IFN-? and CD38 in Hyperprogressive Cancer Development

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
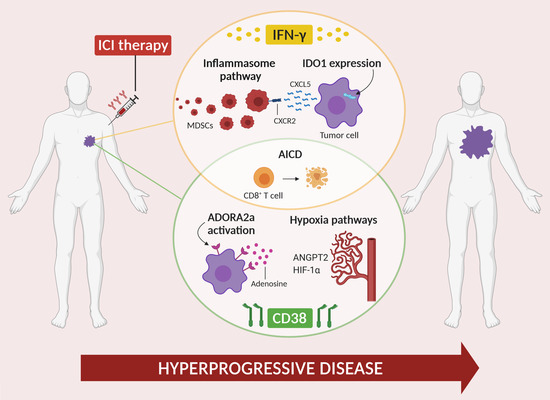
1. Introduction
1.1. Clinical Evidence of Hyperprogression and Associated Factors
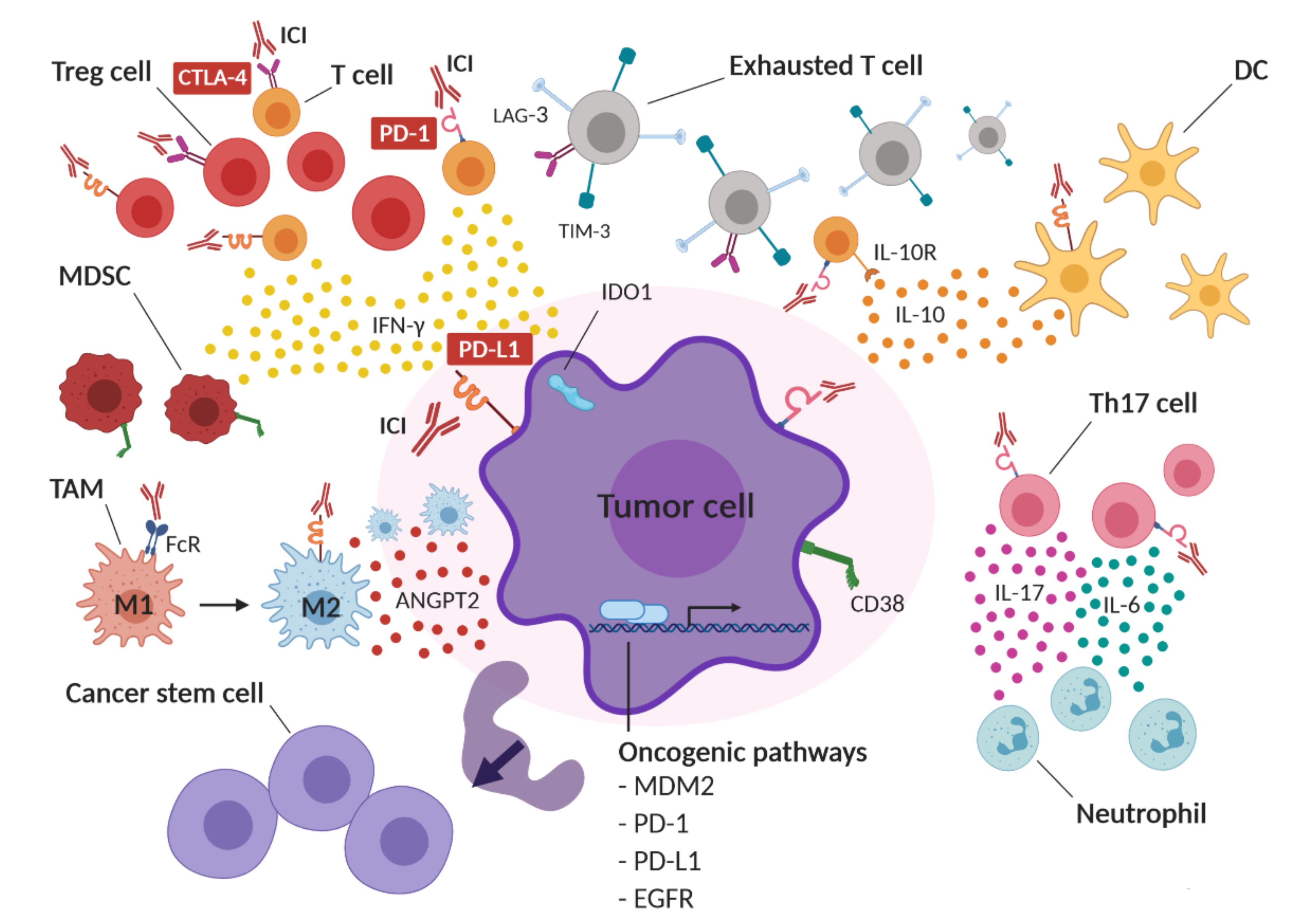
1.2. Hypothesized Mechanisms of HPD
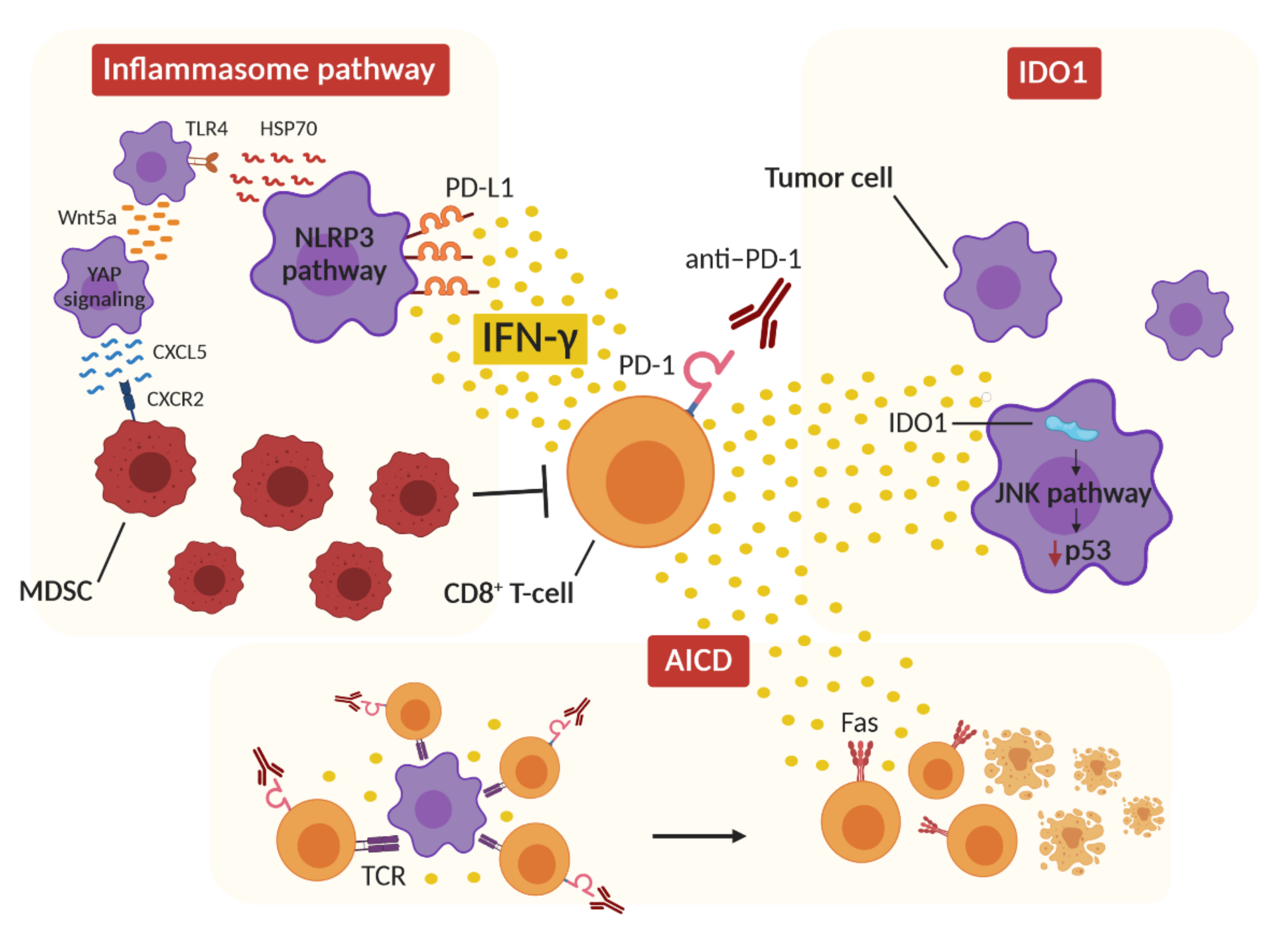
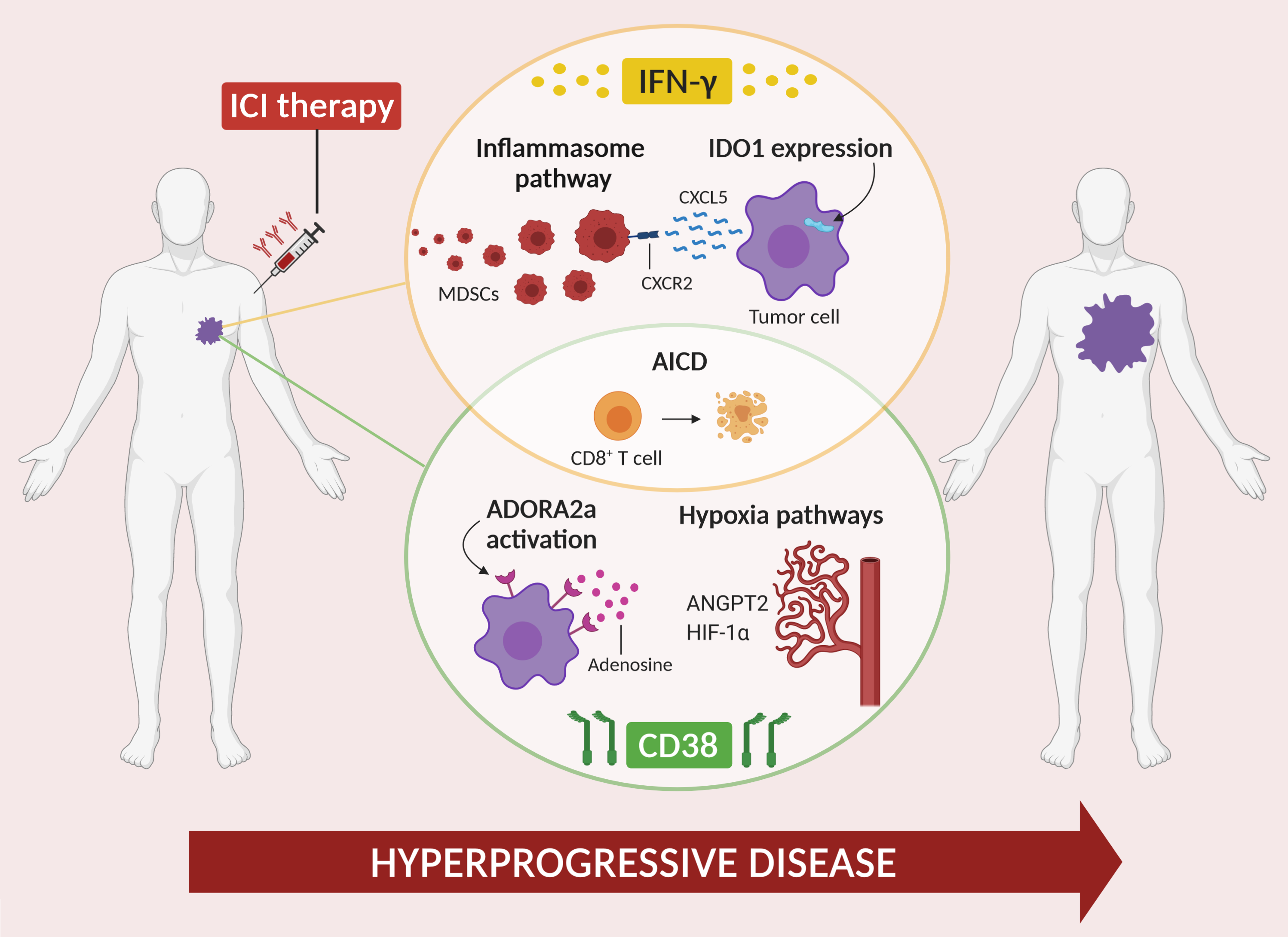
2. Role of IFN-γ in HPD
2.1. IFN-γ and Inflammasome
2.2. IFN-γ and IDO1
2.3. IFN-γ and Activation-Induced Cell Death
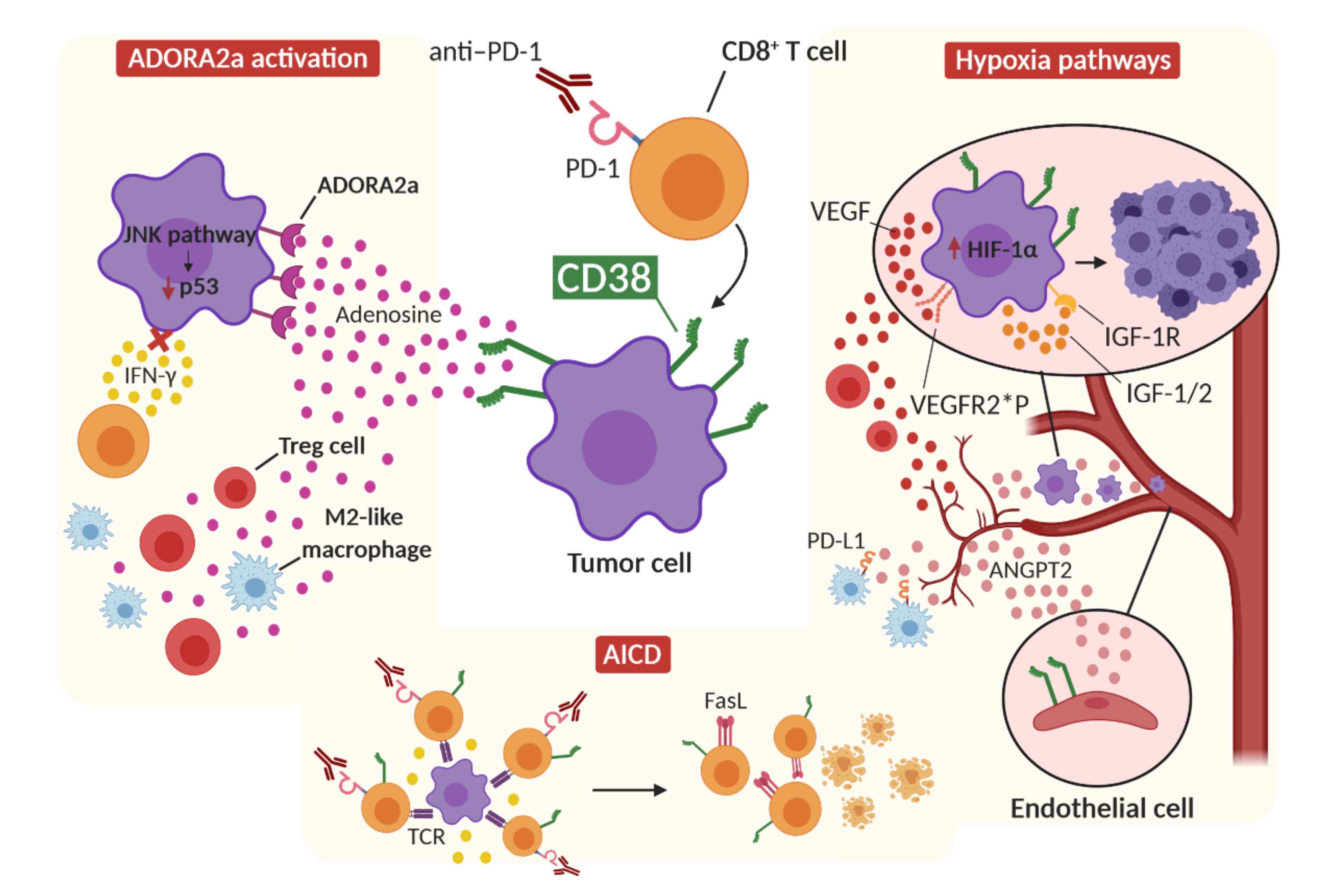
3. Role of CD38 in HPD
3.1. CD38 and Adenosine Receptor Activation
3.2. CD38 and Activation-Induced Cell Death
3.3. CD38 and Hypoxia
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Ahamadi, M.; Freshwater, T.; Prohn, M.; Li, C.; de Alwis, D.; de Greef, R.; Elassaiss-Schaap, J.; Kondic, A.; Stone, J. Model-Based Characterization of the Pharmacokinetics of Pembrolizumab: A Humanized Anti-PD-1 Monoclonal Antibody in Advanced Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 49–57. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef]
- Aoki, M.; Shoji, H.; Nagashima, K.; Imazeki, H.; Miyamoto, T.; Hirano, H.; Honma, Y.; Iwasa, S.; Okita, N.; Takashima, A.; et al. Hyperprogressive disease during nivolumab or irinotecan treatment in patients with advanced gastric cancer. ESMO Open 2019, 4, e000488. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, A.; Nakamura, Y.; Mishima, S.; Kawazoe, A.; Kuboki, Y.; Bando, H.; Kojima, T.; Doi, T.; Ohtsu, A.; Yoshino, T.; et al. Predictive factors for hyperprogressive disease during nivolumab as anti-PD1 treatment in patients with advanced gastric cancer. Gastric Cancer 2019, 22, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; le Moulec, S.; et al. Hyperprogressive Disease in Patients with Advanced Non-Small Cell Lung Cancer Treated with PD-1/PD-L1 Inhibitors or with Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Saâda-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, K.H.; Pyo, K.-H.; Xin, C.-F.; Hong, M.H.; Ahn, B.-C.; Kim, Y.; Choi, S.J.; Yoon, H.I.; Lee, J.G.; et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1104–1113. [Google Scholar] [CrossRef]
- Okan Cakir, M.; Kirca, O.; Gunduz, S.; Ozdogan, M. Hyperprogression after immunotherapy: A comprehensive review. JBUON 2019, 24, 2232–2241. [Google Scholar]
- Kas, B.; Talbot, H.; Ferrara, R.; Richard, C.; Lamarque, J.-P.; Pitre-Champagnat, S.; Planchard, D.; Balleyguier, C.; Besse, B.; Mezquita, L.; et al. Clarification of Definitions of Hyperprogressive Disease During Immunotherapy for Non–Small Cell Lung Cancer. JAMA Oncol. 2020, 6, 1039–1046. [Google Scholar] [CrossRef]
- Kanjanapan, Y.; Day, D.; Wang, L.; Al-Sawaihey, H.; Abbas, E.; Namini, A.; Siu, L.L.; Hansen, A.; Razak, A.A.; Spreafico, A.; et al. Hyperprogressive disease in early-phase immunotherapy trials: Clinical predictors and association with immune-related toxicities. Cancer 2019, 125, 1341–1349. [Google Scholar] [CrossRef]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef] [Green Version]
- Matos, I.; Martin-Liberal, J.; Hierro, C.; Ochoa De Olza, M.; Viaplana, C.; Costa, M.; Felip-Falg’s, E.; Mur-Bonet, G.; Vieito, M.; Brana, I.; et al. Incidence and clinical implications of a new definition of hyperprogression (HPD) with immune checkpoint inhibitors (ICIs) in patients treated in phase 1 (Ph1) trials. J. Clin. Oncol. 2018, 36, 3032. [Google Scholar] [CrossRef]
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin. Cancer Res. 2019, 25, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Hakim, F.T.; Flomerfelt, F.A.; Boyiadzis, M.; Gress, R.E. Aging, immunity and cancer. Curr. Opin. Immunol. 2004, 16, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Hurez, V.; Padrón, S.; Svatek, R.S.; Curiel, T.J. Considerations for successful cancer immunotherapy in aged hosts. Clin. Exp. Immunol. 2017, 187, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Fields, E.C.; McGuire, W.P.; Lin, L.; Temkin, S.M. Radiation treatment in women with ovarian cancer: Past, present, and future. Front. Oncol. 2017, 7, 177. [Google Scholar] [CrossRef] [Green Version]
- Singavi, A.K.; Menon, S.; Kilari, D.; Alqwasmi, A.; Ritch, P.S.; Thomas, J.P.; Martin, A.L.; Oxencis, C.; Ali, S.; George, B. Predictive biomarkers for hyper-progression (HP) in response to immune checkpoint inhibitors (ICI) – analysis of somatic alterations (SAs). Ann. Oncol. 2017, 28, v405. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: A retrospective analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Bellosillo, B.; Caux, C.; de Langen, A.; Mazieres, J.; Normanno, N.; Preusser, M.; Provencio, M.; Rojo, F.; Wolf, J.; et al. Immune checkpoint inhibitor treatment in patients with oncogene- addicted non-small cell lung cancer (NSCLC): Summary of a multidisciplinary round-table discussion. Esmo Open 2019, 4, e000498. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Cho, B.-C.; Kim, J.-H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Corral Jaime, J.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521–536. [Google Scholar] [CrossRef]
