Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced?

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Androgen-Based Therapies Retain Activity after Failure of First-Line AR-Based Treatments
1.2. Combination Therapies Which Include Androgen Blockades Can Extend Patient Survival
1.3. Androgen Blockade: A Time-Limited Treatment?
2. Targeting the Androgen Signaling Pathway
- Extracellular provision of testosterone;
- Activation of testosterone by 5α reductase to dihydrotestosterone (DHT);
- Androgen metabolism and receptor engagement in the cell cytoplasm;
- Turnover and metabolism of the AR and co-activator proteins;
- Transcription complex formation and activation of gene expression in the cell nucleus;
- Blockade of AR-stimulated genes and cytokines—second messengers
- Direct binding inhibitors of the AR;
- Testosterone activating 5α- reductase inhibitors and
- Intratumoral and extratumoral testosterone inhibitors.
2.1. Resistance to Androgen Blockade
2.2. Targeting Alternative Pathways
3. Mechanisms of Cell Death after Application of Androgen Signaling Blockade
3.1. The LNCaP Cell Line: Industry Standard Sndrogen-Responsive PCa Cell Model
3.2. Cell Death in Cell Line Models of PCa after Androgen Signaling Blockade
3.3. Cell Death in Animal Models of PCa after Androgen Signaling Blockade
3.4. Cell Death in Human Tissues after Androgen Signaling Blockade
4. Gene Expression Changes in Prostate Epithelial Cells during the Application of an Androgen Signaling Blockade in Patients
4.1. The Dynamic Changes in Gene Expression after ADT in Human Tissues
4.2. The Dynamic Changes in Gene Expression after Castration in Mouse Tissues
5. Modeling Resistance to Androgen Blockade: The Quest for a Defining Mechanism
5.1. Cell Line Studies of the ADT Resistant State
5.2. Complexity and Heterogeneity: Modeling ADT in Three Dimensions
5.3. ADT in Genetically Engineered Mice
5.4. Human Xenografted Cell Models
6. Modeling Pathway Responses to Androgen Signaling Blockade
7. Combination Drug Treatments: Is More Better?
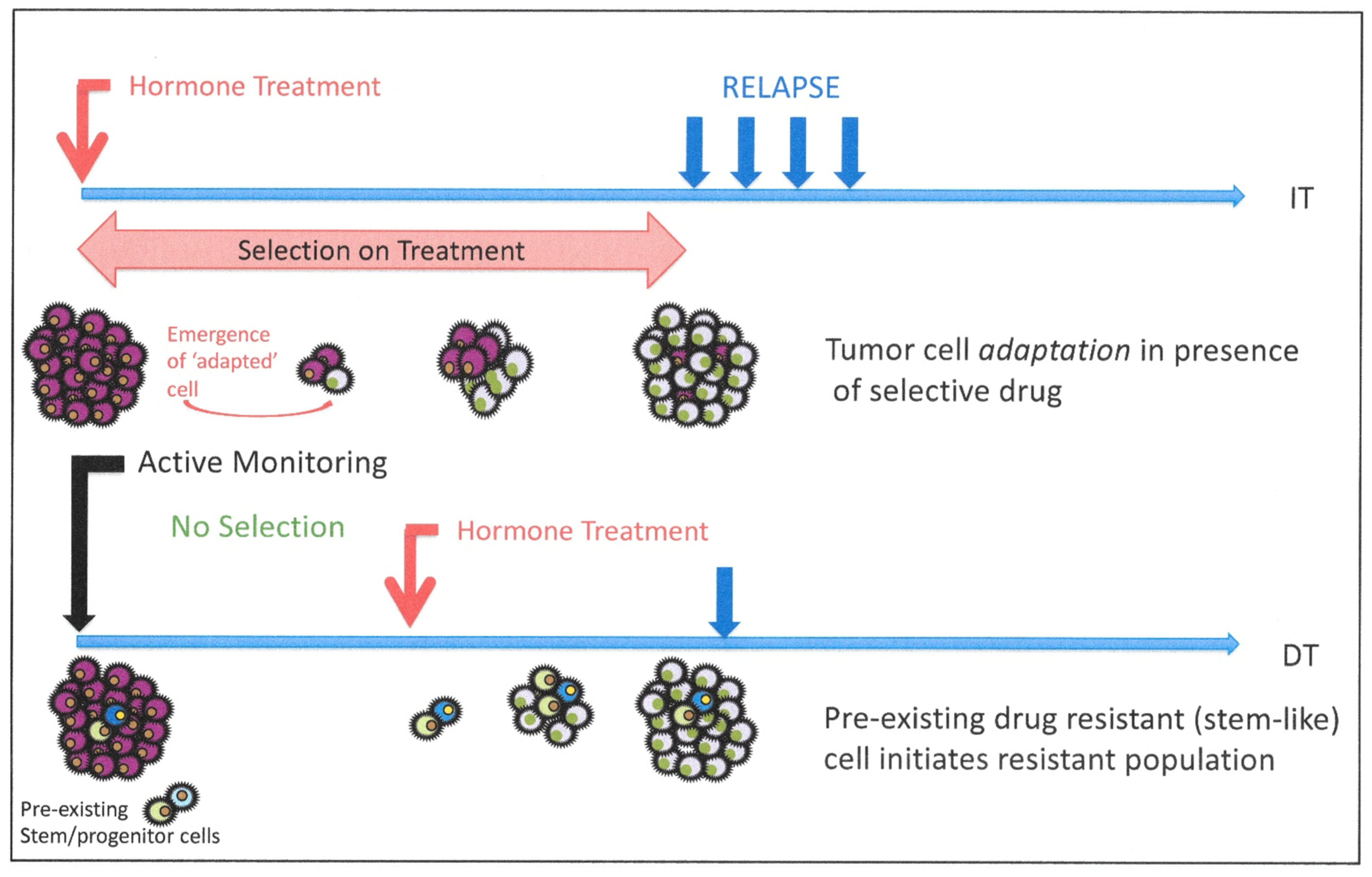
8. Cellular Heterogeneity and Resistance to Androgen Blockade: Is Resistance Intrinsic or Induced?
8.1. Do Prostate Cancer Stem Cells Provide a Treatment-Resistant Reservoir?
- expansion of an AR ± but androgen insensitive population of CAR cells [149]; or
