Simple Summary
AML is a heterogenous malignancy with a variety of underlying genomic abnormalities. Some of the genetic aberrations in AML have led to the development of specific inhibitors which were approved by the Food and Drug Administration (FDA) and are currently used to treat eligible patients. In this review, we describe five gene mutations for which approved inhibitors have been developed, the response of AML patients to these inhibitors, and the known mechanism(s) of resistance. This review also highlights the significance of developing function-based screens for target discovery in the era of personalized medicine.
Abstract
Acute myeloid leukemia (AML) is a highly heterogeneous malignancy characterized by the clonal expansion of myeloid stem and progenitor cells in the bone marrow, peripheral blood, and other tissues. AML results from the acquisition of gene mutations or chromosomal abnormalities that induce proliferation or block differentiation of hematopoietic progenitors. A combination of cytogenetic profiling and gene mutation analyses are essential for the proper diagnosis, classification, prognosis, and treatment of AML. In the present review, we provide a summary of genomic abnormalities in AML that have emerged as both markers of disease and therapeutic targets. We discuss the abnormalities of RARA, FLT3, BCL2, IDH1, and IDH2, their significance as therapeutic targets in AML, and how various mechanisms cause resistance to the currently FDA-approved inhibitors. We also discuss the limitations of current genomic approaches for producing a comprehensive picture of the activated signaling pathways at diagnosis or at relapse in AML patients, and how innovative technologies combining genomic and functional methods will improve the discovery of novel therapeutic targets in AML. The ultimate goal is to optimize a personalized medicine approach for AML patients and possibly those with other types of cancers.
1. Introduction
The introduction of imatinib as a tyrosine kinase inhibitor (TKI) targeting BCR-ABL1 revolutionised the treatment of patients with chronic myeloid leukaemia (CML), paving the path for the development of other targeted inhibitors in various types of cancers [1]. The development of imatinib was based on the concept that targeting the driver of malignancy (the BCR-ABL1 oncoprotein) rather than the consequence (proliferation, the target of standard chemotherapies) should control the disease without damaging normal cells that do not express the driver mutation [2]. Before imatinib, CML was a malignancy associated with early death from progression to acute leukaemia or from side effects associated with bone marrow transplantation, which was the only curative option. However, nearly two decades of treating CML patients with imatinib has been a great success for this novel targeted therapy [3]. The success of managing CML using targeted inhibitors has continued with the development of second and third generation BCR-ABL1 TKIs, which help clinicians to manage emerging resistance to imatinib [3]. In some patients, imatinib is so effective at suppressing leukaemia that patients maintain their deep molecular response even after they discontinue therapy (Stop Imatinib trial) [4].
The promising data from targeting BCR-ABL1 in CML encouraged the exploration of novel inhibitors for other cancers with known oncogenic drivers [5]. However, to date, the development of targeted inhibitors for other malignancies has not proven as successful as imatinib, partly due to the presence of several genomic abnormalities contributing to the development of most other types of cancers. In acute myeloid leukaemia (AML), for instance, an average of 13 genetic abnormalities can be observed per patient, and the heterogeneity of the disease along with ambiguity of the main driver among the detected mutations may explain why the development of novel inhibitors for AML has not been as successful [6]. To date, specific inhibitors have now been developed and used in clinical practice for certain subtypes of AML for which there is a well-known oncogene at the centre of disease pathogenicity, such as promyelocytic leukemia/retinoic acid receptor-alpha (PML/RARα) and FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD), or AML patients with mutations in isocitrate dehydrogenase isozymes 1 or 2 (IDH1, IDH2) [7,8]. In fact, AML patients with PML/RARα are curable with a combination of arsenic trioxide (As2O3) and all-trans retinoic acid (ATRA), which have shown higher success rates compared with other AML subtypes [9]. Although there has been promising data regarding the usage of FLT3 inhibitors in combination with azacytidine for the management of FLT3-ITD+ in AML patients, these inhibitors have not improved survival to the same level as imatinib in managing CML or ATRA in managing acute promyelocytic leukaemia (APL).
Because of the observed heterogeneity, AML has been classified into different subtypes, which provide information regarding probable outcome and treatment response. The World Health Organization (WHO) classification system has classified AML into the following subtypes: AML with recurrent genetic abnormalities, which include subtypes with recurrent cytogenetics or molecular genetics abnormalities such as acute promyelocytic leukemia (APL) with PML-RARA, AML with mutated NPM1, AML with biallelic mutations of CEBPA, etc.; AML with myelodysplasia-related changes; therapy-related myeloid neoplasms; AML not otherwise specified (NOS), which includes cases that do not fall into any of the other groups (such as pure erythroid leukemia), acute monoblastic/monocytic leukemia, etc.; myeloid sarcoma (also known as granulocytic sarcoma or chloroma) and myeloid proliferations related to Down syndrome [10]. Heterogeneity of AML is the main obstacle to developing specific inhibitors for clinical management. Thus, personalised medicine offers an ideal approach for successful management of this disease [11]. The principle of personalised medicine is to identify the main pathway(s) that are essential for survival of the leukaemia cells and to target them in a patient-specific manner. The identification of these pathways can be achieved through genomic techniques such as RNA sequencing or whole exome sequencing. However, without functional studies, genomic techniques only provide information on the genetic aberrations and lack the power to characterise the activated signalling pathways that are responsible for the disease phenotype. In this review, we discuss the underlying genomic abnormalities which make AML patients eligible for targeted therapy and also highlight additional alterations that drive resistance to targeted therapies. The cited AML studies are mainly from adult AML patients. Additionally, we will briefly discuss how functional genomics might be used for identification of potential therapeutic targets in a personalised medicine approach for the clinical management of AML patients.
2. PML-RARA
APL is one subtype of AML that is observed in nearly 12% of AML patients [12]. It is characterised by a translocation between the retinoic acid receptor-alpha (RARA) gene on chromosome 17 with the promyelocytic leukemia (PML) gene on chromosome 15, known as t(15,17)(q24, q12) in nearly 99% of APL cases. The RARA gene has also been shown to fuse with other partner genes in rare cases. The PML-RARA fusion is classified into three main categories depending on the breakage point on PML, and consequently the isoform of the resulting mRNA fusion (Figure 1 and Table 1).
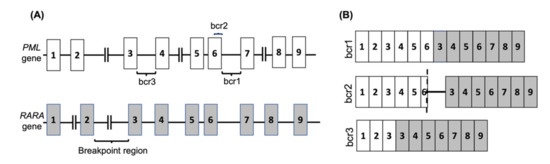
Figure 1.
The PML-RARA fusion gene in APL. (A) The common breakpoint regions on the PML gene are shown, with the most common break occurring between exons 6 and 7 (bcr1) or exons 3 and 4 (bcr3), whereas the RARA gene breakpoint can occur anywhere within the 15kb intronic region between exons 2 and 3. In 5% of PML cases the breakage occurs in exon 6 (bcr2). (B) The common PML-RARA fusion mRNAs are shown. The brc2 variant mRNA contains a variable insertion from RARA intron 2 located between part of PML exon 6 (shown by dash) and RARA exon 3.

Table 1.
The frequency of various fusions of RARA and sensitivity to targeted therapy [13,14,15,16,17,18].
The current standard induction treatment for APL with ATRA and anthracycline, followed by at least two additional cycles of consolidation therapy, has been shown to lead to complete remission (CR) in 95% of APL patients. ATRA simultaneously activates the transcription of genes essential for myeloid differentiation while also inducing the degradation of the PML-RARα protein [7]. The other agent which was shown to be an effective therapy in APL is As2O3. The significance of As2O3 is its effectiveness even in patients who relapse following ATRA/chemotherapy, as up to 80% of such cases have been shown to achieve complete remission following As2O3 therapy [19,20,21]. As2O3 induces the production of reactive oxygen species (ROS), which in turn causes multimerization of PML-RARα through intermolecular disulphide crosslinks at the PML B1-domains; binding of As2O3 to the C-C motif in PML-B2 was shown to be crucial for multimerization. ROS leads to increased ubiquitin carrier protein 9 (UBC9) binding to the PML RING domain, which increases PML-RARα SUMOylation and the recruitment of ring finger protein 4 (RNF4). This ultimately leads to polyubiquitination of PML-RARα protein and its subsequent degradation by the ubiquitin-proteasome system [22,23,24,25]. Response to treatment is usually evaluated by measuring PML-RARA mRNA transcripts using quantitative reverse transcription polymerase chain reaction (qRT-PCR), which is expected to be negative following consolidation therapy. Failure to achieve a negative PCR result post-consolidation (confirmed by two tests with two-week intervals) is considered primary resistance and necessitates additional therapeutic measures.
