor and disease activity in cutaneous T-cell lymphoma. Blood 2019, 134, 1072–1083. [Google Scholar] [CrossRef]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 1–6. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Rui, L.; Drennan, A.C.; Ceribelli, M.; Zhu, F.; Wright, G.W.; Huang, D.W.; Xiao, W.; Li, Y.; Grindle, K.M.; Lu, L.; et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7260–E7267. [Google Scholar] [CrossRef]
- Landires, I.; Núñez-Samudio, V.; Thèze, J. Short communication: Nuclear JAK3 and its involvement in CD4 activation in HIV-infected patients. AIDS Res. Hum. Retrovir. 2013, 29, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.E.; Blumenkopf, T.A.; Brissette, W.H.; Brown, M.F.; Casavant, J.M.; Shang-Poa, C.; Doty, J.L.; Elliott, E.A.; Fisher, M.B.; Hines, M.; et al. Discovery of CP-690,550: A potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J. Med. Chem. 2010, 53, 8468–8484. [Google Scholar] [CrossRef] [PubMed]
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Lovato, P.; Labuda, T.; Eriksen, K.W.; Zhang, Q.; Becker, J.C.; Ødum, N. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia 2006, 20, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Woetmann, A.; Nielsen, M.; Christensen, S.T.; Brockdorff, J.; Kaltoft, K.; Engel, A.-M.; Skov, S.; Brender, C.; Geisler, C.; Svejgaard, A.; et al. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc. Natl. Acad. Sci. USA 1999, 96, 10620–10625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Hwang, B.; Miyamoto, S.; Rui, L. Nuclear import of JAK1 is mediated by a classical NLS and is required for survival of diffuse large B-cell lymphoma. Mol. Cancer Res. 2017, 15, 348–357. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 2009, 106, 10171–10176. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Henderson, B.R.; Eleftheriou, A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp. Cell Res. 2000, 256, 213–224. [Google Scholar] [CrossRef]
- Xu, D.; Farmer, A.; Collett, G.; Grishin, N.V.; Chook, Y.M. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol. Biol. Cell 2012, 23, 3677–3693. [Google Scholar] [CrossRef]
- la Cour, T.; Kiemer, L.; Mølgaard, A.; Gupta, R.; Skriver, K.; Brunak, S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004, 17, 527–536. [Google Scholar] [CrossRef]
- Rottenberg, M.; Carow, B. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef]
- Hutten, S.; Kehlenbach, R.H. CRM1-mediated nuclear export: To the pore and beyond. Trends Cell Biol. 2007, 17, 193–201. [Google Scholar] [CrossRef]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef]
- Senkevitch, E.; Durum, S. The promise of Janus kinase inhibitors in the treatment of hematological malignancies. Cytokine 2017, 98, 33–41. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Harrison, C.N. JAK-STAT signaling in the therapeutic landscape of myeloproliferative neoplasms. Mol. Cell Endocrinol. 2017, 451, 71–79. [Google Scholar] [CrossRef]
- Lokau, J.; Garbers, C. Activating mutations of the gp130/JAK/STAT pathway in human diseases. Adv. Protein Chem. Struct. Biol. 2019, 116, 283–309. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Trivedi, S.; Starz-Gaiano, M. Drosophila Jak/STAT Signaling: Regulation and Relevance in Human Cancer and Metastasis. Int. J. Mol. Sci. 2018, 19, 4056. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Cox, L.; Demeyer, S.; Gielen, O.; Mentens, N.; Jacobs, K.; Geerdens, E.; Gianfelici, V.; Hulselmans, G.; et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014, 124, 3092–3100. [Google Scholar] [CrossRef]
- Gruber, C.N.; Calis, J.J.A.; Buta, S.; Evrony, G.; Martin, J.C.; Uhl, S.A.; Caron, R.; Jarchin, L.; Dunkin, D.; Phelps, R.; et al. Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Kinase Transcriptional and Biochemical Function. Immunity 2020, 53, 672–684. [Google Scholar] [CrossRef]
- Campos, L.W.; Pissinato, L.G.; Yunes, J.A. Deleterious and Oncogenic Mutations in the IL7RA. Cancers 2019, 11, 1952. [Google Scholar] [CrossRef]
- Pérez, C.; González-Rincón, J.; Onaindia, A.; Almaráz, C.; García-Díaz, N.; Pisonero, H.; Curiel-Olmo, S.; Gómez, S.; Cereceda, L.; Madureira, R.; et al. Mutated JAK kinases and deregulated STAT activity are potential therapeutic targets in cutaneous T-cell lymphoma. Haematologica 2015, 100, e450. [Google Scholar] [CrossRef]
- Arora, L.; Kumar, A.P.; Arfuso, F.; Chng, W.J.; Sethi, G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers 2018, 10, 327. [Google Scholar] [CrossRef]
