
Phospho-Ser784-VCP Drives Resistance of Pancreatic Ductal Adenocarcinoma to Genotoxic Chemotherapies and Predicts the Chemo-Sensitizing Effect of VCP Inhibitor

 , , and
, , and 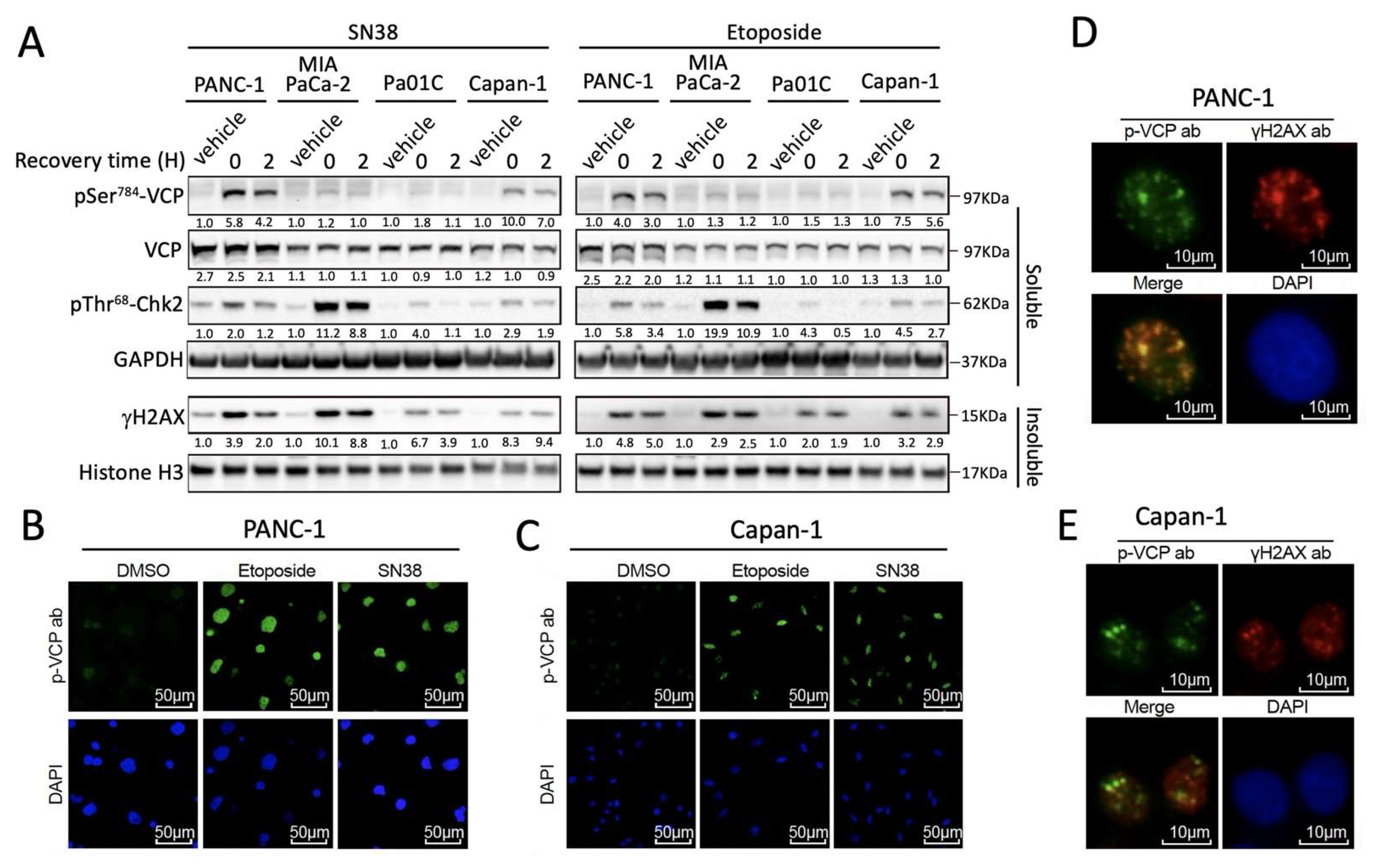{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. DNA and shRNA Constructs
2.3. Human PDAC Tissue Microarray (TMA)
2.4. Immunofluorescence Staining and Image Acquisition
2.5. Immunohistochemistry and Scoring
2.6. Comet Assay
2.7. Cellular Viability and Colony Formation Assays
2.8. Western Blot
2.9. Statistical Analysis
3. Results
3.1. DNA Damage-Induced pSer784-VCP Levels Vary among PDAC Cell Lines
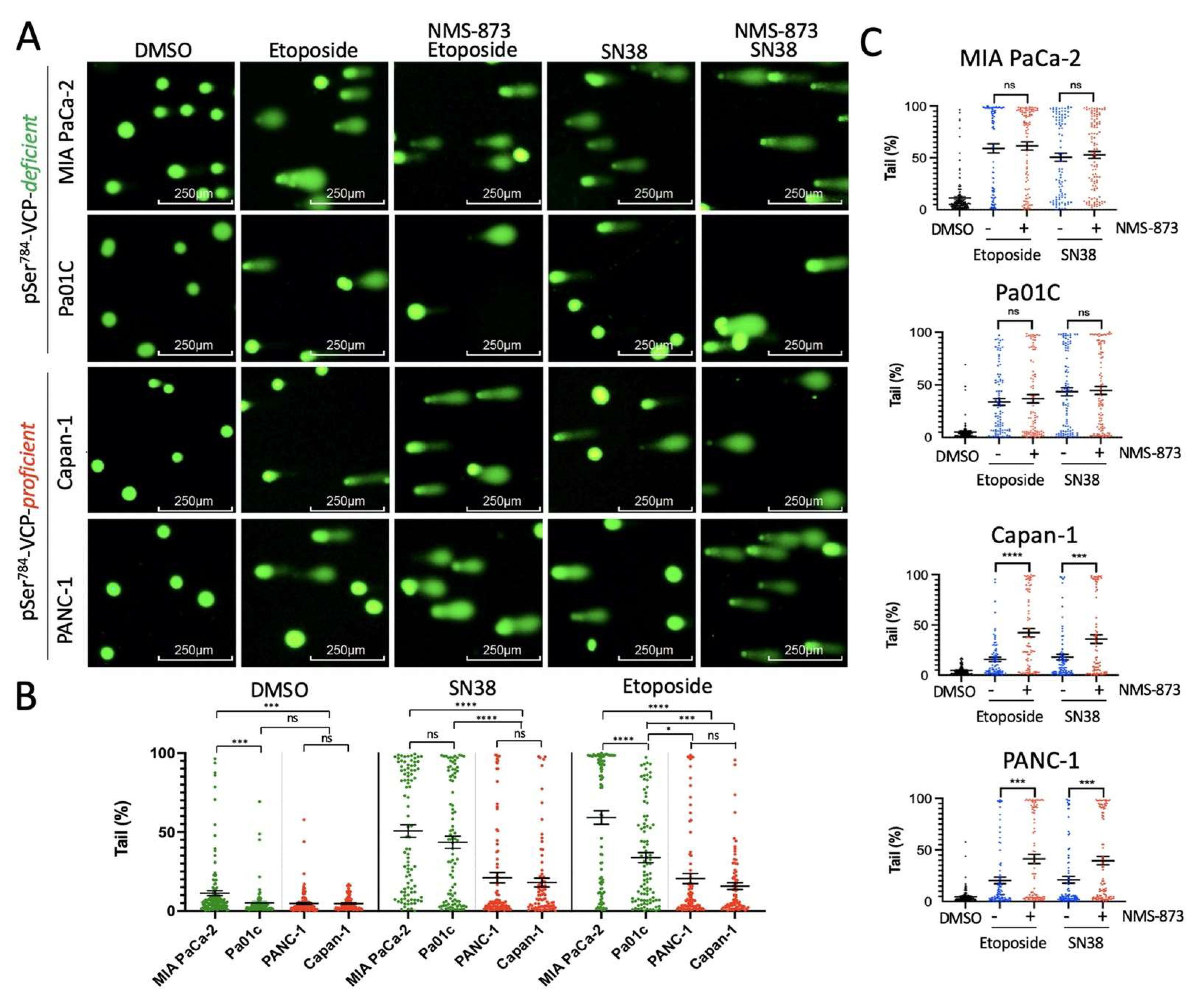
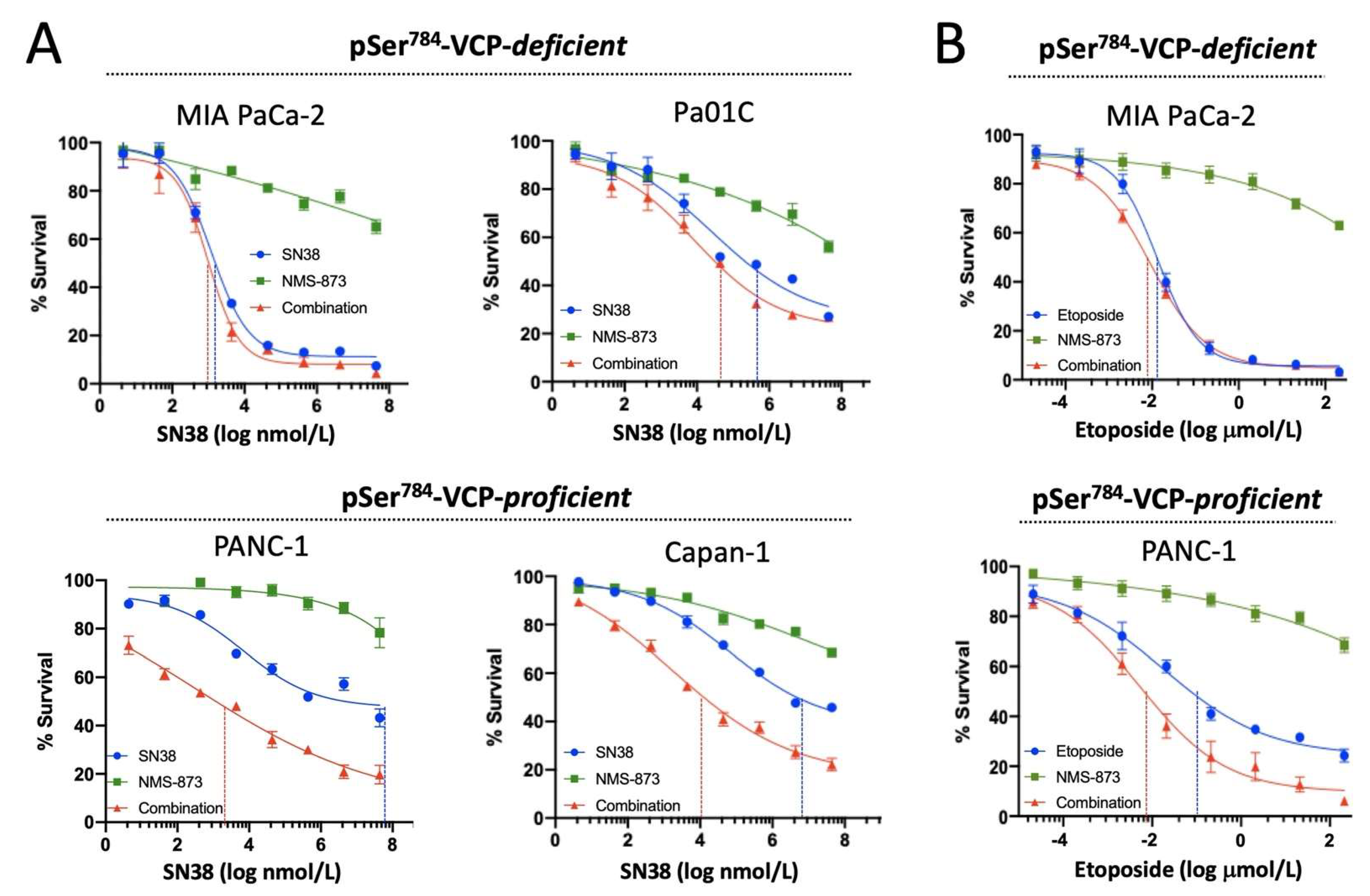
3.2. pSer784-VCP Levels Predict the Chemotherapy Effect on Genome Integrity and Sensitizing Effect of VCP Inhibitor in PDAC Cell Lines
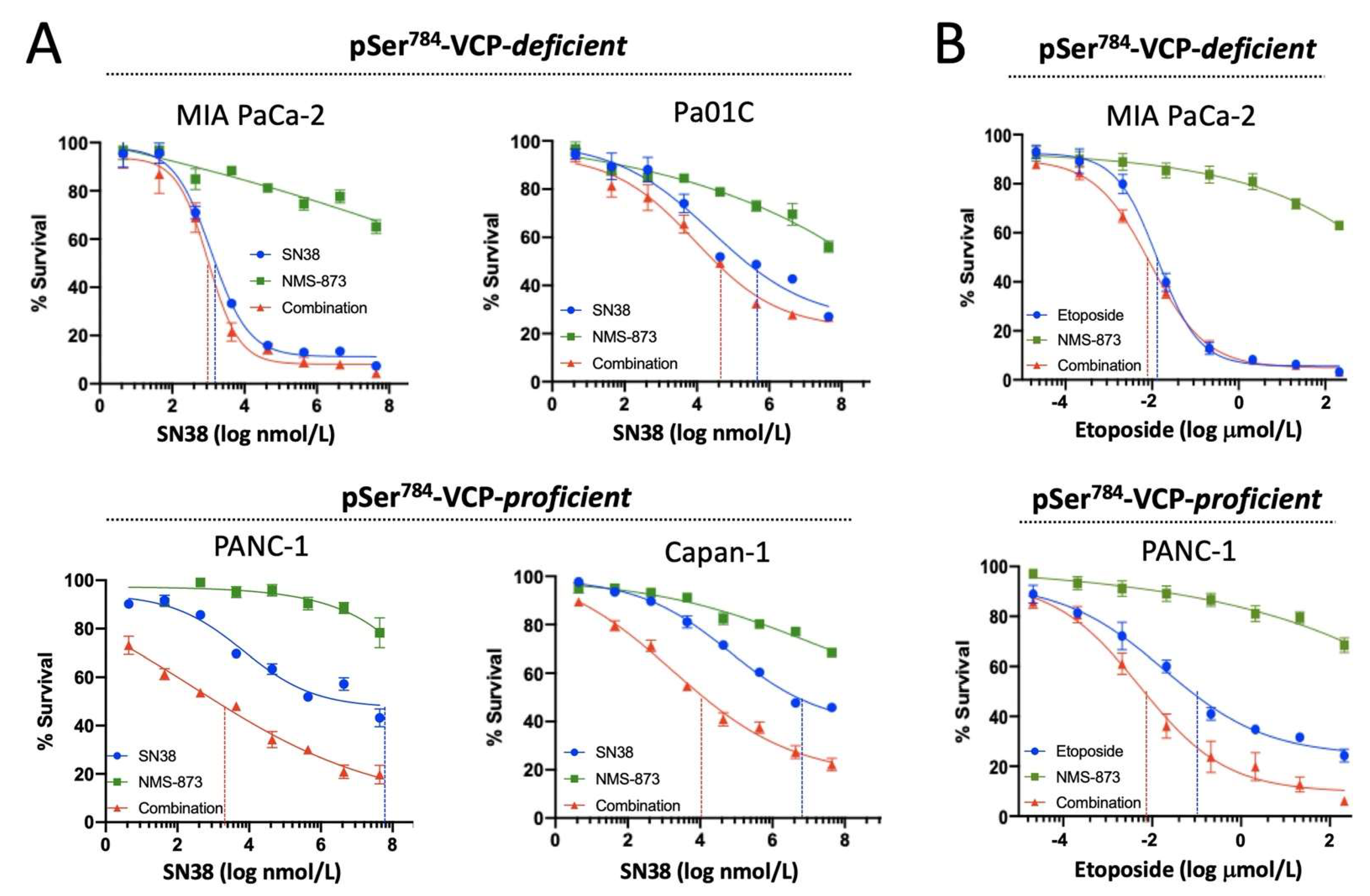
3.3. pSer784-VCP Levels Predict Chemotherapy Effect on Cell Survival and Sensitizing Effect of VCP Inhibitor in PDAC Cell Lines
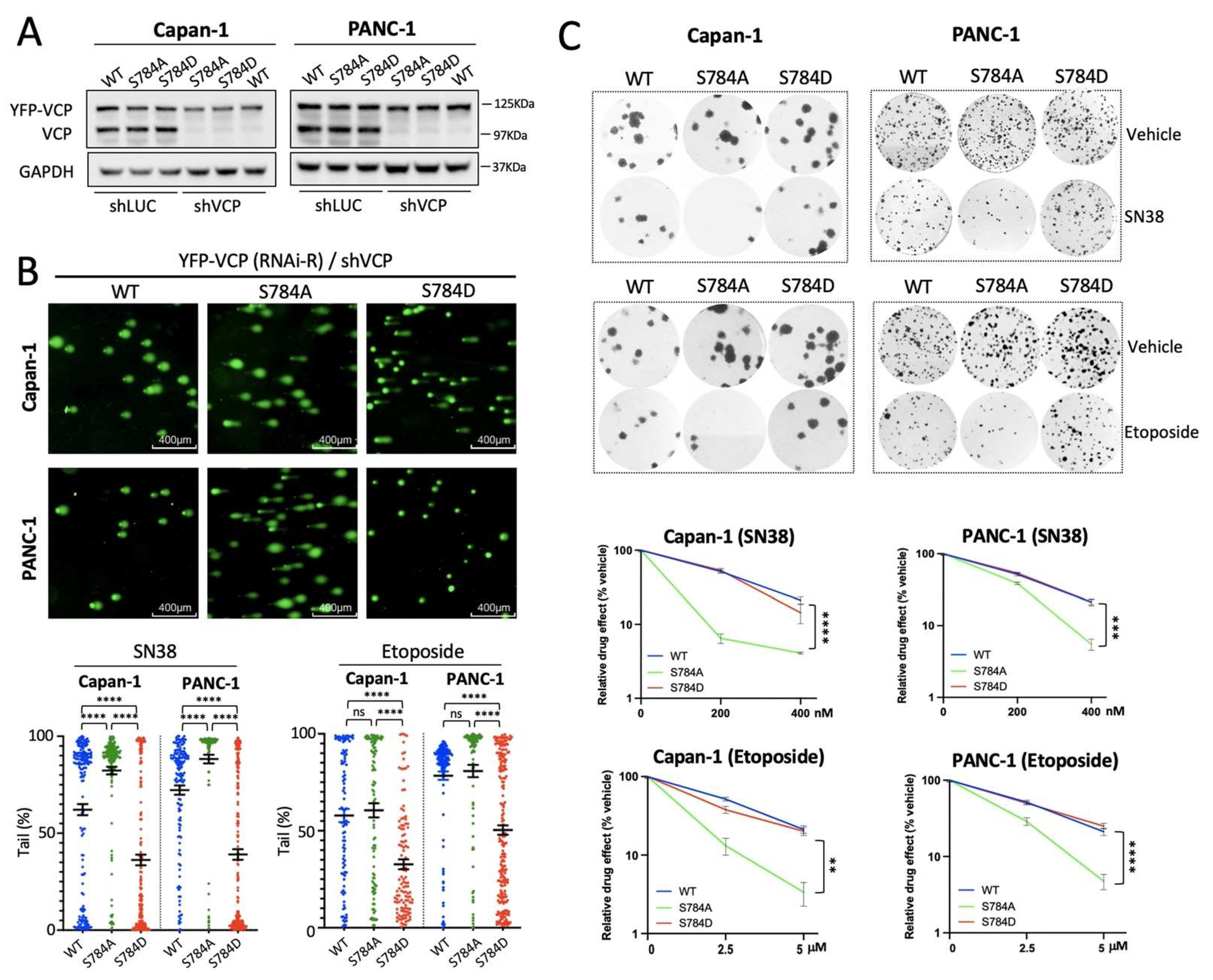
3.4. pSer784-VCP Protects PDAC Cell Lines from Chemotherapy-Induced DNA Damage and Cell Death
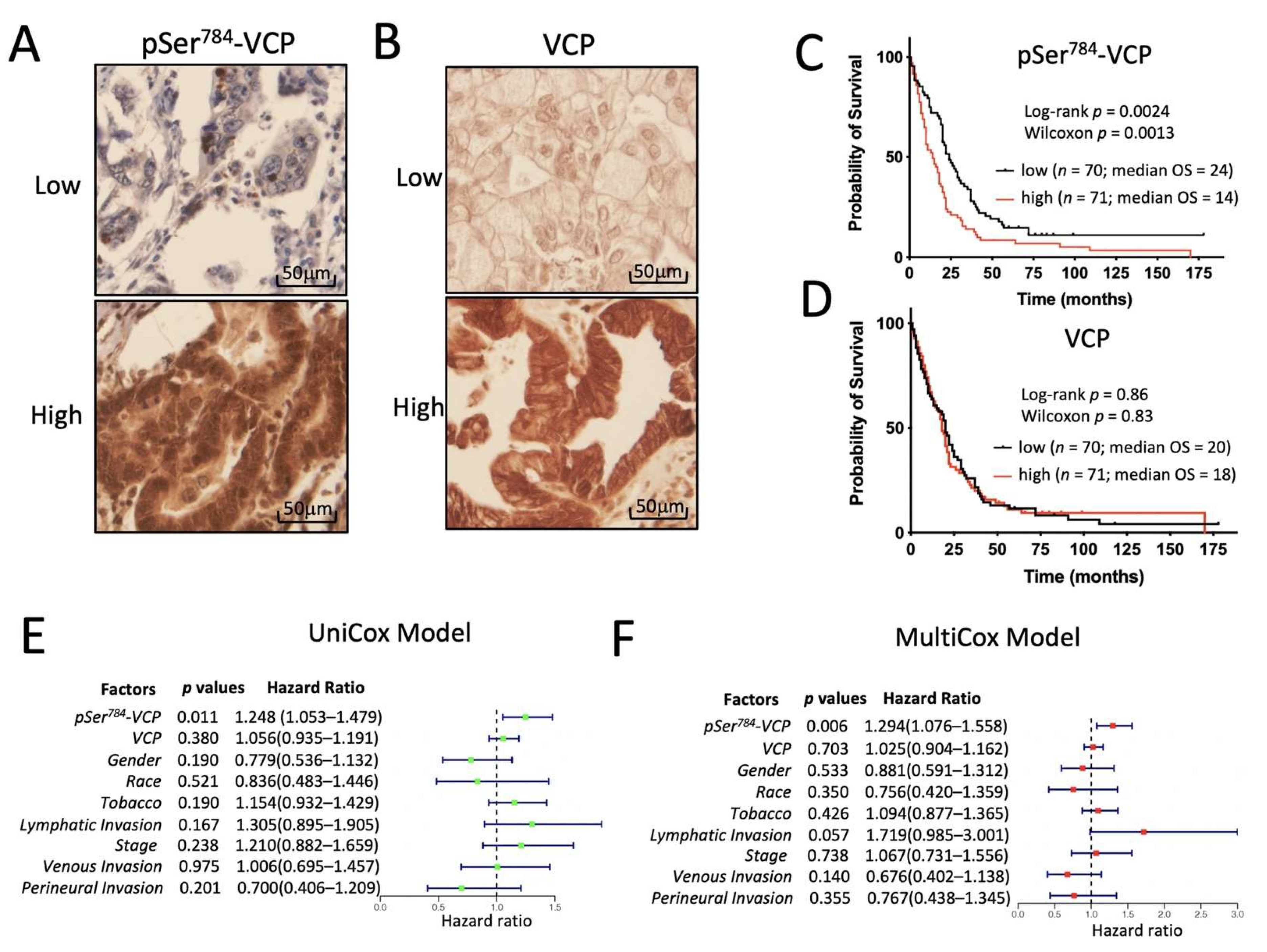
3.5. pSer784-VCP Levels Significantly Associate with Poor Survival of PDAC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- De Dosso, S.; Siebenhüner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef]
- Saung, M.T.; Zheng, L. Current Standards of Chemotherapy for Pancreatic Cancer. Clin. Ther. 2017, 39, 2125–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nat. Cell Biol. 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; de Almeida, F.K.; Simões, C.B.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Van Attikum, H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016, 35, 6–23. [Google Scholar] [CrossRef] [Green Version]