RARα is a transcription factor which, upon activation, binds to a specific element (RARE) in the promoter of its target genes. In the absence of the inducing ligand, RARα forms a heterodimer with the retinoic X receptor (RXR). This heterodimer recruits histone deacetylases (HDACs) to the promoter site, and by deacetylation of the histones, this leads to condensation of the RARα target genes. Under physiological conditions, retinoic acid (RA) can disrupt the RARα-RXR complex and release the HDAC, thereby recruiting activating factors to the promoter site, resulting in the transcriptional activation of target genes. In the event of PML-RARα formation, this complex binds to PML and other chimeric PML-RARα proteins (homo-and hetero-dimerization). In addition to HDACs, it also recruits DNA methylating enzymes such as DNA methyltransferases (DNMT3A and DNMT3B) to the promoter site, which methylates the promoter in addition to deacetylating the histones, leading to stronger target gene suppression. The co-repressors of the PML-RARα complex are not released by physiological levels of RA and are only released by ATRA. In the presence of ATRA, the co-repressor complex loses its ability to bind RARα, providing an opportunity to associate with the co-activator complex for the activation of gene transcription. Figure 2 shows the mechanism of action for ATRA treatment of PML-RARA-positive APL [26,27,28,29,30,31,32].
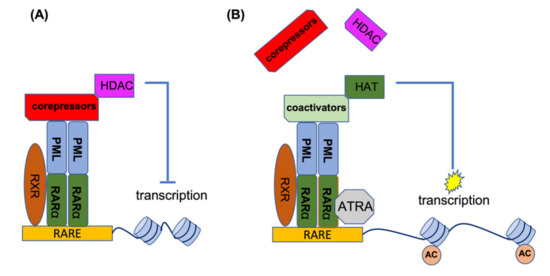
Figure 2.
ATRA mechanism of action for the treatment of APL patients. (A) In the absence of ATRA, PML-RARα binds to RARE elements to recruit co-repressors and HDACs for suppression of gene transcription. (B) ATRA disrupts the co-repressor complex and provides an opportunity for the association of co-activator complexes with RARα, leading to histone acetylation and the activation of gene transcription. AC, acetylation of histones.
Primary clinical resistance, defined as failure to achieve complete remission on ATRA therapy, is very rare. However, secondary resistance has been observed in 10–30% of relapsed cases following complete remission [33]. A variety of mechanisms have been described for resistance to ATRA following relapse. However, for this review, we will only highlight the genomic alterations associated with resistance. As described above, the co-repressor complex is dissociated from PML-RARα in the presence of ATRA. However, mutations in the ligand binding domain (LBD) of the RARA gene can interrupt this interaction and are considered a mechanism of resistance to ATRA. Mutations in the RARA LBD in PML-RARα have been observed through clinical observation in resistant patients, and their resistance to ATRA has been validated through in vitro analysis of cell line models (Table 1) [13,34,35,36]. There are a few studies showing mutated genes at relapse following ATRA therapy. In one study, the APL cells with diagnostic mutations in WT1 (D367_R369del, Q259*R458* K400* H465N), activating mutations of MAP kinase pathway genes (BRAF, KIT, PDGFRA), and inactivating mutations in the genes regulating transcription or epigenetics (NSD1, ASXL1, MED12, KDM6A) were observed in relapsed cells at a higher frequency [37]. At relapse, mutations of these genes were detected along with additional mutations which were not present at diagnosis. These mutations were either acquired during the progression of the disease toward relapse, or were present at low levels at diagnosis as a subclone and selected during the course of therapy (Table 2). In some FLT3-ITD+ APL patients, it was observed that the presenting clone at relapse lost the FLT3-ITD mutations or other passenger mutations that were present at diagnosis, indicating the existence of a pre-leukemic PML-RARA-expressing clone that survived RA/chemotherapy and reinitiated APL [37]. The combination of ATRA and As2O3 has greatly reduced the rate of relapse and improved the lives of APL patients [38], and represents one of the most successful targeted cancer therapies available today, second in line only to TKI treatment in CML.
Table 2.
The mutated genes at relapse in APL patients treated with ATRA [34,37,39].
3. FLT3
The FLT3 gene is located on chromosome 13 and has 24 exons, with translation starting from the middle of exon 1 and continuing through part of exon 24. The resulting human FLT3 protein is composed of 993 amino acids, with three main sections relating to the membrane, extracellular, and intracellular domains. The extracellular domain is composed of the signal peptide (encoded by exons 1 and 2 and is 26 amino acids long), which is cleaved off during processing, and also an extracellular immunoglobulin-like (Ig-like) domain (encoded by exons 3–12). The transmembrane domain (between amino acids 542 and 564) is encoded by exon 13, whereas the intracellular region, including the juxtamembrane and kinase domains (between amino acids 610 and 944, including the kinase insert of nearly 50 amino acids), is encoded by exons 14–23 (Figure 3) [40].
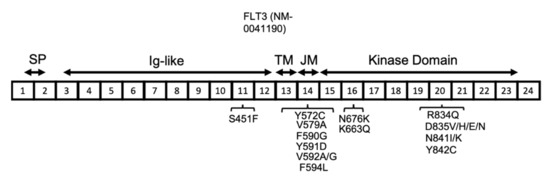
Figure 3.
FLT3 protein structure in AML. FLT3 protein has a signal peptide (SP) at the NH2-terminal, followed by five extracellular Ig-like domains, a transmembrane (TM) domain, a juxta-membrane (JM), and a kinase domain. The JM and kinase domains are separated into two parts by a short region named the kinase insert (KI), which is not shown here. The location of the reported FLT3 activating mutations are shown in association with their related exons (numbered 1–24).
Human FLT3 is predominantly expressed in hematopoietic stem and progenitor cells, both myeloid and lymphoid, and has not been observed in mature cells, highlighting its significance for the early stages of hematopoietic development [40,41,42]. Several transcriptional factors, including HOXA9, MEIS1, PBX1, PBX3, CEBPα, MYB, and PAX5, regulate transcription of FLT3 in both mouse and human cells [40]. The FLT3 protein goes through glycosylation in the endoplasmic reticulum and Golgi apparatus before it trafficks to the membrane [40,43,44]. Additional post-translational modifications known to affect FLT3 protein are phosphorylation and ubiquitination. Several tyrosine residues, including tyrosines 572, 589, 591, 599, 726, 768, 793, 842, 955, and 969 are phosphorylated during the ligand-induced activation of FLT3 [45,46,47]. In the monomeric membrane state, the FLT3 protein remains inactive, and this inactive state is maintained through interactions between the juxtamembrane and kinase domains [48]. This interaction blocks ATP from accessing the kinase domain site, preventing the phosphorylation and activation of tyrosine residues. However, the binding of the FLT3 ligand to FLT3 leads to dimerization and loss of interaction between the juxtamembrane and kinase domains, allowing ATP to access the tyrosine site for autophosphorylation. Tyrosine phosphorylation is the ultimate marker of FLT3 activation [48].
The majority of FLT3 mutations have been observed in the juxtamembrane domain (JMD) and the adjacent tyrosine kinase domain (TKD) of the FLT3 protein, and are mainly comprised of insertion in-frame mutations, known as internal tandem duplication (ITD). Nearly 70% of ITD mutations have been reported to occur within the JMD, and the other 30% were identified mainly within the TKD [49,50]. Comparing the outcome of AML patients with FLT3-ITD suggests differences between the prognostic significance of JMD-ITD and TKD-ITD [51,52,53]. A recent analysis of a large population of patients from the RATIFY trial with FLT3-ITD mutations showed inferior outcomes for patients with TKD-ITD compared with JMD-ITD [53]. The ITD mutations disrupt the interactions between the JMD and the activation loop, leading to availability of ATP to the tyrosine residues and their phosphorylation, resulting in constitutive activation of the protein kinase domain [48,54].
Reports from several groups have shown that mutational load (measured by the ratio of mutant FLT3/non-mutant FLT3) has significant clinical relevance, with a higher ratio associating with poor prognosis [55,56,57]. The allelic ratio for FLT3-ITD refers to the number of ITD-mutated alleles compared with the number of wild-type alleles. AML patients with FLT3-ITDs have been shown to have poor outcomes by several large studies, particularly for patients with a high allelic ratio (≥0.5) [51,58,59]. Most of the FLT3-ITD mutations in the JMD have been reported in a tyrosine-rich region from codon 589 to 599 [40,49,60]. At a much lower frequency, ITD can also occur near the beginning of the TKD (Figure 3) [50]. ITD mutations account for 25% of the mutations in AML patients, with the size of the duplication ranging from 3 to 1236 bp. A large study of FLT3-ITD-positive AML patients showed simple duplication and/or insertions of unknown origin [61]. The same study showed some patients had 2-3 independent FLT3-ITDs in the region of exons 13–15. There is no general agreement on the prognostic significance of the FLT3-ITD, but the level of FLT3-ITD is commonly recognised as a prognostic factor for relapse in AML patients [57]. Recently, a novel mutation of a single amino acid residue in the FLT3 JMD (Q575D) which activated the FLT3 kinase in a manner similar to ITD was reported in AML patients [62]. This is an important finding, as identification of more patients with such mutations will influence the current diagnostic methods for detection of FLT3 mutations, and will recommend screening of the JMD by next generation sequencing in addition to fragment analysis [57]. In most of the AML patients with FLT3-ITD at diagnosis, the FLT3-ITD mutation is detected at a higher allelic burden at relapse [63]. However, other patterns have also been observed during the progression of the disease from diagnosis to relapse. Nearly 20% of patients with AML acquire either a newly detectable FLT3-ITD or FLT3-TKD mutation at relapse or lose the FLT3 mutation from the time of diagnosis [64].