8.2. Evidence for Pre-Existing Resistant Cells in Human Clinical Trials
9. Modeling Pathways to CRPC—Predictions from Mechanism Testing
10. Does Better Androgen Blockade Change the Natural History of Prostate Cancer?
10.1. Long-Term Effects of Low Androgen Levels in Men with Benign and Malignant Prostate Disease
10.2. Androgens Are Not Just Active in Male Reproductive Organs: Extra-Prostatic Effects of Long-Term ADT
11. Future Perspectives: Exploiting Mechanistic Biology for Patient Advantage in An-drogen-Based Therapies
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Huggins, C. Endocrine control of prostate cancer. Science 1943, 97, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.W.; Xie, W.; Regan, M.M.; Pomerantz, M.; Nakabayashi, M.; Daskivich, T.J.; Sartor, O.; Taplin, M.-E.; Kantoff, P.W.; Oh, W.K. Efficacy of androgen deprivation therapy (ADT) in patients with advanced prostate cancer. Cancer 2008, 112, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Mahal, B.A.; Muralidhar, V.; Martin, N.E.; Orio, P.F.; Mouw, K.W.; King, M.T.; Choueiri, T.K.; Trinh, Q.-D.; Hoffman, K.E.; et al. Androgen Deprivation Therapy and Overall Survival for Gleason 8 Versus Gleason 9–10 Prostate Cancer. Eur. Urol. 2019, 75, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Sadar, M.D. Enzalutamide and blocking androgen receptor in advanced prostate cancer: Lessons learnt from the history of drug development of antiandrogens. Res. Rep. Urol. 2018, 10, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Sonpavde, G.; Attard, G.; Bellmunt, J.; Mason, M.D.; Malavaud, B.; Tombal, B.; Sternberg, C.N. The Role of Abiraterone Acetate in the Management of Prostate Cancer: A Critical Analysis of the Literature. Eur. Urol. 2011, 60, 270–278. [Google Scholar] [CrossRef]
- Scher, H.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Tannock, I.F.; De Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Matei, D.V.; Renne, G.; Pimentel, M.; Sandri, M.T.; Zorzino, L.; Botteri, E.; De Cicco, C.; Musi, G.; Brescia, A.; Mazzoleni, F.; et al. Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer: A Systematic Diagnostic Attempt. Clin. Genitourin. Cancer 2012, 10, 164–173. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.-H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rescigno, P.; de Bono, J.S. Immunotherapy for lethal prostate cancer. Nat. Rev. Urol. 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef] [PubMed]
- De Leval, J.; Boca, P.; Youssef, E.; Nicolas, H.; Jeukenne, M.; Seidel, L.; Bouffioux, C.; Coppens, L.; Bonnet, P.; Andrianne, R.; et al. Intermittent versus continuous total androgen blockade in the treatment of patients with advanced hormone-naive prostate cancer: Results of a prospective randomized multicenter trial. Clin. Prostate Cancer 2002, 1, 163–171. [Google Scholar] [CrossRef]
- Abrahamsson, P.-A. Intermittent androgen deprivation therapy in patients with prostate cancer: Connecting the dots. Asian J. Urol. 2017, 4, 208–222. [Google Scholar] [CrossRef]
- Tsao, C.-K.; Galsky, M.D.; Small, A.C.; Yee, T.; Oh, W.K. Targeting the androgen receptor signalling axis in castration-resistant prostate cancer (CRPC). BJU Int. 2012, 110, 1580–1588. [Google Scholar] [CrossRef]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef]
- Ho, Y.; Dehm, S.M. Androgen Receptor Rearrangement and Splicing Variants in Resistance to Endocrine Therapies in Prostate Cancer. Endocrinology 2017, 158, 1533–1542. [Google Scholar] [CrossRef]
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4653–4657. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, N. Mechanisms of Androgen Receptor Activation in Castration-Resistant Prostate Cancer. Endocrinology 2013, 154, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Mostaghel, E.A.; Page, S.T.; Lin, D.W.; Fazli, L.; Coleman, I.M.; True, L.D.; Knudsen, B.; Hess, D.L.; Nelson, C.C.; Matsumoto, A.M. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for cas-tration-resistant prostate cancer. Cancer Res. 2007, 67, 5033–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostaghel, E.A.; Zhang, A.; Hernandez, S.; Marck, B.T.; Zhang, X.; Tamae, D.; Biehl, H.E.; Tretiakova, M.; Bartlett, J.; Burns, J.; et al. Contribution of Adrenal Glands to Intra-tumor Androgens and Growth of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 426–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, Y.; Sadar, M.D. Does increased expression of glucocorticoid receptor support application of antagonists to this receptor for the treatment of castration resistant prostate cancer? AME Med. J. 2018, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Kach, J.; Long, T.M.; Selman, P.; Tonsing-Carter, E.Y.; Bacalao, M.A.; Lastra, R.R.; de Wet, L.; Comiskey, S.; Gillard, M.; VanOpstall, C.; et al. Glucocorticoid receptor Modulators (SGRMs) Delay castrate-resistant prostate cancer growth. Mol. Cancer Ther. 2017, 16, 1680–1692. [Google Scholar] [CrossRef] [Green Version]
- Titus, M.A.; Gregory, C.W.; Ford, O.H.; Schell, M.J.; Maygarden, S.J.; Mohler, J.L. Steroid 5alpha-reductase isozymes I and II in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4365–4371. [Google Scholar] [CrossRef] [Green Version]
- Liss, M.A.; Thompson, I.M. Prostate cancer prevention with 5-alpha reductase inhibitors: Concepts and controversies. Curr. Opin. Urol. 2018, 28, 42–45. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Smith, M.R.; Ryan, C.J.; Berry, W.R.; Shore, N.D.; Liu, G.; Higano, C.S.; Alumkal, J.J.; Hauke, R.; Tutrone, R.F.; et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann. Oncol. 2017, 28, 2264–2271. [Google Scholar] [CrossRef]
- Culig, Z. Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr. Mol. Biol. Rep. 2017, 3, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Sumiyoshi, T.; Mizuno, K.; Yamasaki, T.; Miyazaki, Y.; Makino, Y.; Okasho, K.; Li, X.; Utsunomiya, N.; Goto, T.; Kobayashi, T.; et al. Clinical utility of androgen receptor gene aberrations in circulating cell-free DNA as a biomarker for treatment of castration-resistant prostate cancer. Sci. Rep. 2019, 9, 4030. [Google Scholar] [CrossRef] [Green Version]