- Polo, S.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [Green Version]
- Tobias, F.; Löb, D.; Lengert, N.; Durante, M.; Drossel, B.; Taucher-Scholz, G.; Jakob, B. Spatiotemporal Dynamics of Early DNA Damage Response Proteins on Complex DNA Lesions. PLOS ONE 2013, 8, e57953. [Google Scholar] [CrossRef]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Acs, K.; Luijsterburg, M.S. Should I stay or should I go: VCP/p97-mediated chromatin extraction in the DNA damage response. Exp. Cell Res. 2014, 329, 9–17. [Google Scholar] [CrossRef]
- Acs, K.; Luijsterburg, M.S.; Ackermann, L.; Salomons, F.A.; Hoppe, T.; Dantuma, N. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011, 18, 1345–1350. [Google Scholar] [CrossRef]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hübscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef]
- Puumalainen, M.-R.; Lessel, D.; Rüthemann, P.; Kaczmarek, N.; Bachmann, K.; Ramadan, K.; Naegeli, H. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Boom, J.V.D.; Wolf, M.; Weimann, L.; Schulze, N.; Li, F.; Kaschani, F.; Riemer, A.; Zierhut, C.; Kaiser, M.; Iliakis, G.; et al. VCP/p97 Extracts Sterically Trapped Ku70/80 Rings from DNA in Double-Strand Break Repair. Mol. Cell 2016, 64, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Havens, C.G.; Walter, J.; Harper, J.W. A Genome-wide Screen Identifies p97 as an Essential Regulator of DNA Damage-Dependent CDT1 Destruction. Mol. Cell 