FLT3 point mutations have also been reported among AML patients at a lower frequency compared with ITD (7% of AML patients) [40,61]. In contrast to ITD mutations, the prognosis of AML patients with FLT3 point mutations is more favourable [65]. The most common codon affected by point mutation is D835, located near the activation loop, which can undergo a variety of mutations, including D835V, H, E, and N, all of which lead to FLT3 activation. The other less common activating point mutations in FLT3 include S451F, Y572C, V579A, F590G, Y591D, V592A, V592G, F594L, K663Q, N676K, R834Q, N841I, N841K, and Y842C (Figure 3) [40,66,67,68,69,70,71,72]. The type of mutation influences the downstream signalling pathways that are activated. The FLT3-ITD protein is mainly located in the endoplasmic reticulum (ER), whereas the FLT3 D835Y mutant is mainly located to the plasma membrane. The subcellular localization of FLT3 influences the downstream signalling pathways that are activated. For instance, the FLT3 protein, when located at the plasma membrane, stimulates proteins such as PI3K/AKT and mitogen-activated protein kinase (MAPK), whereas FLT3 with ITD mutations primarily activates the STAT5 signalling pathway [73,74].
Several inhibitors targeting FLT3 have been developed, and based on the underlying mechanism of FLT3 inhibition, they have been categorised into two classes [75]. The class I multi-kinase inhibitors include midostaurin, gilteritinib, and crenolanib, among which only midostaurin has been FDA and EMA (European Medicines Agencies)-approved for treatment of AML patients with activating FLT3 mutations (in combination with cytarabine and daunorubicin) [40]. Gilteritinib, on the other hand, is approved for the treatment of relapse or for refractory AML patients [76]. The common feature of class I inhibitors is their binding to the “gatekeeper” domain adjacent to the activation loop or the ATP-binding domain in both active and inactive receptor conformations [75,77]. Class II inhibitors, such as sorafenib, quizartinib, and ponatinib, bind to the hydrophobic region of FLT3 (ATP-binding domain) in its inactive conformation and were developed to improve drug specificity [78].
Although the response to FLT3 inhibitors has been promising, the duration of response remains short, which is mainly due to the emergence of resistant clones [76,79,80,81]. The mechanism of evolving resistance to FLT3 inhibition has many features in common with the mechanisms of resistance to TKIs in other types of leukaemia. Usually at the time of resistance, there is a dominant clone, shown in several cases to be present as a minor clone at diagnosis, but selected and expanded at the expense of other clones that are suppressed during treatment. However, resistant clones might also develop during treatment as a result of acquiring new mutations [82,83]. Resistance may be due to point mutations that disrupt drug binding, or activation of alternative pro-survival signalling pathways in spite of FLT3 inhibition. According to a recent model proposed by Joshi et al., the mechanism of resistance to the FLT3 inhibitor gilteritinib changes during the evolution of resistance. Early resistance is mainly due to protective factors from the bone marrow microenvironment, metabolic adaptation, slower growth, and dependence on Aurora kinase B, while late resistance is due to mainly intrinsic mechanisms such as mutations in the RAS pathway and the continuation of altered metabolism. Available data from different studies suggests the involvement of various factors in the development of resistance, including both intrinsic and extrinsic factors [84].
Alterations of the FLT3 gene, such as certain FLT3 kinase domain mutations, have been predicted to be resistant to some of the FLT3 inhibitors based on in vitro models, or have been observed in the resistant samples while they were absent from the diagnostic sample [85]. The mutations causing resistance to a drug of one class are highly likely to demonstrate resistance to other drugs of the same class due to common features of drug binding, but may remain sensitive to the drugs from the other class due to the difference in the binding site [86]. Class I FLT3 inhibitors are less specific for FLT3 and can bind the active and inactive conformation of the FLT3 protein. This explains the low frequency of FLT3 mutations as a mechanism of resistance to this class of FLT3 inhibitors. This is in contrast with the pattern of resistance observed in the class II FLT3 inhibitor quizartinib, which has low inhibitory activity against FLT3-TKD mutations at therapeutic doses [78,87]. Mutations of the FLT3-TKD D835 or I836 amino acid were shown to be the main FLT3 mutations associated with resistance to class II FLT3 inhibitors [88]. The available data suggests that nearly one third of patients who developed resistance to FLT3 inhibition have these mutations in FLT3 at the time of relapse [85,88,89]. A recent investigation of 67 FLT3-ITD+ AML patients who were treated with class I or II inhibitors demonstrated that more than half of the patients had detectable mutations at relapse, from which 26% had mutations in the TKD of FLT3 (D835). Interestingly, mutations of the FLT3 TKD were more common in those treated with class II inhibitors [89]. A study by the Austrian-German Acute Myeloid Leukemia Study Group (AGAMLS) of FLT3-ITD+ AML patients who were treated with midostaurin and either relapsed or had refractory response, showed loss of FLT3-ITD mutation in nearly half of the patients. In patients who were still positive for FLT3-ITD, the clone was different from the dominant diagnostic clone, as demonstrated by mutations in other genes, suggesting that mechanisms other than FLT3 kinase activity contribute to relapse or refractory responses to midostaurin [83].
In more than 50% of cases, no FLT3 mutation is detected upon the onset of clinical TKI resistance, suggesting that mutations in other genes or pro-survival signals from the bone marrow microenvironment provides alternative survival pathways. Mutations in epigenetic modifiers, the RAS/MAPK pathways WT1, and TP53, have been observed in resistant clones following FLT3 targeted therapy [89,90,91]. The FLT3 molecule can also be inhibited or inactivated through mechanisms that are alternative to the inhibition of FLT3 kinase activity, and these mechanisms might serve as the basis for future treatment of AML patients [40]. As glycosylation of FLT3 is required for its maturation, inhibition of this process using agents such as Fluvastatin have been shown to inhibit FLT3 activity using in vitro and in vivo models [92]. Inhibiting HSP90, which is required for stabilization of FLT3-ITD, might be another approach, as inhibition of HSP90 has been shown to degrade FLT3-ITD through polyubiquitination and the proteasome machinery [93]. Another approach for suppressing FLT3-ITD and not wild-type FLT3 involves the proteasome pathway through stimulation of the E2 ubiquitin ligase, UBCH8, using the HDAC inhibitor LBH589 [46]. These recent developments in understanding the mechanisms of resistance to FLT3 inhibitors provides the opportunity for developing new potential therapies to improve the outcome of the FLT3-ITD AML patients.
4. BCL-2
The B-cell leukemia/lymphoma 2 gene (BCL-2) is located on the long arm of chromosome 18, has three exons, and encodes a 26 KD protein consisting of 239 amino acids. The C-terminus of the protein is highly hydrophobic, which leads to its dominant localization within the mitochondrial outer membrane. However, it also localises to the nuclear envelope and the membrane of the endoplasmic reticulum [94]. The BCL-2 protein is expressed by hematopoietic lineages and various other normal tissues such as the epithelium and neurons [95].
BCL-2 belongs to the BCL-2 family of proteins, which is divided into pro-apoptotic and anti-apoptotic families. The pro-apoptotic proteins can be BH3-only proteins or pro-apoptotic multi-domain effector proteins (BAX and BAK). The mechanism of action for the pro-apoptotic family members is through inducing mitochondrial outer membrane permeabilization (MOMP). The BH3-only proteins include BID, BIM, BMF, PUMA, BAD, BIK, HRK, and NOXA [96,97,98]. The BH3 domain of these proteins binds to a hydrophobic groove on anti-apoptotic members of the BCL-2 family to negate their anti-apoptotic action [99]. Binding of the BH3 domain to anti-apoptotic proteins releases BAX and BAK (effectors), which enable them to oligomerise within the mitochondrial outer membrane for MOMP. The other mechanism of apoptosis induction by the BH3-only proteins is to activate BAX and BAK directly [100,101]. The anti-apoptotic protein members include BCL-2, BCL-XL, myeloid cell leukemia sequence 1 (MCL-1), BCL-w, and BFL-1/A1. BCL-2 prevents MOMP by sequestering the pro-apoptotic proteins [102]. This action of BCL-2 and other anti-apoptotic members of this family blocks the release of cytochrome c, a hallmark of mitochondrial apoptosis [103], which consequently prevents apoptosis activation and increases cell survival [104,105]. This anti-apoptotic activity contributes to cancer development and also resistance to chemotherapy, as cancer cells often control the death mechanisms by elevating the expression of BCL-2, BCL-XL, and MCL-1, which neutralises the apoptotic action of the BH3-only proteins of the BCL-2 family [106].