- Merson, S.; Yang, Z.H.; Brewer, D.; Olmos, D.; Eichholz, A.; McCarthy, F.; Fisher, G.; Kovacs, G.; Berney, D.M.; Foster, C.S.; et al. Focal amplification of the androgen receptor gene in hormone-naive human prostate cancer. Br. J. Cancer 2014, 110, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Takeda, D.Y.; Spisak, S.; Seo, J.-H.; Pomerantz, M.M.; Hahn, W.C.; Freedman, M.L. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2018, 129, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K.; et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res. 2015, 75, 3663–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicher, W.; Luedeke, M.; Azoitei, A.; Zengerling, F.; Herweg, A.; Genze, F.; Schrader, M.G.; Schrader, A.J.; Cronauer, M.V. Stilbene Induced Inhibition of Androgen Receptor Dimerization: Implications for AR and ARΔLBD-Signalling in Human Prostate Cancer Cells. PLoS ONE 2014, 9, e98566. [Google Scholar] [CrossRef] [Green Version]
- Dalala, K.; Bana, F.; Lia, H.; Morin, H.; Roshan-Moniri, M.; Tam, K.J.; Shepherd, A.; Sharma, A.; Peacock, J.; Carlson, M.L.; et al. Selectively targeting the dimerization interface of human androgen receptor with small-molecules to treat castration-resistant prostate cancer. Cancer Lett. 2018, 437, 35–43. [Google Scholar] [CrossRef]
- Shah, K.; Bradbury, N.A. Kinase modulation of androgen receptor signaling: Implications for prostate cancer. Cancer Cell Microenviron. 2015, 2, e123. [Google Scholar]
- Patek, S.; Willder, J.; Heng, J.; Taylor, B.; Horgan, P.; Leung, H.; Underwood, M.; Edwards, J. Androgen receptor phosphorylation status at serine 578 predicts poor outcome in prostate cancer patients. Oncotarget 2017, 8, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a Novel Androgen Receptor Exon Generates a Constitutively Active Androgen Receptor that Mediates Prostate Cancer Therapy Resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [Green Version]
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef]
- Burd, C.J.; Morey, L.M.; Knudsen, K.E. Androgen receptor corepressors and prostate cancer. Endocr. Relat. Cancer 2006, 13, 979–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Kumari, S.; Hu, Q.; Senapati, D.; Venkadakrishnan, V.B.; Wang, D.; DePriest, A.D.; Schlanger, S.E.; Ben-Salem, S.; Valenzuela, M.M.; et al. A comprehensive analysis of coregulator recruitment, androgen receptor function and gene expression in prostate cancer. eLife 2017, 6, e28482. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.-L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 2008, 15, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, A.; Larsson, P.; Sarwar, M.; Semenas, J.; Khaja, A.S.S.; Persson, J.L. The interplay between AR, EGF receptor and MMP-9 signaling pathways in invasive prostate cancer. Mol. Med. 2018, 24, 34. [Google Scholar] [CrossRef] [PubMed]
- Hoy, J.J.; Kallifatidis, G.; Smith, D.K.; Lokeshwar, B.L. Inhibition of androgen receptor promotes CXC-chemokine receptor 7-mediated prostate cancer cell survival. Sci. Rep. 2017, 7, 3058. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; An, J.; Yang, Y.; Wu, D.; Bai, Y.; Cao, W.; Ma, L.; Chen, J.; Yu, Z.; He, Y.; et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol. Med. 2018, 10, e8478. [Google Scholar] [CrossRef]
- Azad, A.A.; Zoubeidi, A.; Gleave, M.E.; Chi, K.N. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nature Rev. Urol. 2015, 12, 26–36. [Google Scholar] [CrossRef]
- Kita, K.; Shiota, M.; Tanaka, M.; Otsuka, A.; Matsumoto, M.; Kato, M.; Tamada, S.; Iwao, H.; Miura, K.; Nakatani, T.; et al. Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCaP95 prostate cancer cells. Cancer Sci. 2017, 108, 1820–1827. [Google Scholar] [CrossRef] [Green Version]
- Centenera, M.M.; Carter, S.L.; Gillis, J.L.; Marrocco-Tallarigo, D.L.; Grose, R.H.; Tilley, W.D.; Butler, L.M. Co-targeting AR and HSP90 suppresses prostate cancer cell growth and prevents resistance mechanisms. Endocr. Relat. Cancer 2015, 22, 805–818. [Google Scholar] [CrossRef] [Green Version]
- Neklesa, T.K.; Crews, C.M. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Chen, X.; Shen, M.; Yang, D.-R.; Fang, L.; Weng, G.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol. Oncol. 2018, 12, 269–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.D.; Haugk, K.; Woodke, L.; Nelson, P.; Coleman, I.M.; Plymate, S.R. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J. Cell. Biochem. 2006, 99, 392–401. [Google Scholar] [CrossRef]
- Ao, M.; Williams, K.; Bhowmick, N.A.; Hayward, S.W. Transforming growth factor-beta promotes invasion in tumorigenic but not in nontumorigenic human prostatic epithelial cells. Cancer Res. 2006, 66, 8007–8016. [Google Scholar] [CrossRef] [Green Version]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S.; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β Signaling in a Subpopulation of Human Stromal Cells Promotes Prostatic Carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef]
- Pal, S.K.; Patel, J.; He, M.; Foulk, B.; Kraft, K.; Smirnov, D.A.; Twardowski, P.W.; Kortylewski, M.; Bhargava, V.; Jones, J.O. Identification of mechanisms of resistance to treatment with abiraterone acetate or enzalutamide in patients with castration-resistant prostate cancer (CRPC). Cancer 2017, 124, 1216–1224. [Google Scholar] [CrossRef]
- Bungaro, M.; Buttigliero, C.; Tucci, M. Overcoming the mechanisms of primary and acquired resistance to new generation hormonal therapies in advanced prostate cancer: Focus on androgen receptor independent pathways. Cancer Drug Resist. 2020, 3. [Google Scholar] [CrossRef]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid bio-synthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Rane, J.K.; Scaravilli, M.; Ylipää, A.; Pellacani, D.; Mann, V.M.; Simms, M.S.; Nykter, M.; Collins, A.T.; Visakorpi, T.; Maitland, N.J. MicroRNA Expression Profile of Primary Prostate Cancer Stem Cells as a Source of Biomarkers and Therapeutic Targets. Eur. Urol. 2015, 67, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sun, Y.; Li, L.; Niu, Y.; Lin, W.; Lin, C.; Antonarakis, E.S.; Luo, J.; Yeh, S.; Chang, C. Preclinical Study using Malat1 Small Interfering RNA or Androgen Receptor Splicing Variant 7 Degradation Enhancer ASC-J9 ® to Suppress Enzalutamide-resistant Prostate Cancer Progression. Eur. Urol. 2017, 72, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ark, A.V.; Cao, J.; Li, X. Mechanisms and Approaches for Overcoming Enzalutamide Resistance in Prostate Cancer. Front. Oncol. 2018, 8, 180. [Google Scholar] [CrossRef]