2011, 44, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, M.; Ruan, H.; Weiner, J.; Clauser, K.; Strack, P.; Jin, S.; Williams, A.; Greulich, H.; Gardner, J.; Venere, M.; et al. Valosin-Containing Protein Phosphorylation at Ser784 in Response to DNA Damage. Cancer Res. 2005, 65, 7533–7540. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Stokes, M.P.; Rush, J.; MacNeill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Rogers, A.; Asleh, K.; Won, J.; Gao, D.; Leung, S.; Li, S.; Vij, K.R.; Zhu, J.; Held, J.M.; et al. Phospho-Ser784-VCP Is Required for DNA Damage Response and Is Associated with Poor Prognosis of Chemotherapy-Treated Breast Cancer. Cell Rep. 2020, 31, 107745. [Google Scholar] [CrossRef] [PubMed]
- Shao, J. Ser784 phosphorylation: A clinically relevant enhancer of VCP function in the DNA damage response. Mol. Cell. Oncol. 2020, 7, 1796179. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodhiawala, P.B.; Khurana, N.; Zhang, D.; Cheng, Y.; Li, L.; Wei, Q.; Seehra, K.; Jiang, H.; Grierson, P.M.; Wang-Gillam, A.; et al. TPL2 enforces RAS-induced inflammatory signaling and is activated by point mutations. J. Clin. Investig. 2020, 130, 4771–4790. [Google Scholar] [CrossRef]
- Diamond, M.I.; Cai, S.; Boudreau, A.; Carey, C.J.; Lyle, N.; Pappu, R.V.; Swamidass, S.J.; Bissell, M.; Piwnica-Worms, H.; Shao, J. Subcellular Localization and Ser-137 Phosphorylation Regulate Tumor-suppressive Activity of Profilin-1. J. Biol. Chem. 2015, 290, 9075–9086. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kim, S.