- Lamberti, G.; Andrini, E.; Sisi, M.; Rizzo, A.; Parisi, C.; di Federico, A.; Gelsomino, F.; Ardizzoni, A. Beyond EGFR, ALK and ROS1: Current evidence and future perspectives on newly targetable oncogenic drivers in lung adenocarcinoma. Crit. Rev. Oncol./Hematol. 2020, 156, 103119. [Google Scholar] [CrossRef] [PubMed]
- Tunali, I.; Gray, J.E.; Qi, J.; Abdalah, M.; Jeong, D.K.; Guvenis, A.; Gillies, R.J.; Schabath, M.B. Novel clinical and radiomic predictors of rapid disease progression phenotypes among lung cancer patients treated with immunotherapy: An early report. Lung Cancer 2019, 129, 75–79. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Bougioukas, G.; Didilis, V.; Gatter, K.C.; Harris, A.L. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br. J. Cancer 2003, 89, 877–885. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Teixidó, C.; González-Cao, M.; Karachaliou, N.; Rosell, R. Predictive factors for immunotherapy in melanoma. Ann. Transl. Med. 2015, 3, 208. [Google Scholar] [CrossRef]
- Passiglia, F.; Bronte, G.; Bazan, V.; Natoli, C.; Rizzo, S.; Galvano, A.; Listì, A.; Cicero, G.; Rolfo, C.; Santini, D.; et al. PD-L1 expression as predictive biomarker in patients with NSCLC: A pooled analysis. Oncotarget 2016, 7, 19738–19747. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- De Simone, M.; Arrigoni, A.; Rossetti, G.; Gruarin, P.; Ranzani, V.; Politano, C.; Bonnal, R.J.P.; Provasi, E.; Sarnicola, M.L.; Panzeri, I.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016, 45, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowther, D.E.; Goods, B.A.; Lucca, L.E.; Lerner, B.A.; Raddassi, K.; van Dijk, D.; Hernandez, A.L.; Duan, X.; Gunel, M.; Coric, V.; et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight 2016, 1, e85935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology 2017, 6, e1249561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [Green Version]
- Zuazo-Ibarra, M.; Arasanz, H.; Fernández-Hinojal, G.; María, G.-C.; Hernández-Marín, B.; Martínez-Aguillo, M.; Jose Lecumberri, M.; Fernández, A.; Teijeira, L.; Vera, R.; et al. Highly differentiated CD4 T cells Unequivocally Identify Primary Resistance and Risk of Hyperprogression to PD-L1/PD-1 Immune Checkpoint Blockade in Lung Cancer. bioRxiv 2018, 320176. [Google Scholar] [CrossRef]
- Lamichhane, P.; Karyampudi, L.; Shreeder, B.; Krempski, J.; Bahr, D.; Daum, J.; Kalli, K.R.; Goode, E.L.; Block, M.S.; Cannon, M.J.; et al. IL10 Release upon PD-1 Blockade Sustains Immunosuppression in Ovarian Cancer. Cancer Res. 2017, 77, 6667–6678. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Fourcade, J.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Kirkwood, J.M.; Zarour, H.M. IL10 and PD-1 Cooperate to Limit the Activity of Tumor-Specific CD8+ T Cells. Cancer Res. 2015, 75, 1635–1644. [Google Scholar] [CrossRef] [Green Version]
- Scholz, A.; Lang, V.; Henschler, R.; Czabanka, M.; Vajkoczy, P.; Chavakis, E.; Drynski, J.; Harter, P.N.; Mittelbronn, M.; Dumont, D.J.; et al. Angiopoietin-2 promotes myeloid cell infiltration in a β2-integrin–dependent manner. Blood 2011, 118, 5050–5059. [Google Scholar] [CrossRef]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol. Res. 2017, 5, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.A.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.J.; Mulders, P.; Punt, C.J.A.; et al. PD-1 Blockade Augments Th1 and Th17 and Suppresses Th2 Responses in Peripheral Blood From Patients With Prostate and Advanced Melanoma Cancer. J. Immunother. 2012, 35, 169–178. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Song, X.; Xu, L.; Ma, J.; Zhang, Y.; Gong, W.; Zhang, Y.; Zhou, X.; Wang, Z.; Wang, Y.; et al. The binding of an anti-PD-1 antibody to FcγRΙ has a profound impact on its biological functions. Cancer Immunol. Immunother. 2018, 67, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.G.; Ebert, S.; Schlahsa, L.; Scholz, C.J.; Braun, M.; Hauck, P.; Horn, E.; Monoranu, C.-M.; Thiemann, V.J.; Wustrow, M.P.; et al. Cognate Nonlytic Interactions between CD8 + T Cells and Breast Cancer Cells Induce Cancer Stem Cell–like Properties. Cancer Res. 2019, 79, 1507–1519. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.X.; Lee, C.H.; Qi, C.F.; Wang, H.; Naghashfar, Z.; Abbasi, S.; Morse, H.C. IFN Regulatory Factor 8 Regulates MDM2 in Germinal Center B Cells. J. Immunol. 2009, 183, 3188–3194. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Wang, H.; Li, C.; Fang, J.-Y.; Xu, J. Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy. Front. Immunol. 2018, 9, 1774. [Google Scholar] [CrossRef]