- Farah, E.; Li, C.; Cheng, L.; Kong, Y.; Lanman, N.A.; Pascuzzi, P.; Lorenz, G.R.; Zhang, Y.; Ahmad, N.; Li, L.; et al. Notch signalling is activated in and contributes to resistance on enzalutamide-resistant prostate cancer cells. J. Biol Chem. 2019, 294, 8543–8554. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pascuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/Beta catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line--a new model for studies on human prostatic carcinoma. Prog. Clin. Boil. Res. 1980, 37, 115–132. [Google Scholar]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Schalpers, C.; Kuiper, G.G.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Veldscholte, J.; Berrevoets, C.A.; Brinkmann, A.O.; Grootegoed, J.A.; Mulder, E. Anti-androgens and the mutated androgen receptor of LNCaP cells: Differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry 1992, 31, 2393–2399. [Google Scholar] [CrossRef]
- Spans, L.; Helsen, C.; Clinckemalie, L.; Broeck, T.V.D.; Prekovic, S.; Joniau, S.; Lerut, E.; Claessens, F. Comparative Genomic and Transcriptomic Analyses of LNCaP and C4-2B Prostate Cancer Cell Lines. PLoS ONE 2014, 9, e90002. [Google Scholar] [CrossRef] [PubMed]
- Gibas, Z.; Becher, R.; Kawinski, E.; Horoszewicz, J.; Sandberg, A.A. A high-resolution study of chromosome changes in a human prostatic carcinoma cell line (LNCaP). Cancer Genet. Cytogenet. 1984, 11, 399–404. [Google Scholar] [CrossRef]
- King, K.J.; Nicholson, H.D.; Assinder, S.J. Effect of increasing ratio of estrogen: Androgen on proliferation of normal human prostate stromal and epithelial cells, and the malignant cell line LNCaP. Prostate 2006, 66, 105–114. [Google Scholar] [CrossRef]
- Lee, E.C.Y.; Zhan, P.; Schallhom, R.; Packman, K.; Tenniswood, M. Antiandrogen-induced cell death in LNCaP human prostate cancer cells. Cell Death Differ. 2003, 10, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCourt, C.; Maxwell, P.; Mazzucchelli, R.; Montironi, R.; Scarpelli, M.; Salto-Tellez, M.; O’Sullivan, J.M.; Longley, D.B.; Waugh, D.J. Elevation of c-FLIP in Castrate-Resistant Prostate Cancer Antagonizes Therapeutic Response to Androgen Receptor–Targeted Therapy. Clin. Cancer Res. 2012, 18, 3822–3833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Church, D.R.; Yang, B.; Huang, W.; Laurila, T.A.; Jarrard, D.F. Androgen deprivation induces senescence characteristics in prostate cancer cells in vitro and in vivo. Prostate 2012, 73, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, A.; Sharrard, R.; Berthon, P.; Maitland, N.; Maitland, N.J. Androgen receptor localisation and turnover in human prostate epithelium treated with the antiandrogen, casodex. J. Mol. Endocrinol. 2000, 24, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-H.; Lee, S.O.; Niu, Y.; Xu, D.; Liang, L.; Li, L.; Yeh, S.D.; Fujimoto, N.; Yeh, S.; Chang, C. Differential androgen deprivation therapies with anti-androgens casodex/bicalutamide or MDV3100/Enzalutamide versus anti-androgen receptor ASC-J9(R) Lead to promotion versus suppression of prostate cancer metastasis. J. Biolog. Chem. 2013, 288, 19359–19369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltering, K.K.; Helenius, M.A.; Sahu, B.; Manni, V.; Linja, M.J.; Jänne, O.A.; Visakorpi, T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009, 69, 8141–8149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Guerrer, J.; Alfaro, I.E.; Gómez, F.; Protte, A.A.; Bernales, S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate 2013, 73, 1291–1305. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kregel, S.; Chen, J.L.; Tom, W.; Krishnan, V.; Kach, J.; Brechka, H.; Fessenden, T.B.; Isikbay, M.; Paner, G.P.; Szmulewitz, R.Z.; et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 2016, 7, 26259–26274. [Google Scholar] [CrossRef] [PubMed]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Griend, D.J.V.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerasuolo, M.; Paris, D.; Iannotti, F.A.; Melck, D.; Verde, R.; Mazzarella, E.; Motta, A.; Ligresti, A. Neuroendocrine Transdifferentiation in Human Prostate Cancer Cells: An Integrated Approach. Cancer Res. 2015, 75, 2975–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentini, A.; Biancolella, M.; Amati, F.; Gravina, P.; Miano, R.; Chillemi, G.; Farcomeni, A.; Bueno, S.; Vespasiani, G.; Desideri, A.; et al. Valproic Acid Induces Neuroendocrine Differentiation and UGT2B7 Up-Regulation in Human Prostate Carcinoma Cell Line. Drug Metab. Dispos. 2007, 35, 968–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Patrawala, L.; Calhoun, T.; Schneiderbroussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [Green Version]
- Birnie, R.; Bryce, S.D.; Roome, C.; Dussupt, V.; Droop, A.P.; Lang, S.; Berry, A.P.; Hyde, C.F.; Lewis, J.L.; Stower, M.J.; et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. 2008, 9, R83. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.E.; Marlein, C.; Walker, H.F.; Frame, F.M.; Mann, V.M.; Simms, M.S.; Davies, B.R.; Collins, A.T.; Maitland, N.J. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017, 8, 56698–56713. [Google Scholar] [CrossRef] [Green Version]
- Polson, E.S.; Lewis, J.; Celik, H.; Mann, V.M.; Stower, M.J.; Simms, M.S.; Rodrigues, G.; Collins, A.T.; Maitland, N.J. Monoallelic Ex-pression of TMPRSS2/ERG in Prostate Cancer Stem Cells. Nature Commun. 2013, 4, 1623. [Google Scholar] [CrossRef] [Green Version]
- Pellacani, D.; Droop, A.P.; Frame, F.M.; Simms, M.S.; Mann, V.M.; Collins, A.T.; Eaves, C.J.; Maitland, N.J. Phenotype-independent DNA methylation changes in prostate cancer. Br. J. Cancer 2018, 119, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyytinen, E.-R.; Thalmann, G.; Zhau, H.; Karhu, R.; Kallioniemi, O.; Chung, L.; Visakorpi, T. Genetic changes associated with the acquisition of androgen-independent growth, tumorigenicity and metastatic potential in a prostate cancer model. Br. J. Cancer 1997, 75, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyprianou, N.; Isaacs, J.T. Activation of Programmed Cell Death in the Rat Ventral Prostate after Castration. Endocrinology 1988, 122, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Kyprianou, N.; English, H.F.; Isaacs, J.T. Programmed cell death during regression of PC-82 human prostate cancer following androgen ablation. Cancer Res. 1990, 50, 3748–3753. [Google Scholar]