The viability of AML cells depends on BCL-2, and inhibition of BCL-2 results in the death of the AML cells [107]. BCL-2 has been shown to be overexpressed in CD34+ AML cells [108] and associated with poor prognosis and resistance to chemotherapy [109,110]. The higher dependency of AML CD34+ cells on BCL-2 has led to therapies based on targeting BCL-2, which might spare normal haematopoietic stem cells (HSC) which were shown to depend more on MCL-1 for their survival [111,112,113]. ‘BH3-mimetic’ inhibitors directly bind to BCL-2 family anti-apoptotic proteins by mimicking the BH3 domain of pro-apoptotic proteins, and as a consequence of this action they antagonise BCL-2 family anti-apoptotic proteins [114]. Venetoclax is a potent and selective inhibitor of BCL-2 and was approved in 2018 by the FDA and later by EMA in combination with either DNA methyltransferase inhibitors (DNMTi’s) or low-dose cytarabine (LDAC) in older or unfit AML patients [115,116]. Venetoclax specifically targets BCL-2 and has a very low affinity to other members of the anti-apoptotic BCL-2 family, like BCL-XL and BCL-W; therefore, this compound does not have any adverse action on platelets [117]. Through a different mechanism, venetoclax in combination with azacytidine has been shown to reduce the leukemia stem cell population in AML by decreasing amino acid uptake and consequently reducing oxidative phosphorylation (OXPHOS), which is essential for LSC survival [118,119,120]. Despite promising responses in various studies, primary and adaptive resistance to venetoclax monotherapy has been observed in AML patients [115]. The success of achieving complete remission with venetoclax monotherapy in relapse or refractory AML was reported to be 19% and in combination therapy between 30–54%; in newly diagnosed patients, nearly 30% do not achieve remission with combination therapy [118,121].
Expression of BCL-2 is essential for the efficacy of venetoclax, and the expression shift from BCL-2 to the other anti-apoptotic family members MCL-1 or BCL2L1 can cause venetoclax resistance [113,115,122]. The reliance of monocytic AML cells on MCL-1 rather than BCL-2 seems to be the underlying reason for selection of a pre-existing monocytic subpopulation at the time of relapse following ven/aza therapy [123]. The functional genomics screen of the AML cell line model, MOLM13, has shown the inactivation of the TP53 and BAX genes as key elements of venetoclax resistance, explaining venetoclax insensitivity in samples from AML patients who have low expression of these two genes [124]. These data suggest that the mechanism of resistance to venetoclax is through replacement of BCL-2 activity by another anti-apoptotic family member or by inactivation of some major apoptotic genes.
Resistance has also been shown to occur through altering the metabolic target of venetoclax. Untreated LSCs rely on amino acid metabolism as the main source of OXPHOS, and shifting the metabolism toward fatty acid oxidation is considered a mechanism of developing resistance in LSCs at relapse [118]. Mutations of the RAS pathway (PTPN11, KRAS, NRAS) were found to be associated with poor response to ven/aza therapy [118], and this association might be due to their role in enhancing fatty acid metabolism, as demonstrated in lung cancer [125]. The altered metabolism in relapsed AML LSCs compared with de novo AML seems to be responsible for poor or lack of response to ven/aza [126]. The elevated metabolism of nicotinamide in relapsed AML was demonstrated to be responsible for resistance to ven/aza, as it enhances the activity of tricarboxylic acid (TCA) cycle enzymes, including isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, and malate dehydrogenase, leading to increased metabolism of not only amino acids but also fatty acids. The uptake of nicotinamide in relapsed LSC was demonstrated to be increased and converted to NAD+ by nicotinamide phosphoribosyltransferase (NAMPT), and the inhibition of NAMPT overcame resistance to ven/aza in relapsed LSCs in vitro [126]. A summary of molecular markers associated with response or resistance to venetoclax is summarised in Table 3.
Table 3.
Molecular markers associated with development of resistance or prediction of response to venetoclax [115,118,121].
In summary, BCL2 seems to be a novel therapeutic target for AML patients with promising findings through clinical studies. The recent data on the mechanism of venetoclax action highlights the diversity in the pathways through which this drug inhibits AML cells, how metabolic alterations play a role in development of resistance and the requirement for combining venetoclax with other inhibitors to improve the clinical outcome and reduce the rate of relapse.
5. IDH1/IDH2
The isoforms 1 and 2 of isocitrate dehydrogenase (IDH1 and IDH2 genes), located on chromosomes 2q33 and 16q26, encode for IDH1 and IDH2 catalytic enzymes, respectively [127]. IDHs are a group of homodimeric enzymes involved in cellular metabolism and epigenetic regulation, including adaptation to hypoxia, histone demethylation, and deoxyribonucleic acid (DNA) modification, known to result in altered function in certain tumour cells [128]. IDH1 and IDH2 function as homodimers with two active sites per dimer and each subunit is composed of a large domain, a small domain, and a clasp domain [129]. IDH1 and IDH2 are important components of the TCA cycle, and are responsible for the first of two decarboxylations and dehydrogenations in this process [128]. Under normal physiological conditions, these enzymes catalyse the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) to produce reduced nicotinamide adenine dinucleotide phosphate (NADPH) from NADP+ [128,130,131]. Several dioxygenases rely on this process, as they require sufficient cellular levels of α-KG for metabolic and epigenetic regulation [129]. The evolutionarily conserved arginine residues at Arg132 (IDH1) and Arg172 (IDH2) in the active site of these enzymes are critical for the binding of isocitrate within the catalytic pocket [129].
Somatic mutations in IDH1 and IDH2 have been described firstly in glioma and later in AML [127,132]. Together, they are detected in up to 20% of AML patients (6–16% for IDH1 and 8–19% for IDH2), and are enriched in patients with normal karyotypes, with increased prevalence in elderly patients [6,133,134]. IDH1/2 mutations are early clonal events in disease evolution and occur as heterozygous missense variants with an oncogenic gain of function [128,135]. All IDH1/2 variants described in AML affect three codons within exon 4 (R132 of IDH1, and R140 or R172 of IDH2) [128,132]. These mutations have been described to affect the active site of the IDH enzyme, which confer a gain-of-function activity that causes reduction of α-KG to an oncometabolite, the (R) enantiomer of 2-hydroxyglutarate (2-HG) [128,130,131]. Increased levels of 2-HG can competitively inhibit α-KG-dependent dioxygenases, such as the ten eleven translocation (TET) enzyme family and histone lysine demethylases, leading to a ‘hypermethylation signature’ of the downstream target genes, which then leads to a block of myeloid differentiation and the accumulation of immature hematopoietic cells that is characteristic of AML [128,131]. Additionally, altered expression of genes including PU1, RUNX1, GATA1, and CEBPA leads to overexpression of the MAPK cell signalling pathway and HOXA genes, which together with activation of the extracellular-signal-regulated kinase (ERK)-NF-κB signalling pathway result in impaired differentiation, leukemic cell proliferation, and DNA damage [132].
The prognostic implication of IDH mutations in AML is controversial, but seems to be influenced by the specific location of the mutation, other co-occurring mutations, and the risk groups according to genomic profile [128,132,136]. They have been reported to frequently co-occur with NPM1 and SRSF2 mutations, while being almost mutually exclusive with both TET2 and WT1 mutations [128,130]. Another important aspect of these mutations is the fact that they remain stable during disease progression [132,137,138]. Because they usually occur as early genomic events in disease pathogenesis, they are typically present in dominant clones and persist even after chemotherapy treatment [132]. The prognostic significance of IDH mutations in AML is not well known. Long-term persistence of IDH mutations using highly sensitive techniques was seen in AML patients who were in molecular remission for NPM1 mutation following chemotherapy. Their disappearance following allogenic stem cell transplantation concluded their presence in a pre-leukaemia clone and resulted in them not being a suitable marker for monitoring minimal residual disease in AML [139,140].
Over recent years, multiple novel small molecule inhibitors which target mutated IDH by binding to the active site of the enzyme and preventing reduction of α-KG to 2-HG have been emerging [128,132,141,142]. In 2013, the first IDH2 inhibitor was developed by Agios Pharmaceuticals. Years later, AG-221 (Enasidenib), an allosteric and non-competitive enzyme inhibitor, was approved for pre-clinical studies [143]. Following the successful preclinical studies of this inhibitor in mouse models, an international, open-label, phase 1/2 trial was launched to investigate the safety and tolerability of enasidenib in patients with relapsed or refractory IDH2-mutant AML [143]. These clinical trials were deemed successful, as the overall response rate (ORR) of the drug was 40% when compared with controls [143,144]. IDH1 inhibitors, including AG-120 (Ivosidenib, Agios), a reversible, allosteric and competitive enzyme inhibitor, and IDH305 (Novartis Oncology) also showed evidence of efficacy. Ivosidenib trials were successful for the treatment of IDH1-mutated relapsed or refractory AML, with an ORR of 41.6%, and later on also for the treatment of newly diagnosed IDH1-mutant AML, with ORR of 54.5% [145,146]. Currently, there are two IDH inhibitors that are FDA-approved for the treatment of AML: enasidenib and ivosidenib. These two agents are not currently approved by EMA. Enasidenib was approved for the treatment of adult patients with relapsed or refractory AML with an IDH2 mutation in 2017. Ivosidenib was approved by the FDA for patients with relapsed or refractory IDH1-mutated AML in 2018, and also as a front-line therapy for newly diagnosed elderly patients 75 years or older or who are ineligible to receive intensive chemotherapy in 2019 [132].