- Ludin, A.; Zon, L.I. The dark side of PD-1 receptor inhibition. Nature 2017, 552, 41–42. [Google Scholar] [CrossRef] [Green Version]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Ratner, L.; Waldmann, T.A.; Janakiram, M.; Brammer, J.E. Rapid Progression of Adult T-Cell Leukemia–Lymphoma after PD-1 Inhibitor Therapy. N. Engl. J. Med. 2018, 378, 1947–1948. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, S.; McCall, N.; Park, K.; Guan, Q.; Fontina, P.; Ertel, A.; Zhan, T.; Dicker, A.P.; Lu, B. Blockade of Tumor-Expressed PD-1 promotes lung cancer growth. OncoImmunology 2018, 7, e1408747. [Google Scholar] [CrossRef]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [Green Version]
- Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; Ibañez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.E.; Kitagawa, M.; Kuida, K.; Flavell, R.A.; Fu, X.Y. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell. Biol. 1997, 17, 5328–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Fu, X.-Y.; Plate, J.; Chong, A.S.-F. IFN-y Induces Cell Growth Inhibition by Fas-mediated Apoptosis: Requirement of STATI Protein for Up-Regulation of Fas and FasL Expression. Cancer Res. 1998, 58, 2832–2837. [Google Scholar]
- Hobeika, A.C.; Etienne, W.; Torres, B.A.; Johnson, H.M.; Subramaniam, P.S. IFN-gamma Induction of p21WAF1 Is Required for Cell Cycle Inhibition and Suppression of Apoptosis. J. Interferon Cytokine Res. 1999, 19, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.-M. IFNγ sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene 2002, 21, 2295–2308. [Google Scholar] [CrossRef] [Green Version]
- Kammertoens, T.; Friese, C.; Arina, A.; Idel, C.; Briesemeister, D.; Rothe, M.; Ivanov, A.; Szymborska, A.; Patone, G.; Kunz, S.; et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 2017, 545, 98–102. [Google Scholar] [CrossRef]
- Lollini, P.-L.; de Giovanni, C.; del Re, B.; Nicoletti, G.; Prodi, G.; Nanni, P. Interferon-mediated enhancement of metastasis. Are MHC antigens involved? Clin. Exp. Metastasis 1987, 5, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Apilado, R.; Coleman, J.; Ben-Sasson, S.; Tsang, S.; Hu-Li, J.; Paul, W.E.; Huang, H. Interferon γ Stabilizes the T Helper Cell Type 1 Phenotype. J. Exp. Med. 2001, 194, 165–172. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Agarwal, P.; Lins, D.C.; Mescher, M.F. Autocrine IFN-γ Promotes Naive CD8 T Cell Differentiation and Synergizes with IFN-α To Stimulate Strong Function. J. Immunol. 2012, 189, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-γ Drives T reg Fragility to Promote Anti-tumor Immunity. Cell 2017, 169, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lollini, P.-L.; Nanni, P.; de Giovanni, C.; Nicoletti, G.; Landuzzi, L. Re: Randomized Trial of Adjuvant Human Interferon Gamma Versus Observation in High-Risk Cutaneous Melanoma: A Southwest Oncology Group Study. JNCI J. Natl. Cancer Inst. 1996, 88, 926–927. [Google Scholar] [CrossRef] [Green Version]
- Lollini, P.-L.; Bosco, M.C.; Cavallo, F.; de Giovanni, C.; Giovarelli, M.; Landuzzi, L.; Musiani, P.; Modesti, A.; Nicoletti, G.; Palmieri, G.; et al. Inhibition of tumor growth and enhancement of metastasis after transfection of the γ-interferon gene. Int. J. Cancer 1993, 55. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Butler, M.; Lutzky, J.; Lawrence, D.P.; Robert, C.; Miller, W.; Linette, G.P.; Ascierto, P.A.; Kuzel, T.; Algazi, A.P.; et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J. Clin. Oncol. 2015, 33, 3003. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberti, G.; Sisi, M.; Andrini, E.; Palladini, A.; Giunchi, F.; Lollini, P.-L.; Ardizzoni, A.; Gelsomino, F. The Mechanisms of PD-L1 Regulation in Non-Small-Cell Lung Cancer (NSCLC): Which Are the Involved Players? Cancers 2020, 12, 3129. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Gu, X.; Chen, L.; Yao, Z.; Song, J.; Niu, X.; Xiang, R.; Cheng, T.; Qin, Z.; Deng, W.; et al. Interferon-γ produced by tumor-infiltrating NK cells and CD4+ T cells downregulates TNFSF15 expression in vascular endothelial cells. Angiogenesis 2014, 17, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Jauch, D.; Martin, M.; Schiechl, G.; Kesselring, R.; Schlitt, H.J.; Geissler, E.K.; Fichtner-Feigl, S. Interleukin 21 controls tumour growth and tumour immunosurveillance in colitis-associated tumorigenesis in mice. Gut 2011, 60, 1678–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A.A. Interleukin-35 Limits Anti-Tumor Immunity. Immunity 2016, 44, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Chen, X.; Hao, S.; Jia, R.; Wang, N.; Chen, S.; Li, M.; Wang, C.; Mao, H. Increased interleukin-35 expression in tumor-infiltrating lymphocytes correlates with poor prognosis in patients with breast cancer. Cytokine 2017, 89, 76–81. [Google Scholar] [CrossRef]
- Zou, J.-M.; Qin, J.; Li, Y.-C.; Wang, Y.; Li, D.; Shu, Y.; Luo, C.; Wang, S.-S.; Chi, G.; Guo, F.; et al. IL-35 induces N2 phenotype of neutrophils to promote tumor growth. Oncotarget 2017, 8, 33501–33514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+ CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Nishibori, T.; Tanabe, Y.; Su, L.; David, M. Impaired Development of CD4+ CD25+ Regulatory T Cells in the Absence of STAT1. J. Exp. Med. 2004, 199, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Hong, J.; Sun, W.; Xu, G.; Li, N.; Chen, X.; Liu, A.; Xu, L.; Sun, B.; Zhang, J.Z. Role of IFN-g in induction of Foxp3 and conversion of CD4+CD25- T cells to CD4+ Tregs. J. Clin. Investig. 2006, 116, 2434–2441. [Google Scholar] [CrossRef] [Green Version]