- Westin, P.; Bergh, A.; Damber, J.-E. Castration rapidly results in a major reduction in epithelial cell numbers in the rat prostate, but not in the highly differentiated dunning R3327 prostatic adenocarcinoma. Prostate 1993, 22, 65–74. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Lundmo, I.P.; Berges, R.; Martikainen, P.; Kyprianou, N.; English, H.F. Androgen regulation of programmed death of normal and malignant prostatic cells. J. Androl. 1992, 13, 457–464. [Google Scholar]
- Lamb, D.J.; Zhang, L. Challenges in Prostate Cancer Research: Animal Models for Nutritional Studies of Chemoprevention and Disease Progression. J. Nutr. 2005, 135, 3009S–3015S. [Google Scholar] [CrossRef] [Green Version]
- Newhall, K.R.; Isaacs, J.T.; Wright, G.L. Dunning rat prostate tumors and cultured cell lines fail to express human prostate carcinoma-associated antigens. Prostate 1990, 17, 317–325. [Google Scholar] [CrossRef]
- Murphy, W.M.; Soloway, M.S.; Barrows, G.H. Pathologic changes associated with androgen deprivation therapy for prostate cancer. Cancer 1991, 68, 821–828. [Google Scholar] [CrossRef]
- Westin, P.; Stattin, P.; Damber, J.E.; Bergh, A. Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am. J. Pathol. 1995, 146, 1368–1375. [Google Scholar]
- Epstein, J.I.; Zelefsky, M.J.; Sjoberg, D.D.; Nelson, J.B.; Egevad, L.; Magi-Galluzzi, C.; Vickers, A.J.; Parwani, A.V.; Reuter, V.E.; Fine, S.W.; et al. A Contemporary Prostate Cancer Grading System: A Validated Alternative to the Gleason Score. Eur. Urol. 2016, 69, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.S.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Keto, C.J.; Aronson, W.J.; Terris, M.K.; Presti, J.C.; Kane, C.J.; Amling, C.L.; Freedland, S.J. Detectable prostate-specific antigen Nadir during androgen-deprivation therapy predicts adverse prostate cancer-specific outcomes: Results from the SEARCH database. Eur. Urol. 2014, 65, 620–627. [Google Scholar] [CrossRef] [Green Version]
- Kruithof-Dekker, I.G.; Têtu, B.; Janssen, P.J.; Van der Kwast, T.H. Elevated estrogen receptor expression in human prostatic stromal cells by androgen ablation therapy. J. Urol. 1996, 156, 1194–1197. [Google Scholar] [CrossRef]
- Al-Ubaidi, F.L.T.; Schultz, N.; Loseva, O.; Egevad, L.; Granfors, T.; Helleday, T. Castration Therapy Results in Decreased Ku70 Levels in Prostate Cancer. Clin. Cancer Res. 2013, 19, 1547–1556. [Google Scholar] [CrossRef] [Green Version]
- Thompson, T.C.; Li, L. Connecting androgen receptor signaling and the DNA damage response: Development of new therapies for advanced prostate cancer. Mol. Cell. Oncol. 2017, 4, e1321167. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.-M.; Robinson, D.; Lonigro, R.J.; et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results From NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene Expression Analysis of Human Prostate Carcinoma during Hormonal Therapy Identifies Androgen-Responsive Genes and Mechanisms of Therapy Resistance. Am. J. Pathol. 2010, 164, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Lehmusvaara, S.; Erkkilä, T.; Urbanucci, A.; Waltering, K.; Seppälä, J.; Larjo, A.; Tuominen, V.J.; Isola, J.; Kujala, P.; Lähdesmäki, H.; et al. Chemical castration and anti-androgens induce differential gene expression in prostate cancer. J. Pathol. 2012, 227, 336–345. [Google Scholar] [CrossRef]
- Shaw, G.L.; Whitaker, H.C.; Corcoran, M.; Dunning, M.J.; Luxton, H.J.; Kay, J.; Massie, C.E.; Miller, J.L.; Lamb, A.D.; Ross-Adams, H.; et al. The Early Effects of Rapid Androgen Deprivation on Human Prostate Cancer. Eur. Urol. 2016, 70, 214–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinowska, K.; Neuwirt, H.; Cavarretta, I.T.; Bektic, J.; Steiner, H.; Dietrich, H.; Moser, P.L.; Fuchs, D.; Hobisch, A.; Culig, Z. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr. Relat. Cancer 2009, 16, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.K.; Erb, H.H.H.; Nappo, G.; Mann, V.M.; Simms, M.S.; Collins, A.T.; Visakorpi, T.; Maitland, N.J. Inhibition of the glucocorticoid receptor results in an enhanced miR-99a/100-mediated radiation response in stem-like cells from human prostate cancers. Oncotarget 2016, 7, 51965–51980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Hoogen, C.; Van Der Horst, G.; Cheung, H. High aldehyde dehydrogenase activity identifies tumor-initiating and me-tastasis-initiating cells in human prostate cancer. Cancer Res. 2010, 70, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, L.K.; Frame, F.M.; Maitland, N.J. Stem cells and the role of ETS transcription factors in the differentiation hierarchy of normal and malignant prostate epithelium. J. Steroid Biochem. Mol. Biol. 2017, 166, 68–83. [Google Scholar] [CrossRef]
- Jing, C. Tazarotene-Induced Gene 1 (TIG1) Expression in Prostate Carcinomas and Its Relationship to Tumorigenicity. J. Natl. Cancer Inst. 2002, 94, 482–490. [Google Scholar] [CrossRef]
- Nagpal, S.; Patel, S.; Asano, A.T.; Johnson, A.T.; Duvic, M.A.; Chandraratna, R.A. Tazarotene-Induced Gene 1 (TIG1), a Novel Retinoic Acid Receptor-Responsive Gene in Skin. J. Investig. Dermatol. 1996, 106, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Oldridge, E.E.; Walker, H.F.; Stower, M.J.; Simms, M.S.; Mann, V.M.; Collins, A.T.; Pellacani, D.; Maitland, N.J. Retinoic acid represses invasion and stem cell phenotype by induction of the metastasis suppressors RARRES1 and LXN. Oncogenesis 2013, 2, e45. [Google Scholar] [CrossRef] [Green Version]
- Nash, S.H.; Till, C.; Song, X.; Lucia, M.S.; Parnes, H.L.; Thompson, I.M., Jr.; Lippman, S.M.; Platz, E.A.; Schenk, J. Serum Retinol and Carotenoid Concentrations and Prostate Cancer Risk: Results from the Prostate Cancer Prevention Trial. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1507–1515. [Google Scholar] [CrossRef] [Green Version]