- Ahmed, C.M.; Noon-Song, E.N.; Kemppainen, K.; Pascalli, M.P.; Johnson, H.M. Type I IFN receptor controls activated TYK2 in the nucleus: Implications for EAE therapy. J. Neuroimmunol. 2013, 254, 101–109. [Google Scholar] [CrossRef]
- Mishra, J.; Waters, C.M.; Kumar, N. Molecular mechanism of interleukin-2-induced mucosal homeostasis. Am. J. Physiol. Cell Physiol. 2012, 302, C735–C747. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Kim, S.J.; Yoon, S. Activated Rac1 regulates the degradation of IκBα and the nuclear translocation of STAT3-NFκB complexes in starved cancer cells. Exp. Mol. Med. 2016, 48, e231. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Schindler, C. Regulation of Stat3 nuclear export. J. Clin. Investig. 2003, 111, 553–559. [Google Scholar] [CrossRef]
- Strebovsky, J.; Walker, P.; Lang, R.; Dalpke, A.H. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J. 2011, 25, 863–874. [Google Scholar] [CrossRef]
- Brender, C.; Lovato, P.; Sommer, V.H.; Woetmann, A.; Mathiesen, A.M.; Geisler, C.; Wasik, M.; Ødum, N. Constitutive SOCS-3 expression protects T-cell lymphoma against growth inhibition by IFNα. Leukemia 2005, 19, 209–213. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Paludo, J.; Nowakowski, K.E.; Stenson, M.J.; King, R.L.; Wellik, L.E.; Wu, X.; Witzig, T.E. The effect of CRM1 inhibition on human non-Hodgkin lymphoma cells. Blood Cancer J. 2019, 9, 1–4. [Google Scholar] [CrossRef]
- Kuruvilla, J.; Savona, M.; Baz, R.; Mau-Sorensen, P.M.; Gabrail, N.; Garzon, R.; Stone, R.; Wang, M.; Savoie, L.; Martin, P.; et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017, 129, 3175–3183. [Google Scholar] [CrossRef]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 1–9. [Google Scholar] [CrossRef]
- Borden, K.L.B. The Nuclear Pore Complex and mRNA Export in Cancer. Cancers 2020, 13, 42. [Google Scholar] [CrossRef]
- Verbeke, D.; Demeyer, S.; Prieto, C.; de Bock, C.E.; De Bie, J.; Gielen, O.; Jacobs, K.; Mentens, N.; Verhoeven, B.M.; Uyttebroeck, A.; et al. The XPO1 Inhibitor KPT-8602 Synergizes with Dexamethasone in Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 5747–5758. [Google Scholar] [CrossRef]
- Vercruysse, T.; De Bie, J.; Neggers, J.E.; Jacquemyn, M.; Vanstreels, E.; Schmid-Burgk, J.L.; Hornung, V.; Baloglu, E.; Landesman, Y.; Senapedis, W.; et al. The Second-Generation Exportin-1 Inhibitor KPT-8602 Demonstrates Potent Activity against Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 2528–2541. [Google Scholar] [CrossRef]
- Woetmann, A.; Lovato, P.; Eriksen, K.W.; Krejsgaard, T.; Labuda, T.; Zhang, Q.; Mathiesen, A.M.; Geisler, C.; Svejgaard, A.; Wasik, M.A.; et al. Nonmalignant T cells stimulate growth of T-cell lymphoma cells in the presence of bacterial toxins. Blood 2007, 109, 3325–3332. [Google Scholar] [CrossRef]
- Kaltoft, K.; Bisballe, S.; Dyrberg, T.; Boel, E.; Rasmussen, P.B.; Thestrup-Pedersen, K. Establishment of two continuous T-cell strains from a single plaque of a patient with mycosis fungoides. In Vitro Cell. Dev. Biol. J. Tissue Cult. Assoc. 1992, 28, 161–167. [Google Scholar] [CrossRef]
- Kaltoft, K.; Bisballe, S.; Rasmussen, H.F.; Thestrup-Pedersen, K.; Thomsen, K.; Sterry, W. A continuous T-cell line from a patient with Sézary syndrome. Arch. Dermatol. Res. 1987, 279, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Starkebaum, G.; Loughran, T.P.; Waters, C.A.; Ruscetti, F.W. Establishment of an IL-2 independent, human T-cell line possessing only the p70 IL-2 receptor. Int. J. Cancer 1991, 49, 246–253. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadivel, C.K.; Gluud, M.; Torres-Rusillo, S.; Boding, L.; Willerslev-Olsen, A.; Buus, T.B.; Nielsen, T.K.; Persson, J.L.; Bonefeld, C.M.; Geisler, C.; et al. JAK3 Is Expressed in the Nucleus of Malignant T Cells in Cutaneous T Cell Lymphoma (CTCL). Cancers 2021, 13, 280. https://doi.org/10.3390/cancers13020280
Vadivel CK, Gluud M, Torres-Rusillo S, Boding L, Willerslev-Olsen A, Buus TB, Nielsen TK, Persson JL, Bonefeld CM, Geisler C, et al. JAK3 Is Expressed in the Nucleus of Malignant T Cells in Cutaneous T Cell Lymphoma (CTCL). Cancers. 2021; 13(2):280. https://doi.org/10.3390/cancers13020280
Chicago/Turabian StyleVadivel, Chella Krishna, Maria Gluud, Sara Torres-Rusillo, Lasse Boding, Andreas Willerslev-Olsen, Terkild B. Buus, Tea Kirkegaard Nielsen, Jenny L. Persson, Charlotte M. Bonefeld, Carsten Geisler, and et al. 2021. "JAK3 Is Expressed in the Nucleus of Malignant T Cells in Cutaneous T Cell Lymphoma (CTCL)" Cancers 13, no. 2: 280. https://doi.org/10.3390/cancers13020280