-J.; Mooradian, A.; Wang, F.; Li, Z.; Holohan, S.; Collins, P.L.; Wang, K.; Guo, Z.; Hoog, J.; et al. Cancer-associated exportin-6 upregulation inhibits the transcriptionally repressive and anticancer effects of nuclear profilin-1. Cell Rep. 2021, 34, 108749. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, C.; Cai, S.; Boudreau, A.; Kim, S.-J.; Bissell, M.; Shao, J. Ser71 Phosphorylation Inhibits Actin-Binding of Profilin-1 and Its Apoptosis-Sensitizing Activity. Front. Cell Dev. Biol. 2021, 9, 692269. [Google Scholar] [CrossRef]
- Feng, Y.; Nie, L.; Das Thakur, M.; Su, Q.; Chi, Z.; Zhao, Y.; Longmore, G.D. A Multifunctional Lentiviral-Based Gene Knockdown with Concurrent Rescue that Controls for Off-Target Effects of RNAi. Genom. Proteom. Bioinform. 2010, 8, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.-H.; Langley, E.; Gao, F.; Luo, J.; Li, L.; Meyer, G.; Kim, P.; Singh, S.; Kushnir, V.M.; Early, D.S.; et al. A clinically feasible multiplex proteomic immunoassay as a novel functional diagnostic for pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 24250–24261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.-P.; Wang, M.-J.; Zeng, X.; Chen, G.G.; Huang, R.-Y. Effects of Glycyrrhizin in a Mouse Model of Lung Adenocarcinoma. Cell. Physiol. Biochem. 2017, 41, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wu, X.; Xu, Y.; Yang, G.; Yan, M. Immunohistochemical study of STAT3, HIF-1α and VEGF in pterygium and normal conjunctiva: Experimental research and literature review. Mol. Vis. 2020, 26, 510–516. [Google Scholar] [PubMed]
