
Treatment of Patients with Monoclonal Gammopathy of Clinical Significance

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathophysiology of MGCS
3. Skin Disorders
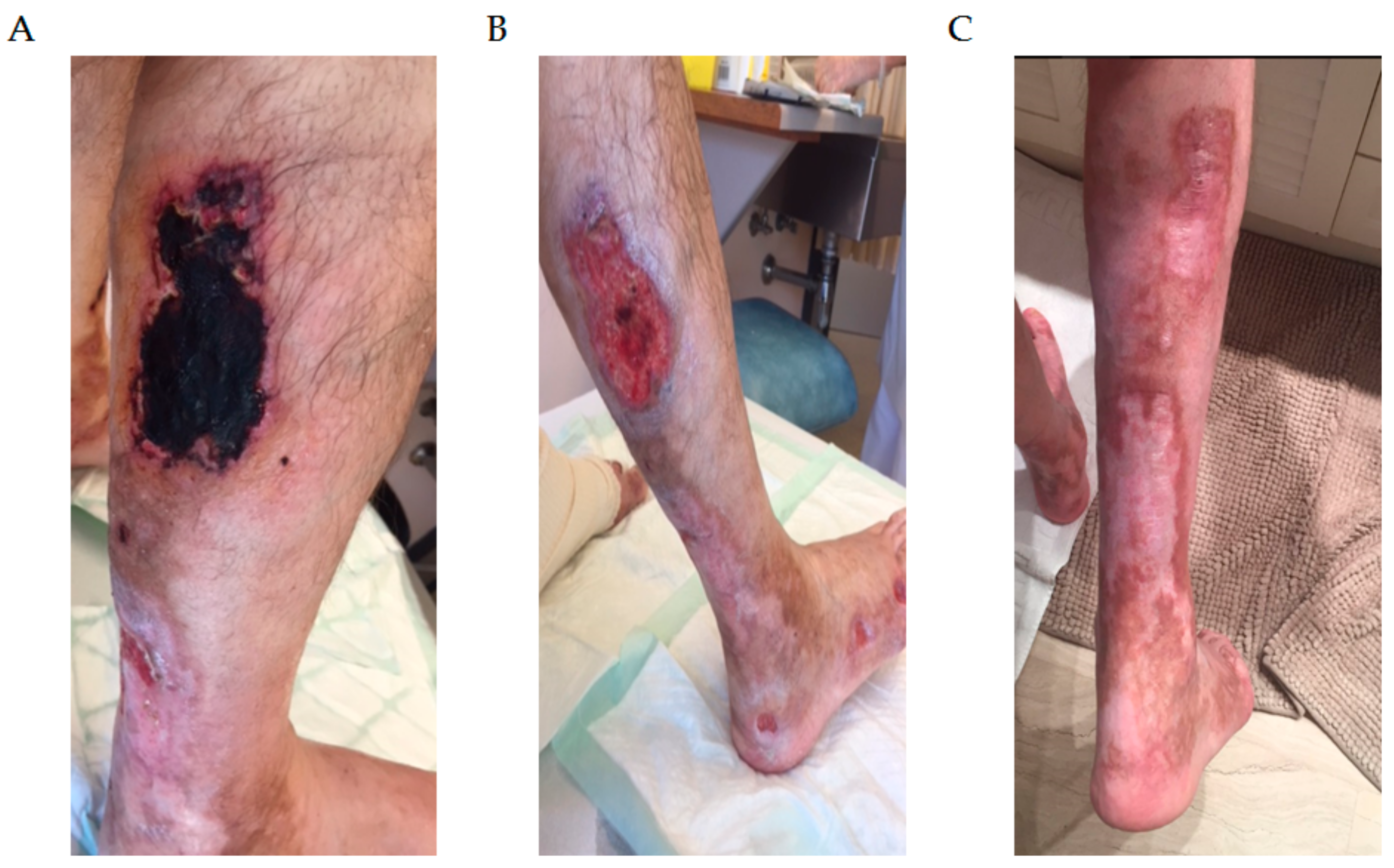
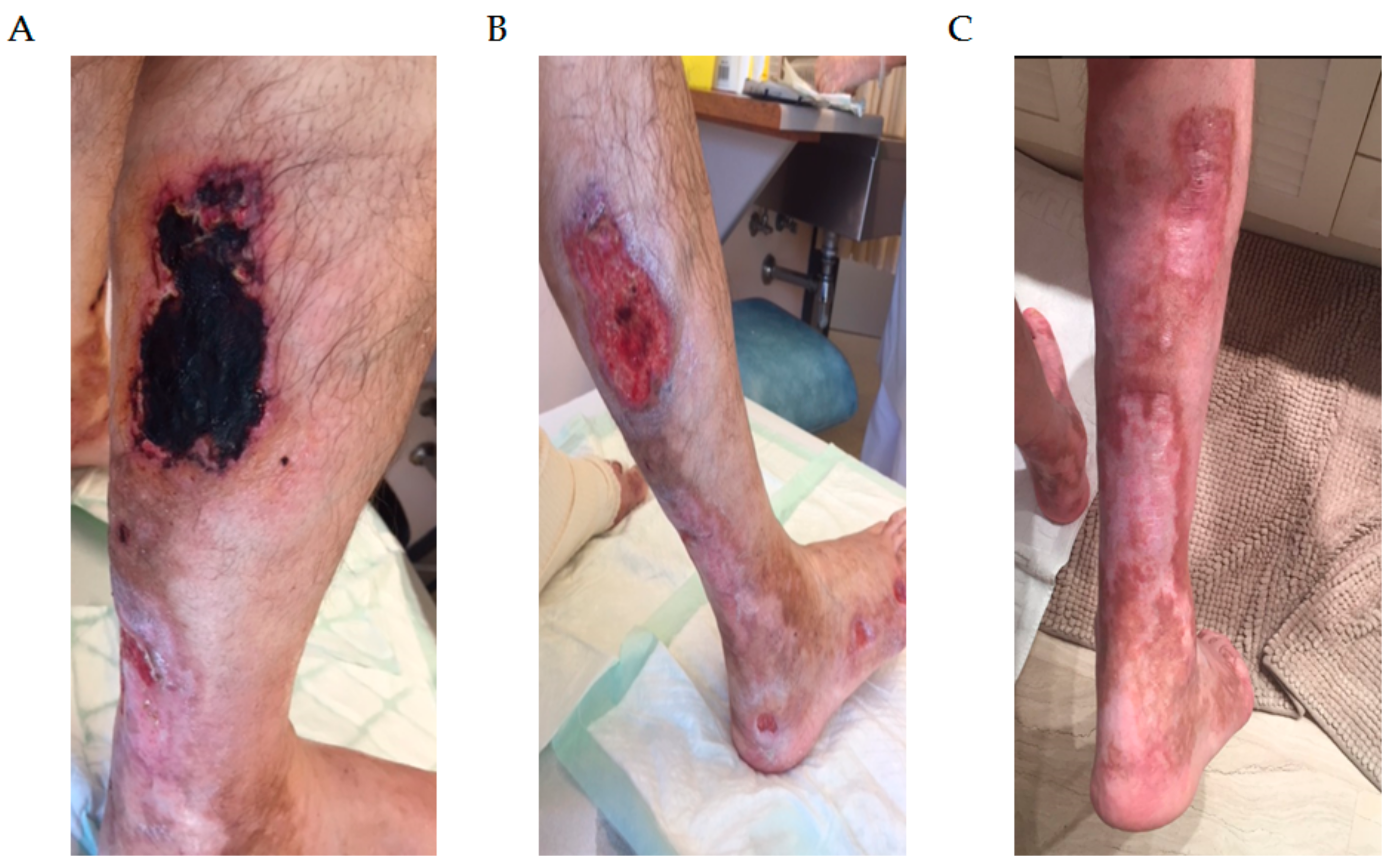
3.1. Type 1 Cryoglobulinemia
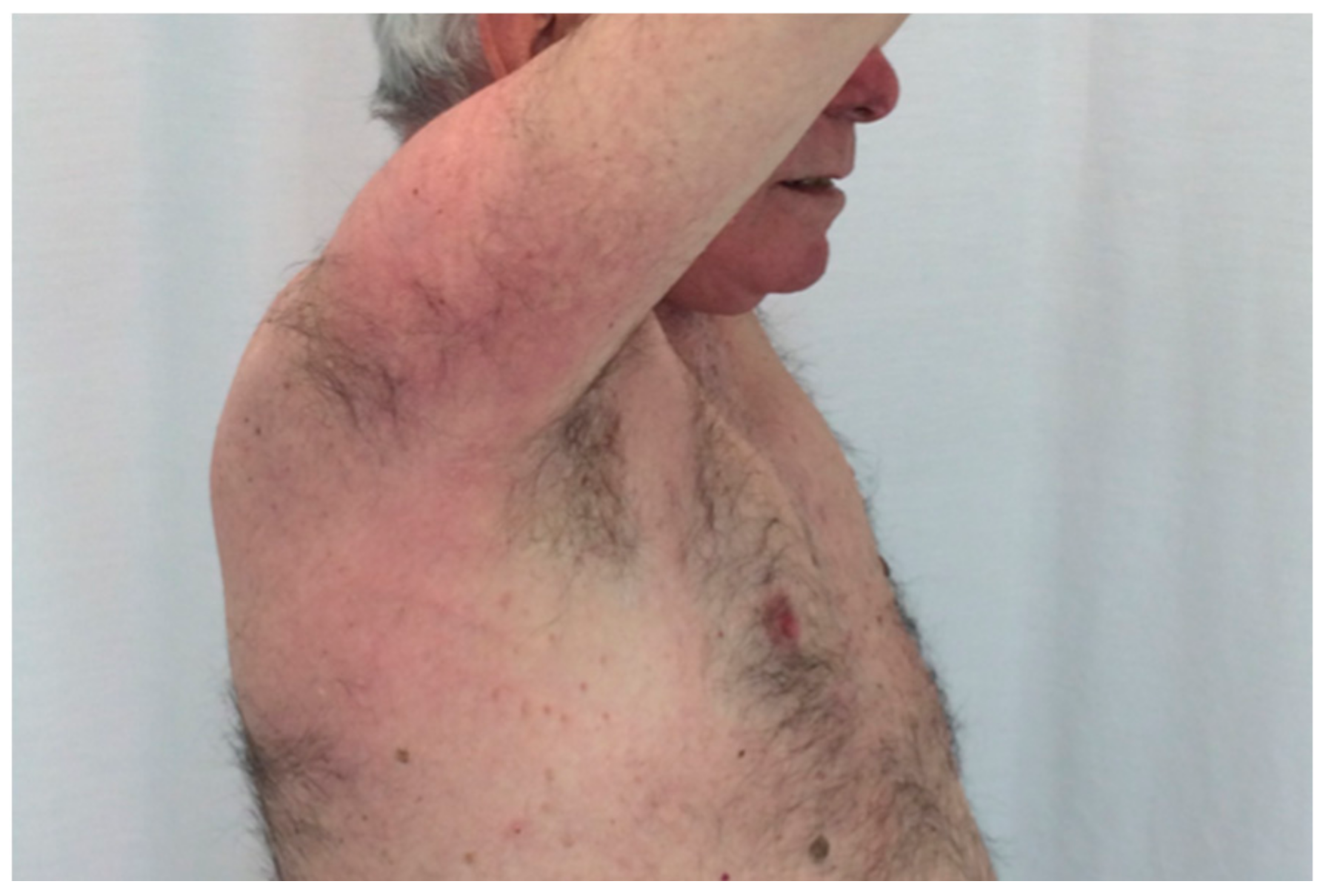
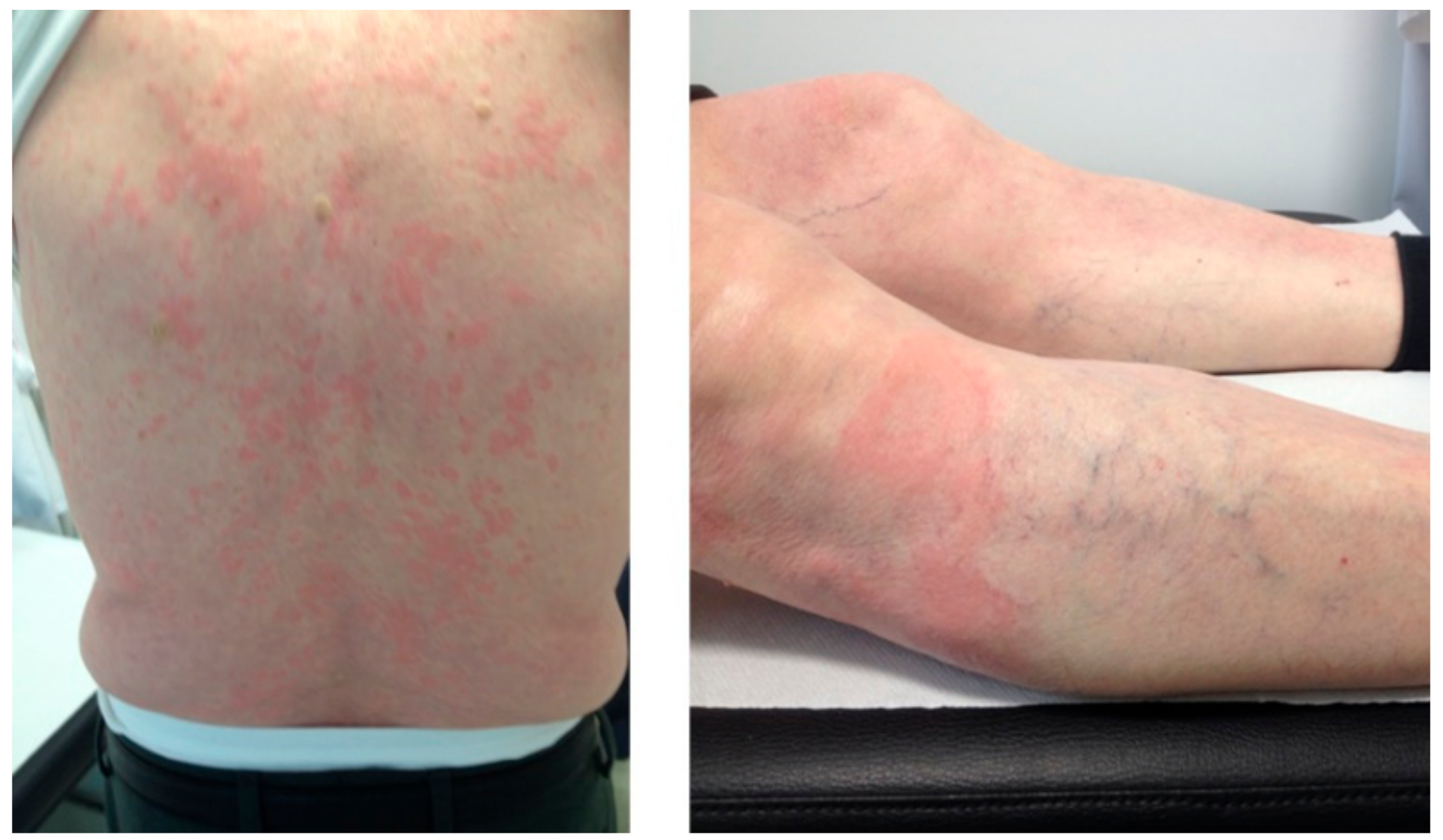
3.2. Schnitzler Syndrome
3.3. Pyoderma Gangrenosum
3.4. Scleromyxedema
3.5. Acquired Generalized Cutis Laxa
4. M-Protein Related Bleeding Disorders
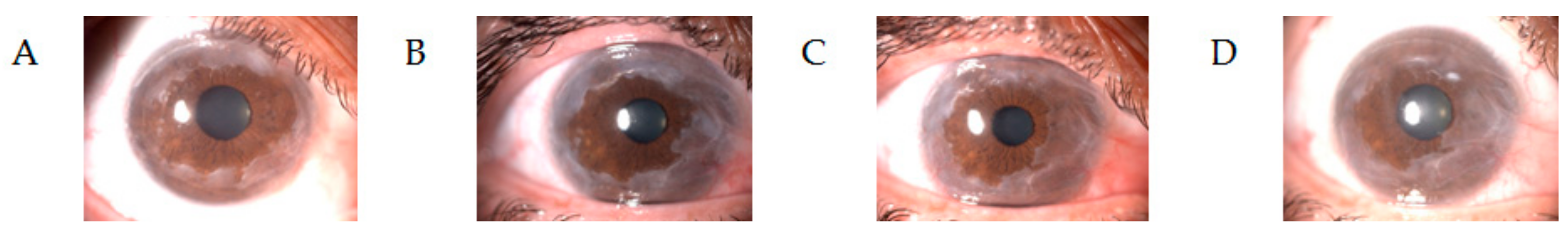
5. Ocular M-Protein Related Diseases
6. Neurologic M-Protein Diseases
IgM Peripheral Neuropathy
7. Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bladé, J. Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 355, 2765–2770. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L.; Morra, E.; Pangalis, G.A.; San Miguel, J.F.; Branagan, A.R.; et al. Clinicopathological Definition of Waldenstrom’s Macroglobulinemia: Consensus Panel Recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Waldenström Macroglobulinemia: 2019 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2019, 94, 