Even though IDH inhibitors have shown promising results, the emergence of resistant subclones has been observed [128]. Co-occurring mutations in the receptor tyrosine kinase (RTK) pathway are associated with both primary and secondary therapeutic resistance to both enasidenib and ivosidenib [147,148]. Baseline mutations in the RTK pathway genes (NRAS, KRAS, PTPN11, KIT, NF1, BRAF, or FLT3) were enriched in patients with mutant IDH1 AML who did not achieve disease remission with ivosidenib treatment [148]. In addition to RTK pathway mutations, a higher mutational burden (≥6 mutations) and/or the presence of a FLT3 co-mutation were also associated with primary resistance to enasidenib [130,147,148]. Another therapeutic resistance mechanism is the emergence of second-site IDH mutations affecting the binding site of the enzyme (i.e., S280F and R119P in IDH1; Q316E and I319M in IDH2) [148,149,150]. The majority of these recently described second-site IDH mutations were associated with a concurrent increase in 2-HG levels that was resistant to targeted therapy [148]. Lastly, secondary resistance was also associated with “isoform switching”, where patients that initially presented with an IDH1 mutation then acquire an IDH2 mutation at the time of relapse, and vice versa [148,151].
Recent studies have revealed new insights into using poly-ADP ribose polymerase (PARP) inhibitors to target IDH mutant cells. The accumulation of 2-HG in cells with IDH mutations inhibits the function of αKG-dependent dioxygenases (KDM4A and KDM4B) that are critical for the homologous recombination (HR) DNA repair pathway [152,153]. This means that IDH1/2 mutations induce an HR defect and the consequent vulnerability of the tumour cells to PARP inhibition. In vivo studies using mouse models have demonstrated that PARP inhibitors are effective against IDH mutant myeloproferative syndrome (MDS)/AML and can overcome resistance to targeted IDH inhibitors [152]. A proof of concept, biomarker-driven, multi-institution, phase II open label clinical trial is currently investigating the effectiveness of PARP inhibitor monotherapy (olaparib) to treat IDH mutant relapsed/refractory AML and MDS [154] (ClinicalTrials.gov Identifier: NCT03953898). The introduction of IDH inhibitors and their promising results from clinical studies along with understanding the mechanisms of resistance to these inhibitors and the exploration of potential agents for overcoming resistance is another example of how target therapy has found its way into management of AML patients for better clinical outcomes.
6. Functional Genomics and Target Discovery
The presence of several mutated genes in AML cells and the heterogeneity of these mutations makes the predicted impact of these genomic abnormalities on the activated signalling pathways difficult. The pathologically activated genes may be several steps downstream of the identified driver mutations and are not recognised or easily predicted. An example of the latter is the activation of MKNK1 in JAK2-mutated neoplasms [155]. One approach to overcome the complexity of identifying the essential genes or pathways in cancer cells is through inhibiting the function of the genes and assessing the viability of the cancer cells. The genes and signalling pathways whose suppression reduces the viability or proliferation of the cancer cells play major roles in pathogenesis and therefore are potential therapeutic targets. These targets may not necessarily be mutated themselves. Interestingly, some of these genes and signalling pathways may be activated due to the interaction of the cancer cells with the surrounding microenvironment and the signals they received from neighbouring cells [156].
The inhibition of genes or pathways to identify those that are essential for viability or proliferation can be done through chemical inhibition, such as applying a panel of inhibitors targeting different molecules [157,158,159]. For instance, the Beat AML clinical trial was initiated based on the concept of using genomic technology to identify each patient’s cancer-driving genetic mutations, followed by matching patients with the most promising targeted treatment, and the initial observations have been promising [11]. Alternatively, one can use inhibition using small hairpin RNA (shRNA) or CRISPR technology libraries [159,160,161,162]. For the last few years, pooled shRNA libraries have been applied to various cancer cell lines in combination with various drugs to identify the genes that are essential for the viability of the cancer cells, and those whose inhibition demonstrates synthetic lethality with the investigated inhibitor [160,163]. The majority of these studies were performed using cell lines because of their easier manipulation and their in vitro viability. The principle of a pooled shRNA library screen is the transduction of the cancer cells by a pool of various shRNAs targeting a certain number of genes, and culturing the cells for a selection period of 8 to 10 cell divisions. During the selection period, the shRNAs targeting the genes essential for survival of the investigated cancer cells are depleted. The depleted shRNAs are then characterised using next generation sequencing techniques [160,161,163].
This technique and similar methods, such as pooled CRISPR interference (CRISPRi), which similarly works by targeting various genes in a population of cancer cells, provide information on the genes, and consequently pathways, which are essential for cancer cell survival, without necessarily having the knowledge about the underlying genomic abnormalities. These functional genomic techniques measure the outcome of all the interactions between the dysregulated genes (intrinsic) and also the interaction of the cancer cells with their microenvironment (extrinsic), if the screen is performed in the presence of a bone marrow-mimicking microenvironment. Therefore, the generated information is expected to be more comprehensive compared with genomic sequencing alone. However, the validity of the findings from functional genomics depends on the condition under which the screen is performed and how close the experimental condition is to the patients’ bone marrow microenvironment. Although pooled shRNA screens have been used within the last decade by several groups, these studies have been limited to cell line models rather than primary cancer cells. The main challenges on screening primary cells are the low efficiency of the current techniques at transducing primary cells with shRNA or CRISPR vectors, and also the short in vitro survival of the primary cells, which is particularly true for AML [164]. We were able to screen primary AML cells in two independent investigations using a selective pooled shRNA library (selected from reviewing literature on the genes involved in AML pathogenesis and also from high throughput sequencing data) [165], and then a pooled library targeting genes known to be involved in major signalling pathways [161], which identified a few essential genes for the viability in some AML cases. However, further modification to the methodology and larger numbers of AML cases with various subtypes are required before validation of this method as a clinically approved tool for therapeutic target discovery in patients. Development of assays for enrichment and in vitro expansion of leukaemia stem cells (LSC) from patient samples is one of the requirements for improving the pooled shRNA or CRISPRi screening technique. Because of the small number of AML LSCs which can be enriched, and the requirement for a 200-1000-fold incrase of the library complex, a library with the smallest but most relevant shRNAs or guide RNAs (gRNAs) will be required for optimization of the screen. To mimic the bone marrow microenvironment, a standardised three-dimensional culture containing mesenchymal stromal cells and other components of the bone marrow niche [166,167,168] must be developed and optimised to standardise the screen across different samples. CRISPR technology has grown rapidly within the last decade and has become the mainstream technique for functional genomics screens. This technology has revolutionised gene editing, and by providing a faster and more efficient method of gene manipulation, has led to significant progress in understanding the function of genes and how they lead to cancer development [169,170]. Development of new single vector constructs containing both gRNAs and the CRISPR-associated endonuclease (Cas protein, active or inactive) [171] provide an opportunity to screen primary leukaemia cells, as this bypasses the requirement for transducing the cells with two constructs. In summary, new developments in genomics technology such as gene editing/targeting, and cell culture techniques such artificial bone marrow models, are expected to convert the pooled functional genomics screen for target discovery into a routine clinical tool for management of AML patients in the era of personalised medicine.
7. Conclusions
The clinical practice of AML is changing mainly due to fast moving genomic technologies which have provided a more detailed picture of the underlying causes of AML, as well as innovations in the design and production of targeted and specific inhibitors. These advances are shifting the treatment of AML from non-specific chemotherapy toward personalized medicine, where AML patients are treated with specific inhibitors based on the specific genomic abnormalities of their leukaemia cells. Development of resistance, however, is a common finding, and identification of new targets to overcome resistance is essential for improving the survival of AML patients. While genomic sequencing provides information on the new acquired mutations at relapse, they may not provide the information on the pathways activated by all the detected mutations. Various functional investigations such as drug screens or functional genomics techniques might provide such information. However, further development in the cell cultures of primary AML cells in a condition mimicking the bone marrow microenvironment will be required before these techniques can be used in clinical practice.
Author Contributions
S.R., A.M.E. and J.S.K. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Elsa U. Pardee Foundation (A.M.E.) and the National Cancer Institute of the National Institutes of Health under Award Number K22CA216008 (A.M.E.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare no conflict of interest.