- Chen, J.; Zeng, W.; Pan, W.; Peng, C.; Zhang, J.; Su, J.; Long, W.; Zhao, H.; Zuo, X.; Xie, X.; et al. Symptoms of systemic lupus erythematosus are diagnosed in leptin transgenic pigs. PLoS Biol. 2018, 16, e2005354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.; Hsu, D.; Clement, M.-V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Guntuku, S.; Cui, X.-S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar]
- Zhao, H.; Piwnica-Worms, H. ATR-Mediated Checkpoint Pathways Regulate Phosphorylation and Activation of Human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [Green Version]
- Takai, H.; Naka, K.; Okada, Y.; Watanabe, M.; Harada, N.; Saito, S.; Anderson, C.W.; Appella, E.; Nakanishi, M.; Suzuki, H.; et al. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002, 21, 5195–5205. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX Is Phosphorylated in an ATR-dependent Manner in Response to Replicational Stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [Green Version]
- Helt, C.E.; Cliby, W.A.; Keng, P.C.; Bambara, R.A.; O’Reilly, M.A. Ataxia Telangiectasia Mutated (ATM) and ATM and Rad3-related Protein Exhibit Selective Target Specificities in Response to Different Forms of DNA Damage. J. Biol. Chem. 2005, 280, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, E.L.; Osheroff, E.L.B.A.N. Etoposide, Topoisomerase II and Cancer. Curr. Med. Chem. Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Magnaghi, P.; D’Alessio, R.; Valsasina, B.; Avanzi, N.; Rizzi, S.; Asa, D.; Gasparri, F.; Cozzi, L.; Cucchi, U.; Orrenius, C.; et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Her, N.-G.; Toth, J.I.; Ma, C.