266–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, D.F.; Pereira, A.; Tovar, N.; Cibeira, M.T.; Magnano, L.; Rozman, M.; López-Guerra, M.; Colomer, D.; Martín-Antonio, B.; Jiménez-Segura, R.; et al. Defining an Ultra-Low Risk Group in Asymptomatic IgM Monoclonal Gammopathy. Cancers 2021, 13, 2055. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.C.; Mouhieddine, T.H.; Tahri, S.; Ghobrial, I.M. Monoclonal Gammopathy of Undetermined Significance (MGUS)—Not So Asymptomatic after All. Cancers 2020, 12, 1554. [Google Scholar] [CrossRef]
- Merlini, G.; Stone, M.J. Dangerous Small B-Cell Clones. Blood 2006, 108, 2520–2530. [Google Scholar] [CrossRef] [Green Version]
- Fermand, J.-P.; Bridoux, F.; Dispenzieri, A.; Jaccard, A.; Kyle, R.A.; Leung, N.; Merlini, G. Monoclonal Gammopathy of Clinical Significance: A Novel Concept with Therapeutic Implications. Blood 2018, 132, 1478–1485. [Google Scholar] [CrossRef] [Green Version]
- Leung, N.; Bridoux, F.; Hutchison, C.A.; Nasr, S.H.; Cockwell, P.; Fermand, J.-P.; Dispenzieri, A.; Song, K.W.; Kyle, R.A. Monoclonal Gammopathy of Renal Significance: When MGUS Is No Longer Undetermined or Insignificant. Blood 2012, 120, 4292–4295. [Google Scholar] [CrossRef] [Green Version]