- Key, T.J.; Appleby, P.N.; Travis, R.C.; Albanes, D.; Alberg, A.J.; Barricarte, A.; Black, A.; Boeing, H.; Bueno-De-Mesquita, H.B.; Chan, J.M.; et al. Carotenoids, retinol, tocopherols, and prostate cancer risk: Pooled analysis of 15 studies. Am. J. Clin. Nutr. 2015, 102, 1142–1157. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zou, J.X.; Xue, X.; Cai, D.; Zhang, Y.; Duan, Z.; Xiang, Q.; Yang, J.C.; Louie, M.C.; Borowsky, A.D.; et al. ROR-γ drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat. Med. 2016, 22, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 2020, 368, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the Mechanisms of Androgen Deprivation Resistance in Prostate Cancer at the Molecular Level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen Receptor Variant AR-V9 Is co-expressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Mohler, J.L. A brief history of intracrine androgen metabolism by castration-recurrent prostate cancer. Am. J. Clin. Exp. Urol. 2018, 6, 101–106. [Google Scholar]
- Visakorpi, T.; Hyytinen, E.R.; Koivisto, A.P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.L.J.; Isola, J.; Kallioniemi, O.-P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Gonzalez, G.C.; Droop, A.P.; Rippon, H.J.; Tiemann, K.; Pellacani, D.; Georgopoulos, L.J.; Maitland, N.J. Retinoic acid and androgen receptors combine to achieve tissue specific control of human prostatic transglutaminase expression: A novel regulatory network with broader significance. Nucleic Acids Res. 2012, 40, 4825–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, D.A.; Rowley, D.R. The reactive stromal environment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricke, E.A.; Williams, K.; Lee, Y.-F.; Couto, S.; Wang, Y.; Hayward, S.W.; Cunha, G.R.; Ricke, W.A. Androgen hormone action in prostatic carcinogenesis: Stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 2012, 33, 1391–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb. Perspect. Med. 2018, 8, a030510. [Google Scholar] [CrossRef]
- Eder, T.; Weber, A.; Neuwirt, H.; Grünbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-Associated Fibroblasts Modify the Response of Prostate Cancer Cells to Androgen and Anti-Androgens in Three-Dimensional Spheroid Culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef] [Green Version]
- Nash, C.; Boufaied, N.; Badescu, D.; Wang, Y.C.; Paliouras, M.; Trifiro, M.; Ragoussis, I.; Thopmson, A.A. Genome-wide analysis of androgen receptor binding and transcriptomic analysis in mesenchymal subsets during prostate development. Dis. Models Mech. 2019, 12, dmm039297. [Google Scholar] [CrossRef] [Green Version]
- Cioni, B.; Zwart, W.; Bergman, A.M. Androgen receptor moonlighting in the prostate cancer microenvironment Endocrine related. Cancer 2018, 25, R331–R349. [Google Scholar]
- Hall, J.A.; Maitland, N.J.; Stower, M.; Lang, S.H. Primary prostate stromal cells modulate the morphology and migration of primary prostate epithelial cells in type 1 collagen gels. Cancer Res. 2002, 62, 58–62. [Google Scholar]
- Rane, J.K.; Droop, A.P.; Maitland, N.J. A Detailed Analysis of Gene Expression in Human Basal, Luminal, and Stromal Cell Populations from Benign Prostatic Hyperplasia Tissues and Comparisons with Cultured Basal Cells. Eur. Urol. 2017, 72, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.H.; Stark, M.; Collins, A.; Paul, A.B.; Stower, M.J.; Maitland, N.J. Experimental prostate epithelial morphogenesis in response to stroma and three-dimensional matrigel culture. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 2001, 12, 631–640. [Google Scholar]
- Wang, S.; Gao, D.; Chen, Y. The potential of organoids in urological cancer research. Nat. Rev. Urol. 2017, 14, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Sekkingstad, M.; Meloy, B.A. Heterospecific induction of prostatic development in tissue recombinants prepared with mouse, rat, rabbit and human tissues. Differentiation 1983, 24, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; Van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of Multipotent Luminal Progenitor Cells in Human Prostate Organoid Cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid Cultures Derived from Patients with Advanced Prostate Cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Grabowska, M.M.; DeGraff, D.J.; Yu, X.; Jin, R.; Chen, Z.; Borowsky, A.D.; Matusik, R.J. Mouse models of prostate cancer: Picking the best model for the question. Cancer Metastasis Rev. 2014, 33, 377–397. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nat. Cell Biol. 2017, 543, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Julio, M.K.-D.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.-P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nat. Cell Biol. 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Chua, C.W.; Epsi, N.J.; Leung, E.Y.; Xuan, S.; Lei, M.; Li, B.I.; Bergren, S.K.; Hibshoosh, H.; Mitrofanova, A.; Shen, M.M. Differential requirements of androgen receptor in luminal progenitors during prostate regeneration and tumor initiation. eLife 2018, 7, E3506. [Google Scholar] [CrossRef]
- Van Weerden, W.M.; Bangma, C.; De Wit, R. Human xenograft models as useful tools to assess the potential of novel therapeutics in prostate cancer. Br. J. Cancer 2008, 100, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, R.B.; Van Weerden, W.M.; Erkens-Schulze, S.; De Ridder, C.M.; Bangma, C.H.; Trapman, J.; Jenster, G. The Human PC346 Xenograft and Cell Line Panel: A Model System for Prostate Cancer Progression. Eur. Urol. 2006, 49, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Navone, N.M.; van Weerden, W.M.; Vessella, R.L.; Williams, E.D.; Wang, Y.; Isaacs, J.T.; Nguyen, H.M.; Culig, Z.; van der Pluijm, G.; Rentsch, C.A.; et al. Movember GAP1 PDX project: An international collection of serially transplantable prostate cancer patient-derived xenograft (PDX) models. Prostate 2018, 78, 1262–1282. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 Inhibition with Abiraterone in Castration-Resistant Prostate Cancer: Induction of Steroidogenesis and Androgen Receptor Splice Variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.; Voznesensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.; Troncoso, P.; et al. Abiraterone Treatment in Castration-Resistant Prostate Cancer Selects for Progesterone Responsive Mutant Androgen Receptors. Clin. Cancer Res. 2015, 21, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L Mutation in Androgen Receptor Confers Genetic and Phenotypic Resistance to MDV3100 (Enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [Green Version]