-T.; Wei, Y.; Motamedchaboki, K.; Sergienko, E.; Petroski, M.D. p97 Composition Changes Caused by Allosteric Inhibition Are Suppressed by an On-Target Mechanism that Increases the Enzyme’s ATPase Activity. Cell Chem. Biol. 2016, 23, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, L.; Jiang, H.; Knolhoff, B.L.; Lockhart, A.C.; Wang-Gillam, A.; DeNardo, D.G.; Ruzinova, M.B.; Lim, K.-H. Constitutive IRAK4 Activation Underlies Poor Prognosis and Chemoresistance in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Meijer, T.G.; Verkaik, N.S.; Sieuwerts, A.M.; Van Riet, J.; Naipal, K.A.; Van Deurzen, C.H.; Bakker, M.A.D.; Sleddens, H.F.; Dubbink, H.-J.; Toom, T.D.D.; et al. Functional Ex Vivo Assay Reveals Homologous Recombination Deficiency in Breast Cancer Beyond BRCA Gene Defects. Clin. Cancer Res. 2018, 24, 6277–6287. [Google Scholar] [CrossRef] [Green Version]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; Van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional Ex Vivo Assay to Select Homologous Recombination–Deficient Breast Tumors for PARP Inhibitor Treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.M.; Dobbin, Z.C.; Nowsheen, S.; Wielgos, M.; Katre, A.A.; Alvarez, R.D.; Konstantinopoulos, P.A.; Yang, E.S.; Landen, C.N. An ex vivo assay of XRT-induced Rad51 foci formation predicts response to PARP-inhibition in ovarian cancer. Gynecol. Oncol. 2014, 134, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Oliver, A.G.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD 51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Oliver, A.G.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Graeser, M.; McCarthy, A.; Lord, C.