References
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer. 2012, 12, 513–526. [Google Scholar] [CrossRef]
- O’Dwyer, M.E.; Druker, B.J. STI571: An inhibitor of the BCR-ABL tyrosine kinase for the treatment of chronic myelogenous leukaemia. Lancet Oncol. 2000, 1, 207–211. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.-X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Torres-Ayuso, P.; Brognard, J. Combing the Cancer Genome for Novel Kinase Drivers and New Therapeutic Targets. Cancers 2019, 11, 1972. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Noguera, N.; Catalano, G.; Banella, C.; Divona, M.; Faraoni, I.; Ottone, T.; Arcese, W.; Voso, M. Acute Promyelocytic Leukemia: Update on the Mechanisms of Leukemogenesis, Resistance and on Innovative Treatment Strategies. Cancers 2019, 11, 1591. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.; Brown, C.; Iland, H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: Current perspectives. OncoTargets Ther. 2017, 10, 1585–1601. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: Feasibility and preliminary efficacy of the Beat AML Master Trial. Nat. Med. 2020, 26, 1852–1858. [Google Scholar] [CrossRef]
- Guglielmi, C.; Martelli, M.P.; Diverio, D.; Fenu, S.; Vegna, M.L.; Cantù-Rajnoldi, A.; Biondi, A.; Cocito, M.G.; Del Vecchio, L.; Tabilio, A.; et al. Immunophenotype of adult and childhood acute promyelocytic leukaemia: Correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br. J. Haematol. 1998, 102, 1035–1041. [Google Scholar] [CrossRef]
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725. [Google Scholar] [CrossRef]
- Özpolat, B. Acute promyelocytic leukemia and differentiation therapy: Molecular mechanisms of differentiation, retinoic acid resistance and novel treatments. Turk. J. Hematol. 2009, 26, 47–61. [Google Scholar]
- Sobas, M.; Rodriguez-Veiga, R.; Vellenga, E.; Paluszewska, M.; De La Serna, J.; García-Álvarez, F.; Gil, C.; Brunet, S.; Bergua, J.; González-Campos, J.; et al. Characteristics and outcome of adult patients with acute promyelocytic leukemia and increased body mass index treated with the PETHEMA Protocols. Eur. J. Haematol. 2020, 104, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Dawson, M.A.; Somana, K.; Opat, S.; Schwarer, A.; Campbell, L.J.; Iland, H. The PRKAR1A gene is fused to RARA in a new variant acute promyelocytic leukemia. Blood 2007, 110, 4073–4076. [Google Scholar] [CrossRef]
- Strehl, S.; König, M.; Boztug, H.; Cooper, B.W.; Suzukawa, K.; Zhang, S.-J.; Chen, H.-Y.; Attarbaschi, A.; Dworzak, M.N. All-trans retinoic acid and arsenic trioxide resistance of acute promyelocytic leukemia with the variant STAT5B-RARA fusion gene. Leukemia 2013, 27, 1606–1610. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tsuzuki, S.; Tsuzuki, M.; Handa, K.; Inaguma, Y.; Emi, N. BCOR as a novel fusion partner of retinoic acid receptor alpha in a t(X;17)(p11;q12) variant of acute promyelocytic leukemia. Blood 2010, 116, 4274–4283. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Zhu, J.; Shi, X.G.; Ni, J.H.; Zhong, H.J.; Si, G.Y.; Jin, X.L.; Tang, W.; Li, X.S.; Xong, S.M.; et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 1996, 88, 1052–1061. [Google Scholar] [CrossRef]
- Mi, J.-Q.; Li, J.-M.; Shen, Z.-X.; Chen, S.-J.; Chen, Z. How to manage acute promyelocytic leukemia. Leukemia 2012, 26, 1743–1751. [Google Scholar] [CrossRef][Green Version]
- Fox, E.; Razzouk, B.; Widemann, B.C.; Xiao, S.; O’Brien, M.; Goodspeed, W.; Reaman, G.H.; Blaney, S.M.; Murgo, A.J.; Balis, F.M.; et al. Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood 2008, 111, 566–573. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Yan, X.-J.; Zhou, Z.-R.; Yang, F.-F.; Wu, Z.Y.; Sun, H.-B.; Liang, W.-X.; Song, A.-X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic Trioxide Controls the Fate of the PML-RAR Oncoprotein by Directly Binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef]
- Jeanne, M.; Lallemand, V.; Ferhi, O.; Koken, M.; LE Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA Oxidation and Arsenic Binding Initiate the Antileukemia Response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Kheddache-Atmane, S.; El Asmi, F.; Dianoux, L.; Aubry, M.; Chelbi-Alix, M.K. Requirement of PML SUMO Interacting Motif for RNF4- or Arsenic Trioxide-Induced Degradation of Nuclear PML Isoforms. PLoS ONE 2012, 7, e44949. [Google Scholar] [CrossRef] [PubMed]
- Lång, E.; Grudic, A.; Pankiv, S.; Bruserud, O.; Simonsen, A.; Bjerkvig, R.; Bjørås, M.; Bøe, S.O. The arsenic-based cure of acute promyelocytic leukemia promotes cytoplasmic sequestration of PML and PML/RARA through inhibition of PML body recycling. Blood 2012, 120, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D. The pathogenesis of acute promyelocytic leukaemia: Evaluation of the role of molecular diagnosis and monitoring in the management of the disease. Br. J. Haematol. 1999, 106, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, F.; Fromental-Ramain, C.; Yamamoto, K.; Chambon, P. ATP-Driven Chromatin Remodeling Activity and Histone Acetyltransferases Act Sequentially during Transactivation by RAR/RXR In Vitro. Mol. Cell 2000, 6, 1049–1058. [Google Scholar] [CrossRef]
- Di Croce, L.; Raker, V.A.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef]
- Pandolfi, P.P. In vivo analysis of the molecular genetics of acute promyelocytic leukemia. Oncogene 2001, 20, 5726–5735. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Ammatuna, E. The Biology of Acute Promyelocytic Leukemia and Its Impact on Diagnosis and Treatment. Hematology 2006, 2006, 156–161. [Google Scholar] [CrossRef]
- Saeed, S.; Logie, C.; Stunnenberg, H.G.; Martens, J.H. Genome-wide functions of PML-RARalpha in acute promyelocytic leukaemia. Br. J. Cancer 2011, 104, 554–558. [Google Scholar] [CrossRef]
- Gallagher, R.E. Retinoic acid resistance in acute promyelocytic leukemia. Leukemia 2002, 16, 1940–1958. [Google Scholar] [CrossRef]
- Gallagher, R.E.; Moser, B.K.; Racevskis, J.; Poiré, X.; Bloomfield, C.D.; Carroll, A.J.; Ketterling, R.P.; Roulston, D.; Schachter-Tokarz, E.; Zhou, D.-C.; et al. Treatment-influenced associations of PML-RARα mutations, FLT3 mutations, and additional chromosome abnormalities in relapsed acute promyelocytic leukemia. Blood 2012, 120, 2098–2108. [Google Scholar] [CrossRef]
- Cote, S.; Zhou, D.; Bianchini, A.; Nervi, C.; Gallagher, R.E.; Miller, W.H. Altered ligand binding and transcriptional regulation by mutations in the PML/RARalpha ligand-binding domain arising in retinoic acid-resistant patients with acute promyelocytic leukemia. Blood 2000, 96, 3200–3208. [Google Scholar] [CrossRef]
- Marasca, R.; Zucchini, P.; Galimberti, S.; Leonardi, G.; Vaccari, P.; Donelli, A.; Luppi, M.; Petrini, M.; Torelli, G. Missense mutations in the PML/RARalpha ligand binding domain in ATRA-resistant As(2)O(3) sensitive relapsed acute promyelocytic leukemia. Haematologica 1999, 84, 963–968. [Google Scholar]
- Lehmann-Che, J.; Bally, C.; Letouzé, E.; Berthier, C.; Yuan, H.; Jollivet, F.; Ades, L.; Cassinat, B.; Hirsch, P.; Pigneux, A.; et al. Dual origin of relapses in retinoic-acid resistant acute promyelocytic leukemia. Nat. Commun. 2018, 9, 2047. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Ishikawa, Y.; Kawashima, N.; Akashi, A.; Yamaguchi, Y.; Harada, Y.; Hirano, D.; Adachi, Y.; Miyao, K.; Ushijima, Y.; et al. Identification of the novel deletion-type PML-RARA mutation associated with the retinoic acid resistance in acute promyelocytic leukemia. PLoS ONE 2018, 13, e0204850. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Matthews, W.; Jordan, C.; Wiegand, G.W.; Pardoll, D.; Lemischka, I.R. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 1991, 65, 1143–1152. [Google Scholar] [CrossRef]
- Rappold, I.; Ziegler, B.L.; Köhler, I.; Marchetto, S.; Rosnet, O.; Birnbaum, D.; Simmons, P.J.; Zannettino, A.; Hill, B.; Neu, S.; et al. Functional and phenotypic characterization of cord blood and bone marrow subsets expressing FLT3 (CD135) receptor tyrosine kinase. Blood 1997, 90, 111–125. [Google Scholar] [PubMed]
- Williams, A.; Koch, S.; Christian, T.; Brown, P.; Levis, M.; Small, N. Glycosylation and Surface Localization Are Required for FLT3 Activation but Not for FLT3/ITD. Blood 2009, 114, 2748. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.-E.; Böhmer, A.; Markova, B.; Choudhary, C.; Serve, H.; Böhmer, F.-D. Tyrosine Phosphorylation Regulates Maturation of Receptor Tyrosine Kinases. Mol. Cell. Biol. 2005, 25, 3690–3703. [Google Scholar] [CrossRef] [PubMed]