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.-P.; Gibbs, S.; et al. The Evaluation of Monoclonal Gammopathy of Renal Significance: A Consensus Report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fermand, J.-P.; Bridoux, F.; Kyle, R.A.; Kastritis, E.; Weiss, B.M.; Cook, M.A.; Drayson, M.T.; Dispenzieri, A.; Leung, N. How I Treat Monoclonal Gammopathy of Renal Significance (MGRS). Blood 2013, 122, 3583–3590. [Google Scholar] [CrossRef]
- Bladé, J.; Cibeira, M.T. M-Protein–Related Disorders: MGCS. Blood 2018, 132, 1464–1465. [Google Scholar] [CrossRef] [PubMed]
- Milman, T.; Kao, A.A.; Chu, D.; Gorski, M.; Steiner, A.; Simon, C.Z.; Shih, C.; Aldave, A.J.; Eagle, R.C.; Jakobiec, F.A.; et al. Paraproteinemic Keratopathy. Ophthalmology 2015, 122, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Harel, S.; Mohr, M.; Jahn, I.; Aucouturier, F.; Galicier, L.; Asli, B.; Malphettes, M.; Szalat, R.; Brouet, J.-C.; Lipsker, D.; et al. Clinico-Biological Characteristics and Treatment of Type I Monoclonal Cryoglobulinaemia: A Study of 64 Cases. Br. J. Haematol. 2015, 168, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Xu-Monette, Z.Y.; Miranda, R.N.; Dogan, A.; Zou, D.; Luthra, R.; Weber, D.M.; O’Malley, D.P.; Jorgensen, J.L.; Khoury, J.D.; et