- Sawitzki, B.; Kingsley, C.I.; Oliveira, V.; Karim, M.; Herber, M.; Wood, K.J. IFN-γ production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J. Exp. Med. 2005, 201, 1925–1935. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Baker, S.; Wieckiewicz, J.; Wood, K.J. IFN-γ Triggered STAT1-PKB/AKT Signalling Pathway Influences the Function of Alloantigen Reactive Regulatory T Cells. Am. J. Transplant. 2010, 10, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, T.; Cao, A.T.; Weaver, C.T.; Elson, C.O.; Cong, Y. Interleukin-12 Converts Foxp3+ Regulatory T Cells to Interferon–γ-Producing Foxp3+ T Cells That Inhibit Colitis. Gastroenterology 2011, 140, 2031–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenecke, C.; Lee, C.-W.; Thamm, K.; Föhse, L.; Schafferus, M.; Mittrücker, H.-W.; Floess, S.; Huehn, J.; Ganser, A.; Förster, R.; et al. IFN-γ Production by Allogeneic Foxp3 + Regulatory T Cells Is Essential for Preventing Experimental Graft-versus-Host Disease. J. Immunol. 2012, 189, 2890–2896. [Google Scholar] [CrossRef] [Green Version]
- Greifenberg, V.; Ribechini, E.; Rößner, S.; Lutz, M.B. Myeloid-derived suppressor cell activation by combined LPS and IFN-γ treatment impairs DC development. Eur. J. Immunol. 2009, 39, 2865–2876. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Maruyama, A.; Yoshida, S.; Takeda, Y.; Matsumoto, M.; Seya, T. Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. OncoImmunology 2018, 7, e1373231. [Google Scholar] [CrossRef] [Green Version]
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; di Giacomo, A.M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; et al. Myeloid-Derived Suppressor Cells Predict Survival of Patients with Advanced Melanoma: Comparison with Regulatory T Cells and NY-ESO-1- or Melan-A-Specific T Cells. Clin. Cancer Res. 2014, 20, 1601–1609. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef] [Green Version]
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Faure, M.; Rochigneux, P.; Olive, D.; Taix, S.; Brenot-Rossi, I.; Gilabert, M. Hyperprogressive Disease in Anorectal Melanoma Treated by PD-1 Inhibitors. Front. Immunol. 2018, 9, 797. [Google Scholar] [CrossRef]
- Xiong, D.; Wang, Y.; Singavi, A.K.; Mackinnon, A.C.; George, B.; You, M. Immunogenomic Landscape Contributes to Hyperprogressive Disease after Anti-PD-1 Immunotherapy for Cancer. iScience 2018, 9, 258–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.S.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti–PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Hara, Y.; Kubota, T. CARD8 is a negative regulator for NLRP3 inflammasome, but mutant NLRP3 in cryopyrin-associated periodic syndromes escapes the restriction. Arthritis Res. Ther. 2014, 16, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, D.; Fickentscher, C.; de Moerloose, P.; Brandt, K.J. F-actin dampens NLRP3 inflammasome activity via Flightless-I and LRRFIP2. Sci. Rep. 2016, 6, 29834. [Google Scholar] [CrossRef] [Green Version]
- Clipman, S.J.; Henderson-Frost, J.; Fu, K.Y.; Bern, C.; Flores, J.; Gilman, R.H. Genetic association study of NLRP1, CARD, and CASP1 inflammasome genes with chronic Chagas cardiomyopathy among Trypanosoma cruzi seropositive patients in Bolivia. PLoS ONE 2018, 13, e0192378. [Google Scholar] [CrossRef] [Green Version]
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; di Filippo, M.; French, L.; Beer, H.D. The Crosstalk between Nrf2 and Inflammasomes. Int. J. Mol. Sci. 2018, 19, 562. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Li, C.; Ke, M.; Lu, L.; Busuttil, R.; Ying, Q.; Kupiec-Weglinski, J.; Ke, B. Notch Signal Regulates Inflammasome NLRP3 Activation in Liver Ischemia and Reperfusion Injury. J. Hepatol. 2016, 64, S508–S509. [Google Scholar] [CrossRef]
- Jiang, L.; Ke, M.; Yue, S.; Xiao, W.; Yan, Y.; Deng, X.; Ying, Q.L.; Li, J.; Ke, B. Blockade of Notch signaling promotes acetaminophen-induced liver injury. Immunol. Res. 2017, 65, 739–749. [Google Scholar] [CrossRef]
- Jin, Y.; Li, C.; Xu, D.; Zhu, J.; Wei, S.; Zhong, A.; Sheng, M.; Duarte, S.; Coito, A.J.; Busuttil, R.W.; et al. Jagged1-mediated myeloid Notch1 signaling activates HSF1/Snail and controls NLRP3 inflammasome activation in liver inflammatory injury. Cell. Mol. Immunol. 2019, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A.B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 117864691769193. [Google Scholar] [CrossRef] [Green Version]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Katz, J.B.; Muller, A.J.; Prendergast, G.C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol. Rev. 2008, 222, 206–221. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.M.; Young, G.D.; McMiller, T.L.; Chen, S.; Salas, J.T.; Pritchard, T.S.; Xu, H.; Meeker, A.K.; Fan, J.; Cheadle, C.; et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin. Cancer Res. 2015, 21, 3969–3976. [Google Scholar] [CrossRef] [Green Version]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef]