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Stach, L.; Freemont, P.S. The AAA+ ATPase p97, a cellular multitool. Biochem. J. 2017, 474, 2953–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, K.; Halder, S.; Wiseman, K.; Vaz, B. Strategic role of the ubiquitin-dependent segregase p97 (VCP or Cdc48) in DNA replication. Chromosoma 2016, 126, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, B.; Halder, S.; Ramadan, K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013, 4, 60. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, K. p97/VCP- and Lys48-linked polyubiquitination form a new signaling pathway in DNA damage response. Cell Cycle 2012, 11, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shih, D.J.H.; Lin, S.-Y. Role of DNA repair defects in predicting immunotherapy response. Biomark. Res. 2020, 8, 23. [Google Scholar] [CrossRef]
- Javadrashid, D.; Baghbanzadeh, A.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, V.; Solimando, A.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373. [Google Scholar] [CrossRef]
- Wang, X.; Bai, E.; Zhou, H.; Sha, S.; Miao, H.; Qin, Y.; Liu, Z.; Wang, J.; Zhang, H.; Lei, M.; et al. Discovery of a new class of valosine containing protein (VCP/P97) inhibitors for the treatment of non-small cell lung cancer. Bioorganic Med. Chem. 2019, 27, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.-K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, P.; Higa, A.; Taouji, S.; Bexiga, M.G.; Marza, E.; Arma, D.; Castain, C.; Le Bail, B.; Simpson, J.; Rosenbaum, J.; et al. Sorafenib-Mediated Targeting of the AAA+ ATPase p97/VCP Leads to Disruption of the Secretory Pathway, Endoplasmic Reticulum Stress, and Hepatocellular Cancer Cell Death. Mol. Cancer