al. Crystal-Storing Histiocytosis: A Clinicopathological Study of 13 Cases. Histopathology 2016, 68, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Palladini, G.; Milani, P.; Merlini, G. Management of AL Amyloidosis in 2020. Blood 2020, 136, 2620–2627. [Google Scholar] [CrossRef]
- Joly, F.; Cohen, C.; Javaugue, V.; Bender, S.; Belmouaz, M.; Arnulf, B.; Knebelmann, B.; Nouvier, M.; Audard, V.; Provot, F.; et al. Randall-Type Monoclonal Immunoglobulin Deposition Disease: Novel Insights from a Nationwide Cohort Study. Blood 2019, 133, 576–587. [Google Scholar] [CrossRef] [Green Version]
- Willison, H.J. The Clinical and Laboratory Features of Chronic Sensory Ataxic Neuropathy with Anti-Disialosyl IgM Antibodies. Brain 2001, 124, 1968–1977. [Google Scholar] [CrossRef] [Green Version]
- D’Sa, S.; Kersten, M.J.; Castillo, J.J.; Dimopoulos, M.; Kastritis, E.; Laane, E.; Leblond, V.; Merlini, G.; Treon, S.P.; Vos, J.M.; et al. Investigation and Management of IgM and Waldenström-Associated Peripheral Neuropathies: Recommendations from the IWWM-8 Consensus Panel. Br. J. Haematol. 2017, 176, 728–742. [Google Scholar] [CrossRef]