- Yu, G.; Dai, H.; Chen, J.; Duan, L.; Gong, M.; Liu, L.; Xiong, P.; Wang, C.Y.; Fang, M.; Gong, F. Gene delivery of indoleamine 2,3-dioxygenase prolongs cardiac allograft survival by shaping the types of T-cell responses. J. Gene Med. 2008, 10, 754–761. [Google Scholar] [CrossRef]
- Witkiewicz, A.; Williams, T.K.; Cozzitorto, J.; Durkan, B.; Showalter, S.L.; Yeo, C.J.; Brody, J.R. Expression of Indoleamine 2,3-Dioxygenase in Metastatic Pancreatic Ductal Adenocarcinoma Recruits Regulatory T Cells to Avoid Immune Detection. J. Am. Coll. Surg. 2008, 206, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Heikkila, P.S.; Vaara, A.T.; von Smitten, K.A.J.; Vakkila, J.M.; Leidenius, M.H.K. Simultaneous Foxp3 and IDO expression is associated with sentinel lymph node metastases in breast cancer. BMC Cancer 2009, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO Activates Regulatory T Cells and Blocks Their Conversion into Th17-Like T Cells. J. Immunol. 2009, 183, 2475–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botticelli, A.; Cerbelli, B.; Lionetto, L.; Zizzari, I.; Salati, M.; Pisano, A.; Federica, M.; Simmaco, M.; Nuti, M.; Marchetti, P. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J. Transl. Med. 2018, 16, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Perez, R.P.; Riese, M.J.; Lewis, K.D.; Saleh, M.N.; Daud, A.; Berlin, J.; Lee, J.J.; Mukhopadhyay, S.; Zhou, L.; Serbest, G.; et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. J. Clin. Oncol. 2017, 35, 3003. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Gibney, G.T.; Hamid, O.; Lutzky, J.; Olszanski, A.J.; Mitchell, T.C.; Gajewski, T.F.; Chmielowski, B.; Hanks, B.A.; Zhao, Y.; Newton, R.C.; et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J. Immunother. Cancer 2019, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. J. Clin. Oncol. 2018, 36, 108. [Google Scholar] [CrossRef]
- Van den Eynde, B.J.; van Baren, N.; Baurain, J.F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Yue, L.; Yao, R.; Zhou, L.; Yang, Y.; Lu, L.; Gao, W. P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget 2017, 8, 54548–54557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, J.; Li, M.-Q.; Ding, D.; Li, D.-J.; Jin, L.-P.; Hu, W.-G.; Zhu, X.-Y. Indoleamine 2,3-dioxygenase-1 (IDO1) enhances survival and invasiveness of endometrial stromal cells via the activation of JNK signaling pathway. Int. J. Clin. Exp. Pathol. 2013, 6, 431–444. [Google Scholar] [PubMed]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Hammaker, D.R.; Boyle, D.L.; Inoue, T.; Firestein, G.S. Regulation of the JNK pathway by TGF-beta activated kinase 1 in rheumatoid arthritis synoviocytes. Arthritis Res. Ther. 2007, 9, R57. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Kasisomayajula, K.; Peng, J.; Bancalari, E. Inhibition of JNK Enhances TGF-β1-Activated Smad2 Signaling in Mouse Embryonic Lung. Pediatric Res. 2009, 65, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; Plaen, E.; van den Eynde, B.J. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol. Res. 2017, 5, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, L.; Zheng, L.; Yang, Q.; Chen, M.; Wang, J.; Zhu, G.; Chen, Z.; Sun, J. Hyperprogressive Disease In Cervical Small Cell Carcinoma Treated By Immune Checkpoint Inhibitor. Oncotargets Ther. 2019, 12, 8873–8877. [Google Scholar] [CrossRef] [Green Version]
- Pai, C.-C.S.; Huang, J.T.; Lu, X.; Simons, D.M.; Park, C.; Chang, A.; Tamaki, W.; Liu, E.; Roybal, K.T.; Seagal, J.; et al. Clonal Deletion of Tumor-Specific T Cells by Interferon-γ Confers Therapeutic Resistance to Combination Immune Checkpoint Blockade. Immunity 2019, 50, 477–492. [Google Scholar] [CrossRef] [Green Version]
- Refaeli, Y.; van Parijs, L.; Alexander, S.I.; Abbas, A.K. Interferon γ is Required for Activation-induced Death of T Lymphocytes. J. Exp. Med. 2002, 196, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Ju, S.-T.; Panka, D.J.; Cui, H.; Ettinger, R.; EI-Khatib, M.; Sherr, D.H.; Stanger, B.Z.; Marshak-Rothstein, A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 1995, 373, 444–448. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Ohtani, T.; Khan, O.; Setty, M.; Manne, S.; O’Brien, S.; Gherardini, P.F.; Herati, R.S.; Huang, A.C.; Chang, K.-M.; et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018, 48, 1029–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, K.; Shahid, U.; Malavasi, F. Human CD38, a cell-surface protein with multiple functions. Faseb J. 1996, 10, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Dianzani, U.; Funaro, A.; DiFranco, D.; Garbarino, G.; Bragardo, M.; Redoglia, V.; Buonfiglio, D.; de Monte, L.B.; Pileri, A.; Malavasi, F. Interaction between endothelium and CD4+ CD45RA+ lymphocytes. Role of the human CD38 molecule. J. Immunol. 1994, 153, 952–959. [Google Scholar] [PubMed]