- Razumovskaya, E.; Masson, K.; Khan, R.; Bengtsson, S.; Rönnstrand, L. Oncogenic Flt3 receptors display different specificity and kinetics of autophosphorylation. Exp. Hematol. 2009, 37, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, M.; Pietschmann, K.; Müller, J.P.; Böhmer, F.D.; Heinzel, T.; Krämer, O. Ubiquitin conjugase UBCH8 targets active FMS-like tyrosine kinase 3 for proteasomal degradation. Leukemia 2010, 24, 1412–1421. [Google Scholar] [CrossRef]
- Heiss, E.; Masson, K.; Sundberg, C.; Pedersen, M.; Sun, J.; Bengtsson, S.; Rönnstrand, L. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood 2006, 108, 1542–1550. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The Structural Basis for Autoinhibition of FLT3 by the Juxtamembrane Domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Breitenbuecher, F.; Schnittger, S.; Grundler, R.; Markova, B.; Carius, B.; Brecht, A.; Duyster, J.; Haferlach, T.; Huber, C.; Fischer, T. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood 2009, 113, 4074–4077. [Google Scholar] [CrossRef]
- Kayser, S.; Schlenk, R.F.; Londono, M.C.; Breitenbuecher, F.; Wittke, K.; Du, J.; Groner, S.; Späth, D.; Krauter, J.; Ganser, A.; et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009, 114, 2386–2392. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Kayser, S.; Bullinger, L.; Kobbe, G.; Casper, J.; Ringhoffer, M.; Held, G.; Brossart, P.; Lübbert, M.; Salih, H.R.; et al. Differential impact of allelic ratio and insertion site in FLT3-ITD–positive AML with respect to allogeneic transplantation. Blood 2014, 124, 3441–3449. [Google Scholar] [CrossRef]
- Liu, S.-B.; Qiu, Q.-C.; Bao, X.-B.; Ma, X.; Li, H.-Z.; Liu, Y.-J.; Chen, S.-N.; Song, Y.-H.; Wu, D.-P.; Xue, S.-L. Pattern and prognostic value of FLT 3–ITD mutations in Chinese de novo adult acute myeloid leukemia. Cancer Sci. 2018, 109, 3981–3992. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Du, L.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2021. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Löffler, H.; Sauerland, C.M.; Serve, H.; Büchner, T.; et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef]
- Whitman, S.P.; Archer, K.; Feng, L.; Baldus, C.; Becknell, B.; Carlson, B.D.; Carroll, A.J.; Mrózek, K.; Vardiman, J.W.; George, S.L.; et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: A cancer and leukemia group B study. Cancer Res. 2001, 61, 7233–7239. [Google Scholar] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef]
- Linch, D.C.; Hills, R.K.; Burnett, A.K.; Khwaja, A.; Gale, R.E. Impact of FLT3ITD mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood 2014, 124, 273–276. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Alpermann, T.; Kern, W.; Haferlach, T. Diversity of the juxtamembrane and TKD1 mutations (Exons 13-15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes Chromosom. Cancer 2012, 51, 910–924. [Google Scholar] [CrossRef]
- Young, D.J.; Nguyen, B.; Zhu, R.; Seo, J.; Li, L.; Levis, M.J.; Pratz, K.W.; Duffield, A.S.; Small, D. Deletions in FLT-3 juxtamembrane domain define a new class of pathogenic mutations: Case report and systematic analysis. Blood Adv. 2021, 5, 2285–2293. [Google Scholar] [CrossRef]
- Pratz, K.W.; Sato, T.; Murphy, K.M.; Stine, A.; Rajkhowa, T.; Levis, M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 2010, 115, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.R.; McCormick, M.J.; Grutkoski, P.S.; Ducker, G.S.; Banerji, N.; Higgins, R.R.; Mendiola, J.R.; Reinartz, J.J. FLT3 mutations at diagnosis and relapse in acute myeloid leukemia: Cytogenetic and pathologic correlations, including cuplike blast morphology. Arch. Pathol. Lab. Med. 2010, 134, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Linch, D.C.; Hills, R.; Wheatley, K.; Burnett, A.K.; Gale, R.E. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 2007, 110, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Hyrenius-Wittsten, A.; Pilheden, M.; Sturesson, H.; Hansson, J.; Walsh, M.P.; Song, G.; Kazi, J.U.; Liu, J.; Ramakrishan, R.; Garcia-Ruiz, C.; et al. De novo activating mutations drive clonal evolution and enhance clonal fitness in KMT2A-rearranged leukemia. Nat. Commun. 2018, 9, 1770. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Breitenbuecher, F.; Kasper, S.; Estey, E.; Giles, F.; Feldman, E.; Ehninger, G.; Schiller, G.; Klimek, V.; Nimer, S.D.; et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood 2005, 105, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Paez, J.G.; Lee, J.C.; Bo, R.; Stone, R.M.; DeAngelo, D.J.; Galinsky, I.; Wolpin, B.M.; Jonasova, A.; Herman, P.; et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood 2004, 104, 1855–1858. [Google Scholar] [CrossRef]
- Matsuno, N.; Nanri, T.; Kawakita, T.; Mitsuya, H.; Asou, N. A novel FLT3 activation loop mutation N841K in acute myeloblastic leukemia. Leukemia 2005, 19, 480–481. [Google Scholar] [CrossRef]
- Schittenhelm, M.M.; Yee, K.W.H.; Tyner, J.; McGreevey, L.; Haley, A.D.; Town, A.; Griffith, D.J.; Bainbridge, T.; Braziel, R.M.; O’Farrell, A.-M.; et al. FLT3 K663Q is a novel AML-associated oncogenic kinase: Determination of biochemical properties and sensitivity to Sunitinib (SU11248). Leukemia 2006, 20, 2008–2014. [Google Scholar] [CrossRef]
- Reindl, C.; Bagrintseva, K.; Vempati, S.; Schnittger, S.; Ellwart, J.W.; Wenig, K.; Hopfner, K.-P.; Hiddemann, W.; Spiekermann, K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood 2006, 107, 3700–3707. [Google Scholar] [CrossRef]
- Fröhling, S.; Scholl, C.; Levine, R.L.; Loriaux, M.; Boggon, T.J.; Bernard, O.; Berger, R.; Döhner, H.; Döhner, K.; Ebert, B.L.; et al. Identification of Driver and Passenger Mutations of FLT3 by High-Throughput DNA Sequence Analysis and Functional Assessment of Candidate Alleles. Cancer Cell 2007, 12, 501–513. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef]
- Choudhary, C.; Schwäble, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol. Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Melgar, K.; Bolanos, L.; Hueneman, K.; Walker, M.M.; Jiang, J.-K.; Wilson, K.M.; Zhang, X.; Shen, J.; Jiang, F.; et al. Targeting AML-associated FLT3 mutations with a type I kinase inhibitor. J. Clin. Investig. 2020, 130, 2017–2023. [Google Scholar] [CrossRef]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.; Miller, C.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.L.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.K.; Nechiporuk, T.; Bottomly, D.; Piehowski, P.D.; Reisz, J.A.; Pittsenbarger, J.; Kaempf, A.; Gosline, S.J.; Wang, Y.-T.; Hansen, J.R.; et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell 2021, 39, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.-S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, C.; Konopleva, M.; Chen, Y.; Jacamo, R.O.; Borthakur, G.; Cortes, J.; Ravandi, F.; Ramachandran, A.; Andreeff, M. Reversal of Acquired Drug Resistance in FLT3-Mutated Acute Myeloid Leukemia Cells via Distinct Drug Combination Strategies. Clin. Cancer Res. 2014, 20, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.-Y.; Singh, V.K.; Coumar, M.; Hsu, Y.C.; Wang, W.-C.; Song, J.-S.; Chen, C.-H.; Lin, W.-H.; Wu, S.-H.; Hsu, J.T.A.; et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci. Rep. 2015, 5, srep11702. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, Y.; Kantarjian, H.M.; Luthra, R.; Ravandi, F.; Borthakur, G.; Garcia-Manero, G.; Konopleva, M.; Estrov, Z.; Andreeff, M.; Cortes, J.E. Treatment with FLT3 inhibitor in patients withFLT3-mutated acute myeloid leukemia is associated with development of secondaryFLT3-tyrosine kinase domain mutations. Cancer 2014, 120, 2142–2149. [Google Scholar] [CrossRef]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2020, 2, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.-T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef]
- Al-Jamal, H.; Jusoh, S.A.M.; Hassan, R.; Johan, M.F. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 2015, 15, 869. [Google Scholar] [CrossRef]
- Williams, A.B.; Li, L.; Nguyen, B.; Brown, P.; Levis, M.; Small, D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood 2012, 120, 3069–3079. [Google Scholar] [CrossRef]
- Minami, Y.; Kiyoi, H.; Yamamoto, Y.; Ueda, R.; Saito, H.; Naoe, T. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia 2002, 16, 1535–1540. [Google Scholar] [CrossRef]
- Chen-Levy, Z.; Nourse, J.; Cleary, M.L. The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol. Cell. Biol. 1989, 9, 701–710. [Google Scholar] [CrossRef]