- Attard, G.; Borre, M.; Gurney, H.; Loriot, Y.; Andresen-Daniil, C.; Kalleda, R.; Pham, T.; Taplin, M.-E.; PLATO collaborators. Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment. J. Clin. Oncol. 2018, 36, 2639–2646. [Google Scholar] [CrossRef]
- Labrie, F.; Bélanger, A.; Dupont, A.; Luu-The, V.; Simard, J.; Labrie, C. Science behind total androgen blockade: From gene to combination therapy. Clin. Investig. Med. 1993, 16, 475–492. [Google Scholar]
- Labrie, F. GnRH agonists and the rapidly increasing use of combined androgen blockade in prostate cancer. Endocr. Relat. Cancer 2014, 21, R301–R317. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef]
- Yuan, T.-C.; Veeramani, S.; Lin, M.-F. Neuroendocrine-like prostate cancer cells: Neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr. Relat. Cancer 2007, 14, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitland, N.J.; Frame, F.M.; Polson, E.S.; Lewis, J.L.; Collins, A.T. Prostate Cancer Stem Cells: Do They Have a Basal or Luminal Phenotype? Horm. Cancer 2011, 2, 47–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.; Van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J.; et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251–41264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, J.R.; Hirst, A.M.; Droop, A.P.; Adamson, R.; Simms, M.S.; Mann, V.M.; Frame, F.M.; O’Connell, D.; Maitland, N.J. Notch signalling is a potential resistance mechanism of progenitor cells within patient-derived prostate cultures following ROS-inducing treatments. FEBS Lett. 2020, 594, 209–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.J.; Kosari, F.; Karnes, R.J.; Nasir, A.; Johnson, S.H.; Gaitatzes, A.G.; Smadbeck, J.B.; Rangel, L.J.; Vasmatzis, G.; Cheville, J.C. Retention of Interstitial Genes between TMPRSS2 and ERG Is Associated with Low-Risk Prostate Cancer. Cancer Res. 2017, 77, 6157–6167. [Google Scholar] [CrossRef] [Green Version]
- Studer, U.E.; Whelan, P.; Albrecht, W.; Casselman, J.; De Reijke, T.; Hauri, D.; Loidl, W.; Isorna, S.; Sundaram, S.K.; Debois, M.; et al. Immediate or Deferred Androgen Deprivation for Patients With Prostate Cancer Not Suitable for Local Treatment With Curative Intent: European Organisation for Research and Treatment of Cancer (EORTC) Trial 30891. J. Clin. Oncol. 2006, 24, 1868–1876. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Niu, J.; Huang, J. Neuroendocrine differentiation in prostate cancer. Am. J. Transl. Res. 2009, 1, 148–162. [Google Scholar]
- Szczyrba, J.; Niesen, A.; Wagner, M.; Wandernoth, P.M.; Aumüller, G.; Wennemuth, G. Neuroendocrine Cells of the Prostate Derive from the Neural Crest. J. Biol. Chem. 2017, 292, 2021–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.-M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.J.; et al. Platinum-Based Chemotherapy for Variant Castrate-Resistant Prostate Cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezaro, C.J.; Omlin, A.; Lorente, D.; Rodrigues, D.N.; Ferraldeschi, R.; Bianchini, D.; Mukherji, D.; Riisnaes, R.; Altavilla, A.; Crespo, M.; et al. Visceral Disease in Castration-resistant Prostate Cancer. Eur. Urol. 2014, 65, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Mottet, N.; Van Damme, J.; Loulidi, S.; Russel, C.; Leitenberger, A.; Wolff, J.M.; the TAP22 Investigators Group. Intermittent hormonal therapy in the treatment of metastatic prostate cancer: A randomized trial. BJU Int. 2012, 110, 1262–1269. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Antonarakis, E.S.; Wang, H.; Ajiboye, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci. Transl. Med. 2015, 7, 269ra2. [Google Scholar] [CrossRef] [Green Version]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Dart, D.A.; Waxman, J.; Aboagye, E.O.; Bevan, C.L. Visualising Androgen Receptor Activity in Male and Female Mice. PLoS ONE 2013, 8, e71694. [Google Scholar] [CrossRef] [Green Version]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta 2016, 1863, 1238–1260. [Google Scholar] [CrossRef]
- Marker, P.C.; Donjacour, A.; Dahiya, R.; Cunha, G.R. Hormonal, cellular, and molecular control of prostatic development. Dev. Biol. 2003, 253, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Andriole, G.L.; Bostwick, D.; Brawley, O.; Gomella, L.; Marberger, M.; Tindall, D.; Breed, S.; Somerville, M.; Rittmaster, R.; REDUCE study group. Chemoprevention of prostate cancer in men at high risk: Rationale and design of the reduction by dutasteride of prostate cancer events (REDUCE) trial. J. Urol. 2004, 172, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Unger, J.M.; Till, C.; Thompson, I.M., Jr.; Tangen, C.M.; Goodman, P.J.; Wright, J.D.; Barlow, W.E.; Ramsey, S.D.; Minasian, L.M.; Hershman, D.L. Long-term Consequences of Finasteride vs Placebo in the Prostate Cancer Prevention Trial. JNCI J. Natl. Cancer Inst. 2016, 108, djw168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, F.H.; Roobol, M.J. The REDUCE Trial. Eur. Urol. 2010, 58, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Polascik, T.; Schulman, A.; Hamdy, F.C.; Donovan, J.l.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; et al. Faculty Opinions recommendation of 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. Fac. Opin. Post-Publ. Peer Rev. Biomed. Lit. 2016, 375, 1415–1424. [Google Scholar] [CrossRef]
- Traish, A.M. Negative Impact of Testosterone Deficiency and 5α-Reductase Inhibitors Therapy on Metabolic and Sexual Function in Men. Taurine 6 2017, 1043, 473–526. [Google Scholar] [CrossRef]
- Lam, T.; Birzniece, V.; McLean, M.; Gurney, H.; Hayden, A.; Cheema, B.S. The Adverse Effects of Androgen Deprivation Therapy in Prostate Cancer and the Benefits and Potential Anti-oncogenic Mechanisms of Progressive Resistance Training. Sports Med. Open 2020, 6, 13–14. [Google Scholar] [CrossRef]
- Gong, J.; Payne, D.; Caron, J.; Bay, C.P.; McGregor, B.A.; Hainer, J.; Partridge, A.H.; Neilan, T.G.; Di Carli, M.; Nohria, A.; et al. Reduced Cardiorespiratory Fitness and Increased Cardiovascular Mortality After Prolonged Androgen Deprivation Therapy for Prostate Cancer. JACC CardioOncology 2020, 2, 553–563. [Google Scholar] [CrossRef]