- Deaglio, S.; Dianzani, U.; Horenstein, A.L.; Fernandez, J.E.; van Kooten, C.; Bragardo, M.; Funaro, A.; Garbarino, G.; di Virgilio, F.; Banchereau, J.; et al. Human CD38 ligand. A 120-KDA protein predominantly expressed on endothelial cells. J. Immunol. 1996, 156, 727–734. [Google Scholar]
- Estrada-Figueroa, L.A.; Ramirez-Jimenez, Y.; Osorio-Trujillo, C.; Shibayama, M.; Navarro-Garcia, F.; Garcia-Tovar, C.; Talamas-Rohana, P. Absence of CD38 delays arrival of neutrophils to the liver and innate immune response development during hepatic amoebiasis by Entamoeba histolytica. Parasite Immunol. 2011, 33, 661–668. [Google Scholar] [CrossRef]
- Zubiaur, M.; Izquierdo, M.; Terhorst, C.; Malavasi, F.; Sancho, J. CD38 ligation results in activation of the Raf-1/mitogen-activated protein kinase and the CD3-zeta/zeta-associated protein-70 signaling pathways in Jurkat T lymphocytes. J. Immunol. 1997, 159, 193–205. [Google Scholar]
- Ausiello, C.M.; Urbani, F.; la Sala, A.; Funaro, A.; Malavasi, F. CD38 ligation induces discrete cytokine mRNA expression in human cultured lymphocytes. Eur. J. Immunol. 1995, 25, 1477–1480. [Google Scholar] [CrossRef]
- Deaglio, S.; Zubiaur, M.; Gregorini, A.; Bottarel, F.; Ausiello, C.M.; Dianzani, U.; Sancho, J.; Malavasi, F. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood 2002, 99, 2490–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sconocchia, G.; Titus, J.A.; Mazzoni, A.; Visintin, A.; Pericle, F.; Hicks, S.W.; Malavasi, F.; Segal, D.M. CD38 triggers cytotoxic responses in activated human natural killer cells. Blood 1999, 94, 3864–3871. [Google Scholar] [CrossRef]
- Funaro, A.; Morra, M.; Calosso, L.; Zini, M.G.; Ausiello, C.M.; Malavasi, F. Role of the human CD38 molecule in B cell activation and proliferation. Tissue Antigens 1997, 49, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V Gene Mutation Status and CD38 Expression As Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Matrai, Z. CD38 as a prognostic marker in CLL. Hematology 2005, 10, 39–46. [Google Scholar] [CrossRef]
- Hambley, R.; Payne, T.; Taylor, H.; Fegan, C.; Mills, K.; Pepper, C. CD38+ and CD38-B-CLL subsets from patients with bimodal expression of the CD38 antigen exhibit distinct cell cycle distribution and Bcl-2 family protein expression. Br. J. Haematol. 2004, 125, 10. [Google Scholar]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisitti, T.; Aydin, S.; Rossi, D.; Cottino, F.; Bergui, L.; D’Arena, G.; Bonello, L.; Horenstein, A.L.; Brennan, P.; Pepper, C.; et al. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia 2010, 24, 958–969. [Google Scholar] [CrossRef]
- Lin, P.; Owens, R.; Tricot, G.; Wilson, C.S. Flow Cytometric Immunophenotypic Analysis of 306 Cases of Multiple Myeloma. Am. J. Clin. Pathol. 2004, 121, 482–488. [Google Scholar] [CrossRef]
- Harada, H.; Kawano, M.M.; Huang, N.; Harada, Y.; Iwato, K.; Tanabe, O.; Tanaka, H.; Sakai, A.; Asaoku, H.; Kuramoto, A. Phenotypic difference of normal plasma cells from mature myeloma cells. Blood 1993, 81, 2658–2663. [Google Scholar] [CrossRef]
- McKeage, K. Daratumumab: First Global Approval. Drugs 2016, 76, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Isatuximab: First Approval. Drugs 2020, 80, 905–912. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Donk, N.W.C.J.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Martin, T.G.; Corzo, K.; Chiron, M.; van de Velde, H.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells 2019, 8, 1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckert, J.; Wetzel, M.-C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallee, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, A Novel Humanized CD38-Targeting Antibody, Demonstrates Potent Antitumor Activity in Models of Multiple Myeloma and Other CD38+ Hematologic Malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.-B.; Lee, J.-S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Blacher, E.; Vaknine, H.; Lund, F.E.; Stein, R.; Mayo, L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro-Oncology 2012, 14, 1037–1049. [Google Scholar] [CrossRef]
- Liao, S.; Xiao, S.; Chen, H.; Zhang, M.; Chen, Z.; Long, Y.; Gao, L.; Zhu, G.; He, J.; Peng, S.; et al. CD38 enhances the proliferation and inhibits the apoptosis of cervical cancer cells by affecting the mitochondria functions. Mol. Carcinog. 2017, 56, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.; Kato, J.; Hong, J.A.; Merino, M.J.; Schrump, D.S.; Lund, F.E.; Moss, J. CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis 2018, 39, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.-T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, T.; Deaglio, S.; Franco, L.; Calosso, L.; Badolato, R.; Garbarino, G.; Dianzani, U.; Malavasi, F. CD38 expression and functional activities are up-regulated by IFN-γ on human monocytes and monocytic cell lines. J. Leukoc. Biol. 2001, 69, 605–612. [Google Scholar] [CrossRef]