- Hockenbery, D.M.; Zutter, M.; Hickey, W.; Nahm, M.; Korsmeyer, S.J. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc. Natl. Acad. Sci. USA 1991, 88, 6961–6965. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Ren, D.; Tu, H.-C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. BID, BIM, and PUMA Are Essential for Activation of the BAX- and BAK-Dependent Cell Death Program. Science 2010, 330, 1390–1393. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W. Insights into Programmed Cell Death through Structural Biology. Cell 2000, 103, 273–282. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.; Colman, P.M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Thomadaki, H.; Scorilas, A. BCL2 Family of Apoptosis-Related Genes: Functions and Clinical Implications in Cancer. Crit. Rev. Clin. Lab. Sci. 2006, 43, 1–67. [Google Scholar] [CrossRef]
- Reed, J.C. Proapoptotic multidomain Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006, 13, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.G. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 2008, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Martinez, E.; Fontanella, E.; Aiello, A. Two- and three-color immunofluorescence using aminocoumarin, fluorescein, and phycoerythrin-labelled antibodies and single laser flow cytometry. Cytometry 1991, 12, 537–544. [Google Scholar] [CrossRef]
- Karakas, T.; Maurer, U.; Weidmann, E.; Miething, C.C.; Hoelzer, D.; Bergmann, L. High expression of bcl-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 1998, 9, 159–165. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef]
- Vo, T.-T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; DeAngelo, D.J.; Frattini, M.G.; Letai, A. Relative Mitochondrial Priming of Myeloblasts and Normal HSCs Determines Chemotherapeutic Success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.-I.; Akashi, K.; Korsmeyer, S.J. Obligate Role of Anti-Apoptotic MCL-1 in the Survival of Hematopoietic Stem Cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; DeBose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes On-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Weber, K.; Dinsdale, D.; Schmitz, I.; Schulze-Osthoff, K.; Dyer, M.; Cohen, G.M. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009, 16, 1030–1039. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Rev. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Fathi, A.T.; Dinardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Dinardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Tallman, M.S. Emerging therapeutic drugs for AML. Blood 2016, 127, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; DiNardo, C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018, 14, 979–993. [Google Scholar] [CrossRef]
- Aref, S.; Areida, E.S.K.; Aaal, M.F.A.; Adam, O.M.; El-Ghonemy, M.S.; Elbaiomy, M.; Zeid, T.A. Prevalence and Clinical Effect of IDH1 and IDH2 Mutations Among Cytogenetically Normal Acute Myeloid Leukemia Patients. Clin. Lymphoma Myeloma Leuk. 2015, 15, 550–555. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Ravandi, F.; Agresta, S.; Konopleva, M.; Takahashi, K.; Kadia, T.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am. J. Hematol. 2015, 90, 732–736. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Dohner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901. [Google Scholar] [CrossRef]
- Elnahass, Y.; Badawy, R.H.; Elrefaey, F.A.; Nooh, H.A.; Ibrahiem, D.; Nader, H.A.; Mahmoud, H.K.; ElMetnawy, W.H. IDH Mutations in AML Patients; A higher Association with Intermediate Risk Cytogenetics. Asian Pac. J. Cancer Prev. 2020, 21, 721–725. [Google Scholar] [CrossRef]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Nibourel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the Acute Leukemia French Association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [CrossRef]
- Chou, W.-C.; Lei, W.-C.; Ko, B.-S.; Hou, H.-A.; Chen, C.-Y.; Tang, J.-L.; Yao, M.; Tsay, W.; Wu, S.-J.; Huang, S.-Y.; et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 2011, 25, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.; Simpson, M.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.; Levine, R.L.; Flinn, I.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Stein, E.M.; DE Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib inIDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.; Marteyn, B.; Farnoud, N.R.; DE Botton, S.; Bernard, O.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Intlekofer, A.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Issa, G.C.; DiNardo, C.D. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021, 11, 107. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef]
- Gbyli, R.; Song, Y.; Liu, W.; Gao, Y.; Chandhok, N.S.; Fu, X.; Wang, X.; Patel, A.; Sundaram, R.; Tebaldi, T.; et al. PARP Inhibitors Are Effective in IDH1/2 Mutant MDS and AML Resistant to Targeted IDH Inhibitors. Blood 2019, 134, 4222. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Chandhok, N.S.; Wei, W.; Bindra, R.; Halene, S.; Shyr, Y.; Li, J.; Berens, M.; Karlovich, C.; Ivy, S.P.; Prebet, T. The PRIME Trial: PARP Inhibition in IDH Mutant Effectiveness Trial. A Phase II Study of Olaparib in Isocitrate Dehydrogenase (IDH) Mutant Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2019, 134, 3909. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef]
- Kokkaliaris, K.D.; Scadden, D.T. Cell interactions in the bone marrow microenvironment affecting myeloid malignancies. Blood Adv. 2020, 4, 3795–3803. [Google Scholar] [CrossRef]
- Cucchi, D.; Groen, R.; Janssen, J.; Cloos, J. Ex vivo cultures and drug testing of primary acute myeloid leukemia samples: Current techniques and implications for experimental design and outcome. Drug Resist. Updat. 2020, 53, 100730. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Yang, W.F.; Bankhead, A.; Fan, G.; Fletcher, L.B.; Bryant, J.; Glover, J.M.; Chang, B.H.; Spurgeon, S.E.; Fleming, W.H.; et al. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 2013, 73, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Khorashad, J.S.; Eiring, A.M.; Mason, C.C.; Gantz, K.C.; Bowler, A.D.; Redwine, H.M.; Yu, F.; Kraft, I.L.; Pomicter, A.D.; Reynolds, K.R.; et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood 2015, 125, 1772–1781. [Google Scholar] [CrossRef]
- Mason, C.C.; Fiol, C.R.; Baker, M.J.; Nadal-Melsio, E.; Yebra-Fernandez, E.; Bicalho, L.; Chowdhury, A.; Albert, M.; Reid, A.G.; Claudiani, S.; et al. Identification of genetic targets in acute myeloid leukaemia for designing targeted therapy. Br. J. Haematol. 2021, 192, 137–145. [Google Scholar] [CrossRef]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Pomicter, A.; Tantravahi, S.; Mason, C.C.; Senina, A.V.; Ahmann, J.M.; Wang, Q.; Than, H.; Patel, A.B.; Heaton, W.L.; et al. Nuclear–Cytoplasmic Transport Is a Therapeutic Target in Myelofibrosis. Clin. Cancer Res. 2019, 25, 2323–2335. [Google Scholar] [CrossRef]
- Bhavanasi, D.; Gwenn, D.-D.; Liu, X.; Vergez, F.; Danet-Desnoyers, G.; Carroll, M.; Huang, J.; Klein, P.S. Signaling mechanisms that regulate ex vivo survival of human acute myeloid leukemia initiating cells. Blood Cancer J. 2017, 7, 636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, D.; Franzini, A.; Pomicter, A.D.; Halverson, B.J.; Antelope, O.; Mason, C.C.; Ahmann, J.M.; Senina, A.V.; Vellore, N.A.; Jones, C.L.; et al. SIRT5 Is a Druggable Metabolic Vulnerability in Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 266–287. [Google Scholar] [CrossRef]
- Borella, G.; Da Ros, A.; Borile, G.; Porcù, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting mesenchymal stromal cells plasticity to reroute acute myeloid leukemia course. Blood 2021, 138, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Bray, L.J.; Binner, M.; Körner, Y.; Von Bonin, M.; Bornhäuser, M.; Werner, C. A three-dimensional ex vivo tri-culture model mimics cell-cell interactions between acute myeloid leukemia and the vascular niche. Haematologica 2017, 102, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Cartledge Wolf, D.M.; Langhans, S.A. Moving Myeloid Leukemia Drug Discovery into the Third Dimension. Front. Pediatr. 2019, 7, 314. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Tian, X.; Gu, T.; Patel, S.; Bode, A.M.; Lee, M.-H.; Dong, Z. CRISPR/Cas9—An evolving biological tool kit for cancer biology and oncology. NPJ Precis. Oncol. 2019, 3, 8. [Google Scholar] [CrossRef]
- Hunker, A.C.; Soden, M.E.; Krayushkina, D.; Heymann, G.; Awatramani, R.; Zweifel, L.S. Conditional Single Vector CRISPR/SaCas9 Viruses for Efficient Mutagenesis in the Adult Mouse Nervous System. Cell Rep. 2020, 30, 4303–4316. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).