- Di Mauro, M.; Tozzi, A.; Calabresi, P.; Pettorossi, V.E.; Grassi, S. Neo-synthesis of estrogenic or androgenic neurosteroids determine whether long-term potentiation or depression is induced in hippocampus of male rat. Front. Cell Neurosci. 2015, 9, 376. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Clarke, M.V.; Russell, P.K.; Rana, K.; Seto, J.; Roeszler, K.N.; How, J.M.Y.; Chia, L.Y.; North, K.; Zajac, J.D. Androgen Action via the Androgen Receptor in Neurons Within the Brain Positively Regulates Muscle Mass in Male Mice. Endocrinology 2017, 158, 3684–3695. [Google Scholar] [CrossRef]
- Olsen, N.J.; Olson, G.; Viselli, S.M.; Gu, X.; Kovacs, W.J. Androgen Receptors in Thymic Epithelium Modulate Thymus Size and Thymocyte Development. Endocrinology 2001, 142, 1278–1283. [Google Scholar] [CrossRef]
- Narayanan, R.; Coss, C.C.; Dalton, J.T. Development of selective androgen receptor modulators (SARMs). Mol. Cell. Endocrinol. 2018, 465, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Castration Resistant, Non-metastatic Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Cella, D.; Basch, E.M.; Hadaschik, A.B.; Mainwaring, P.N.; Oudard, S.; Graff, J.N.; McQuarrie, K.; Li, S.; Hudgens, S.; et al. Effect of apalutamide on health-related quality of life in patients with non-metastatic castration-resistant prostate cancer: An analysis of the SPARTAN randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1404–1416. [Google Scholar] [CrossRef]
- Kwon, E.D.; Drake, C.G.; Scher, I.H.; Fizazi, K.; Bossi, A.; Eertwegh, A.J.M.V.D.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Ofengeim, D.; Giagtzoglou, N.; Huh, D.; Zou, C.; Yuan, J. Single-Cell RNA Sequencing: Unraveling the Brain One Cell at a Time. Trends Mol. Med. 2017, 23, 563–576. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class | Molecular Target | Drug Example | Escape Mechanism | Ref. |
|---|---|---|---|---|
| Androgen synthesis: serum testosterone levels | Pituitary gonadotrophin secretion. Luteinizing hormone releasing hormone agonist (LHRH) | Zoladex | Intratumoral androgen synthesis AR gene amplification to utilize low intratumoral androgens | [19,20,21] |
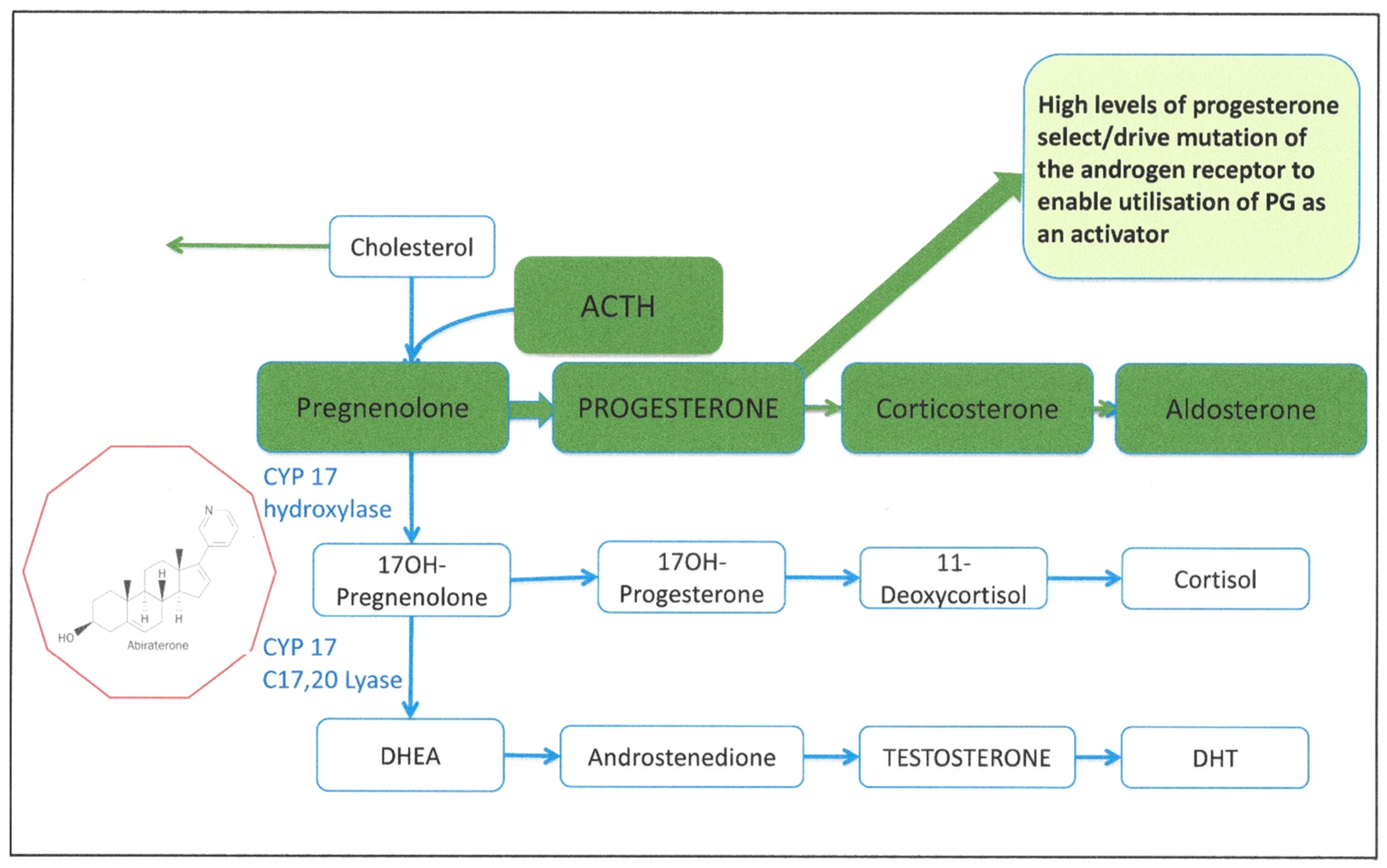
| Androgen synthesis | Intratumoral CYP17 | Abiraterone | Use of glucocorticoid receptor and glucocorticoids | [22,23,24] |
| Testosterone activation to dihydrotestosterone (DHT) | Steroid 5-alpha reductase | Dutasteride/Finasteride | Switch in 5aR isotype or use of testosterone/adrenal androgens | [25,26] |
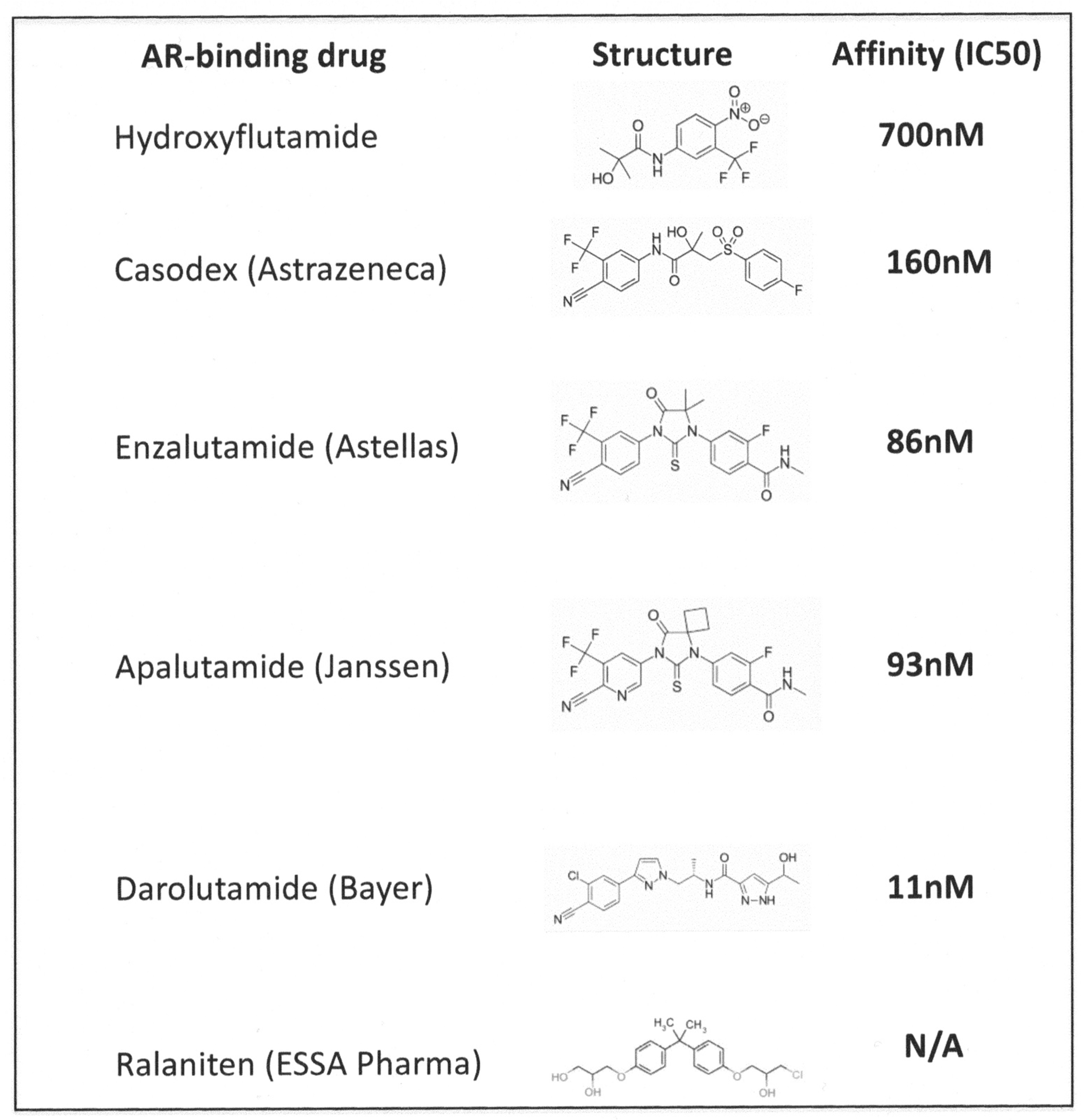
| Androgen Receptor (AR) inhibitor | DHT binding to monomeric androgen receptor | Steroidal: Estrogen, Cyproterone acetate Nonsteroidal: Casodex, Enzalutamide | Gain of function mutations in the androgen receptor gene to enable it to use other steroid hormones. Bypassing of the AR by ligand independent activation of androgen responsive genes | [27,28,29,30,31,32] |
| Dimerization of AR | Androgen receptor | Stilbene, Resveratrol | Mutation of the AR gene and expression of ligand-independent splice variants | [33,34,35] |
| Phosphorylation of androgen receptor | AR S81: CDK1, CDK5 and CDK9 AR S213, S791 and T850: PIM1 and Akt AR S213: CXCR4 | Roscovitine, 5,6-dichloro-1-b-D-ribo-furanosylbenzimidazole (DRB) and flavopiridol SGI-1776, TP-3654 (PIM1) Miltefosine, Perifosine, MK-2206 and GSK-2141795 (Akt) | Redundancy in kinase usage | [36,37] |
| Nuclear translocation of androgen receptor | Androgen receptor | CH5137291, Diallyl Trisulfide Enzalutamide | Expression of AR splice variants which translocate to the nucleus in the absence of androgen. | [38,39] |
| Binding of nuclear androgen receptor to: (1) Recognition sites on DNA; (2) Co-activator molecules | Androgen receptor and co-activators | AR DNA Binding Domain: 4- (4-phenylthiazol-2-yl) morpholine AR interaction interface: LXXLL/FXXLF motifs | Changes in balance between AR co-activator and co-repressor molecules and relative affinity of the receptor by mutation of AR. | [40,41,42] |
| Downstream effects of proteins stimulated by AR activity | Androgen response molecules | Multiple | Activation of alternative salvage pathways which stimulate the signaling molecules downstream from AR activation. | [43,44,45,46] |
| AR stability and degradation | Heat shock proteins (HSP90) Serine proteases, caspases and calpain | LAQ824, a cinnamyl hydroxamatic acid histone deacetylase inhibitor 17-allyamino-17-demethoxygeldanamycin (17-AAG) Multiple | Redundancy in the heat-shock chaperone system Inhibition of AR proteolysis | [47,48,49,50,51] |
| Study | Treatment | Molecular Outcomes | Year |
|---|---|---|---|
| 1 | 3 months Etylamide (LHRH)+Flutamide | 21 patients by immunohistochemistry–only studied (estrogen receptor alpha) ESR1: Intense Stromal ESR+ and normal sporadic basal cells, NOT in CaP cells. No Ku70 expression | 1996 [105] |
| 2 | 3 months Goseralin (GnRH)+Flutamide | Transcriptional study: (290/364 *) AR-regulated genes repressed. No ESR changes (one gene) but SMARCD, ETS2, IL6, ALDH and RARRES1 stimulated. AR expression increased in CRPC only. No Ku70 expression. | 2004 [109] |
| 3 | 12 weeks Goseralin (GnRH) or Bicalutamide | Transcriptional study: (B 97 and G 62) and (B+G 89) changed by >2 fold. 24/128 genes directly AR regulated. Some overlap (16%) within study but little with others, no ESR1 changes, but RARRES1 upregulated. No KU70. | 2012 [110] |
| 4 | 2 months Leuprolerin (GnRH) | Studied DNA damage repair after treatment specific for Ku70 and gamma H2AX. Ku70 decreases with castration mirroring PSA but no grade-specific changes. | 2013 [106] |
| 5 | 7 days Deregalix (LHRH) | Initial transcriptional analysis (749/908). Estrogen receptor upregulation (E+S) in cancers + normal basal cell gene expression (RARRES1). No KU70. | 2016 [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maitland, N.J. Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced? Cancers 2021, 13, 327. https://doi.org/10.3390/cancers13020327
Maitland NJ. Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced? Cancers. 2021; 13(2):327. https://doi.org/10.3390/cancers13020327
Chicago/Turabian StyleMaitland, Norman J. 2021. "Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced?" Cancers 13, no. 2: 327. https://doi.org/10.3390/cancers13020327
APA StyleMaitland, N. J. (2021). Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced? Cancers, 13(2), 327. https://doi.org/10.3390/cancers13020327