- Bürgler, S.; Gimeno, A.; Parente-Ribes, A.; Wang, D.; Os, A.; Devereux, S.; Jebsen, P.; Bogen, B.; Tjønnfjord, G.E.; Munthe, L.A. Chronic Lymphocytic Leukemia Cells Express CD38 in Response to Th1 Cell–Derived IFN-γ by a T-bet–Dependent Mechanism. J. Immunol. 2015, 194, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Snoeck, H.W.; Lardon, F.; Lenjou, M.; Nys, G.; Van, D.B.; Peetermans, M.E. Differential regulation of the expression of CD38 and human leukocyte antigen-DR on CD34+ hematopoietic progenitor cells by interleukin-4 and interferon-gamma. Exp. Hematol. 1993, 21, 1480–1486. [Google Scholar] [PubMed]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Konen, J.M.; Fradette, J.J.; Gibbons, D.L. The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [Green Version]
- Sek, K.; Mølck, C.; Stewart, G.; Kats, L.; Darcy, P.; Beavis, P. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.-R.; Deng, W.-W.; Liu, J.-F.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef]
- Vogt, T.J.; Gevensleben, H.; Dietrich, J.; Kristiansen, G.; Bootz, F.; Landsberg, J.; Goltz, D.; Dietrich, D. Detailed analysis of adenosine A2a receptor (ADORA2A) and CD73 (5′-nucleotidase, ecto, NT5E) methylation and gene expression in head and neck squamous cell carcinoma patients. OncoImmunology 2018, 7, e1452579. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Mirandola, P.; Milani, D.; Varani, K.; Gessi, S.; Klotz, K.-N.; Leung, E.; Baraldi, P.G.; Borea, P.A. Adenosine Receptors as Mediators of Both Cell Proliferation and Cell Death of Cultured Human Melanoma Cells. J. Investig. Dermatol. 2002, 119, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etique, N.; Grillier-Vuissoz, I.; Lecomte, J.; Flament, S. Crosstalk between adenosine receptor (A2A isoform) and ERα mediates ethanol action in MCF-7 breast cancer cells. Oncol. Rep. 2009, 21, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Koszałka, P.; Gołuńska, M.; Urban, A.; Stasiłojć, G.; Stanisławowski, M.; Majewski, M.; Składanowski, A.C.; Bigda, J. Specific Activation of A3, A2A and A1 Adenosine Receptors in CD73-Knockout Mice Affects B16F10 Melanoma Growth, Neovascularization, Angiogenesis and Macrophage Infiltration. PLoS ONE 2016, 11, e0151420. [Google Scholar] [CrossRef] [Green Version]
- Milne, G.R.; Palmer, T.M. Anti-Inflammatory and Immunosuppressive Effects of the A2A Adenosine Receptor. Sci. World J. 2011, 11, 320–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Dev, R.; de Dios Ruiz-Rosado, J.; Partida-Sanchez, S.; Guerau-de-Arellano, M.; Dhakal, P.; Kuivaniemi, H.; Hans, C.P. Pharmacological inhibition of Notch signaling regresses pre-established abdominal aortic aneurysm. Sci. Rep. 2019, 9, 13458. [Google Scholar] [CrossRef] [Green Version]
- Flajolet, M.; Wang, Z.; Futter, M.; Shen, W.; Nuangchamnong, N.; Bendor, J.; Wallach, I.; Nairn, A.C.; Surmeier, D.J.; Greengard, P. FGF acts as a co-transmitter through adenosine A2A receptor to regulate synaptic plasticity. Nat. Neurosci. 2008, 11, 1402–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Jiang, F.; Sluss, H.K.; Zhang, C.; Shokat, K.M.; Flavell, R.A.; Davis, R.J. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 15759–15764. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Bencivenni, S.; Battistello, E.; Vincenzi, F.; Colotta, V.; Catarzi, D.; Varano, F.; Merighi, S.; Borea, P.A.; Varani, K. Inhibition of A2A Adenosine Receptor Signaling in Cancer Cells Proliferation by the Novel Antagonist TP455. Front. Pharmacol. 2017, 8, 888. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Lo, Y.-C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Kamada, T.; Sasaki, A.; Nakamura, Y.; Fukuoka, S.; Tada, Y.; Kawazoe, A.; Shitara, K.; Nishikawa, H. Clinicopathological, genomic and immunological features of hyperprogressive disease during PD-1 blockade in gastric cancer patients. J. Clin. Oncol. 2018, 36, 4106. [Google Scholar] [CrossRef]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef]
- Allard, D.; Turcotte, M.; Stagg, J. Targeting A2 adenosine receptors in cancer. Immunol. Cell Biol. 2017, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.-J.; Lund, F.E.; Stein, R. Dual Role of CD38 in Microglial Activation and Activation-Induced Cell Death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xu, X.; Liu, Y. Activation-Induced Cell Death in T Cells and Autoimmunity. Cell. Mol. Immunol. 2004, 1, 186–192. [Google Scholar] [PubMed]