- Herrendorff, R.; Hänggi, P.; Pfister, H.; Yang, F.; Demeestere, D.; Hunziker, F.; Frey, S.; Schaeren-Wiemers, N.; Steck, A.J.; Ernst, B. Selective in Vivo Removal of Pathogenic Anti-MAG Autoantibodies, an Antigen-Specific Treatment Option for Anti-MAG Neuropathy. Proc. Natl. Acad. Sci. USA 2017, 114, E3689–E3698. [Google Scholar] [CrossRef] [Green Version]
- Dicke, C.; Schneppenheim, S.; Holstein, K.; Spath, B.; Bokemeyer, C.; Dittmer, R.; Budde, U.; Langer, F. Distinct Mechanisms Account for Acquired von Willebrand Syndrome in Plasma Cell Dyscrasias. Ann. Hematol. 2016, 95, 945–957. [Google Scholar] [CrossRef]
- Djunic, I.; Elezovic, I.; Vucic, M.; Srdic-Rajic, T.; Konic-Ristic, A.; Ilic, V.; Milic, N.; Bila, J.; Suvajdzic-Vukovic, N.; Virijevic, M.; et al. Specific Binding of Paraprotein to Platelet Receptors as a Cause of Platelet Dysfunction in Monoclonal Gammopathies. Acta Haematol. 2013, 130, 101–107. [Google Scholar] [CrossRef]
- Szalat, R.; Monsel, G.; Le Goff, W.; Battistella, M.; Bengouffa, D.; Schlageter, M.-H.; Bouaziz, J.-D.; Arnulf, B.; Vignon, M.; Lesnik, P.; et al. The Spectrum of Neutrophilic Dermatoses Associated with Monoclonal Gammopathy: Association with IgA Isotype and Inflammatory Profile. J. Am. Acad. Dermatol. 2015, 73, 809–820. [Google Scholar] [CrossRef]