Ther. 2012, 11, 2610–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, B.; Vaganay, C.; Vargas, J.D.; Alexe, G.; Benaksas, C.; Pardieu, B.; Fenouille, N.; Ellegast, J.M.; Malolepsza, E.; Ling, F.; et al. Targeting acute myeloid leukemia dependency on VCP-mediated DNA repair through a selective second-generation small-molecule inhibitor. Sci. Transl. Med. 2021, 13, eabg1168. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Vij, K.; Li, L.; Dodhiawala, P.; Lim, K.-H.; Shao, J. Phospho-Ser784-VCP Drives Resistance of Pancreatic Ductal Adenocarcinoma to Genotoxic Chemotherapies and Predicts the Chemo-Sensitizing Effect of VCP Inhibitor. Cancers 2021, 13, 5076. https://doi.org/10.3390/cancers13205076
Wang F, Vij K, Li L, Dodhiawala P, Lim K-H, Shao J. Phospho-Ser784-VCP Drives Resistance of Pancreatic Ductal Adenocarcinoma to Genotoxic Chemotherapies and Predicts the Chemo-Sensitizing Effect of VCP Inhibitor. Cancers. 2021; 13(20):5076. https://doi.org/10.3390/cancers13205076
Chicago/Turabian StyleWang, Faliang, Kiran Vij, Lin Li, Paarth Dodhiawala, Kian-Huat Lim, and Jieya Shao. 2021. "Phospho-Ser784-VCP Drives Resistance of Pancreatic Ductal Adenocarcinoma to Genotoxic Chemotherapies and Predicts the Chemo-Sensitizing Effect of VCP Inhibitor" Cancers 13, no. 20: 5076. https://doi.org/10.3390/cancers13205076
APA StyleWang, F., Vij, K., Li, L., Dodhiawala, P., Lim, K.-H., & Shao, J. (2021). Phospho-Ser784-VCP Drives Resistance of Pancreatic Ductal Adenocarcinoma to Genotoxic Chemotherapies and Predicts the Chemo-Sensitizing Effect of VCP Inhibitor. Cancers, 13(20), 5076. https://doi.org/10.3390/cancers13205076