- Vrbacky, F.; Smolej, L.; Vroblova, V.; Pekova, S.; Hrudkova, M.; Cervinka, M.; Pecka, M.; Krejsek, J.; Maly, J. Angiopoietin-2 mRNA expression is increased in chronic lymphocytic leukemia patients with poor prognostic features. Hematology 2010, 15, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Long, Y.; Xiao, S.; Liang, L.; He, Z.; Yue, C.; Wei, X.; Zhou, Y. CD38 affects the biological behavior and energy metabolism of nasopharyngeal carcinoma cells. Int. J. Oncol. 2018, 54, 585–599. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Madu, C.; Li, L.; Lu, Y. Selection, Analysis and Improvement of Anti-Angiogenesis Compounds Identified by an Anti-HIF-1α Screening and Validation System. J. Cancer 2016, 7, 1926–1938. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Han, B.; Luo, K.; Ren, Z.; Cai, L.; Sun, L. NOX2-ROS-HIF-1α signaling is critical for the inhibitory effect of oleanolic acid on rectal cancer cell proliferation. Biomed. Pharmacother. 2017, 85, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Yong, H.; Xia, Y.; Xie, Q.; Gao, G.; Zhou, X. Chemotherapy regimen based on sorafenib combined with 5-FU on HIF-1α and VEGF expression and survival in advanced gastric cancer patients. Oncol. Lett. 2017, 13, 2703–2707. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Jeong, Y.-J.; Cho, H.-J.; Hoe, H.-S.; Park, K.-K.; Park, Y.-Y.; Choi, Y.H.; Kim, C.-H.; Chang, H.-W.; Park, Y.-J.; et al. Delphinidin inhibits angiogenesis through the suppression of HIF-1α and VEGF expression in A549 lung cancer cells. Oncol. Rep. 2017, 37, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Haase, V. The VHL Tumor Suppressor: Master Regulator of HIF. Curr. Pharm. Des. 2009, 15, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.J.; Le, V.H.; Oyama, T.; Ricketts, C.J.; Ho, T.H.; Cheng, E.H. Chromosome 3p Loss–Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 3533–3539. [Google Scholar] [CrossRef]
- Brouwer, N.J.; Wierenga, A.P.A.; Gezgin, G.; Marinkovic, M.; Luyten, G.P.M.; Kroes, W.G.M.; Versluis, M.; van der Velden, P.A.; Verdijk, R.M.; Jager, M.J. Ischemia Is Related to Tumour Genetics in Uveal Melanoma. Cancers 2019, 11, 1004. [Google Scholar] [CrossRef] [Green Version]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [Green Version]
- Perrot-Applanat, M.; di Benedetto, M. Autocrine functions of VEGF in breast tumor cells. Cell Adhes. Migr. 2012, 6, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Refae, S.; Gal, J.; Brest, P.; Giacchero, D.; Borchiellini, D.; Ebran, N.; Peyrade, F.; Guigay, J.; Milano, G.; Saada-Bouzid, E. Hyperprogression under Immune Checkpoint Inhibitor: A potential role for germinal immunogenetics. Sci. Rep. 2020, 10, 3565. [Google Scholar] [CrossRef]
- Glubb, D.M.; Cerri, E.; Giese, A.; Zhang, W.; Mirza, O.; Thompson, E.E.; Chen, P.; Das, S.; Jassem, J.; Rzyman, W.; et al. Novel Functional Germline Variants in the VEGF Receptor 2 Gene and Their Effect on Gene Expression and Microvessel Density in Lung Cancer. Clin. Cancer Res. 2011, 17, 5257–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Li, J.-Y.; Wu, Y.-J.; Yu, H.; Shen, Q.-D.; Tian, T.; Li, L.; Qiu, H.-X. CD38 as a prognostic factor in Chinese patients with chronic lymphocytic leukaemia. Leuk. Res. 2009, 33, 237–243. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Tumor Types | Treatment | Rate of HPD (%) | Criteria Used to Define HPD |
|---|---|---|---|---|
| Champiat et al. (2017) [12] | Multiple tumor types 1 | PD-1/PD-L1 inhibitors | 9.2 |
|
| Kanjanapan et al. (2019) [18] | Multiple tumor types 2 | CTLA-4, PD-1/PD-L1 inhibitors | 7 | |
| Aoki et al. (2019) [9] | AGC | PD-1 inhibitors | 29.4 |
|
| Ferrara et al. (2018) [13] | NSCLC | PD-1/PD-L1 inhibitors | 14 |
|
| Saâda-Bouzid et al. (2017) [14] | HNSCC | PD-1/PD-L1 inhibitors | 29.4 |
|
| Kim et al. (2019) [15] | NSCLC | PD-1/PD-L1 inhibitors | 17 | |
| Sasaki et al. (2019) [10] | AGC | PD-1 inhibitors | 21 |
|
| Kato et al. (2017) [19] | Multiple tumor types 3 | CTLA-4, PD-1/PD-L1 inhibitors | 3.9 |
|
| Kamada et al. (2019) [11] | AGC | PD-1 inhibitors | 10 | |
| Matos et al. (2018) [20] | Multiple tumor types 4 | PD-1/PD-L1 inhibitors | 15 |
|
| Lo Russo et al. (2019) [21] | NSCLC | PD-1/PD-L1 inhibitors | 25.7 |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelicola, S.; Ruzzi, F.; Landuzzi, L.; Scalambra, L.; Gelsomino, F.; Ardizzoni, A.; Nanni, P.; Lollini, P.-L.; Palladini, A. IFN-? and CD38 in Hyperprogressive Cancer Development. Cancers 2021, 13, 309. https://doi.org/10.3390/cancers13020309
Angelicola S, Ruzzi F, Landuzzi L, Scalambra L, Gelsomino F, Ardizzoni A, Nanni P, Lollini P-L, Palladini A. IFN-? and CD38 in Hyperprogressive Cancer Development. Cancers. 2021; 13(2):309. https://doi.org/10.3390/cancers13020309
Chicago/Turabian StyleAngelicola, Stefania, Francesca Ruzzi, Lorena Landuzzi, Laura Scalambra, Francesco Gelsomino, Andrea Ardizzoni, Patrizia Nanni, Pier-Luigi Lollini, and Arianna Palladini. 2021. "IFN-? and CD38 in Hyperprogressive Cancer Development" Cancers 13, no. 2: 309. https://doi.org/10.3390/cancers13020309
APA StyleAngelicola, S., Ruzzi, F., Landuzzi, L., Scalambra, L., Gelsomino, F., Ardizzoni, A., Nanni, P., Lollini, P.-L., & Palladini, A. (2021). IFN-? and CD38 in Hyperprogressive Cancer Development. Cancers, 13(2), 309. https://doi.org/10.3390/cancers13020309