- Rowczenio, D.M.; Pathak, S.; Arostegui, J.I.; Mensa-Vilaro, A.; Omoyinmi, E.; Brogan, P.; Lipsker, D.; Scambler, T.; Owen, R.; Trojer, H.; et al. Molecular Genetic Investigation, Clinical Features, and Response to Treatment in 21 Patients with Schnitzler Syndrome. Blood 2018, 131, 974–981. [Google Scholar] [CrossRef]
- Mahévas, T.; Arnulf, B.; Bouaziz, J.-D.; Bulai Livideanu, C.; Osio, A.; Servy, A.; Cribier, B.; Sassolas, B.; Jachiet, M.; Michel, L.; et al. Plasma Cell-Directed Therapies in Monoclonal Gammopathy-Associated Scleromyxedema. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Allain, J.-S.; Thonier, F.; Pihan, M.; Boulland, M.-L.; de Guibert, S.; Launay, V.; Doncker, A.-V.; Ganard, M.; Aliouat, A.; Pangault, C.; et al. IGHV Segment Utilization in Immunoglobulin Gene Rearrangement Differentiates Patients with Anti-Myelin-Associated Glycoprotein Neuropathy from Others Immunoglobulin M-Gammopathies. Haematologica 2018, 103, e207–e210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.J.; Advani, R.H.; Branagan, A.R.; Buske, C.; Dimopoulos, M.A.; D’Sa, S.; Kersten, M.J.; Leblond, V.; Minnema, M.C.; Owen, R.G.; et al. Consensus Treatment Recommendations from the Tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020, 7, e827–e837. [Google Scholar] [CrossRef]
- Muchtar, E.; Magen, H.; Gertz, M.A. How I Treat Cryoglobulinemia. Blood 2017, 129, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Roccatello, D.; Sciascia, S.; Baldovino, S.; Rossi, D.; Alpa, M.; Naretto, C.; Di Simone, D.; Menegatti, E. Improved (4 Plus 2) Rituximab Protocol for Severe Cases of Mixed Cryoglobulinemia: A 6-Year Observational Study. Am. J. Nephrol. 2016, 43, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel Anti-B-Cell Maturation Antigen Antibody-Drug Conjugate (GSK2857916) Selectively Induces Killing of Multiple Myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Simon, A.; Asli, B.; Braun-Falco, M.; De Koning, H.; Fermand, J.-P.; Grattan, C.; Krause, K.; Lachmann, H.; Lenormand, C.; Martinez-Taboada, V.; et al. Schnitzler’s Syndrome: Diagnosis, Treatment, and Follow-Up. Allergy 2013, 68, 562–568. [Google Scholar] [CrossRef]
- Gusdorf, L.; Asli, B.; Barbarot, S.; Néel, A.; Masseau, A.; Puéchal, X.; Gottenberg, J.-E.; Grateau, G.; Blanchard-Delaunay, C.; Rizzi, R.; et al. Schnitzler Syndrome: Validation and Applicability of Diagnostic Criteria in Real-Life Patients. Allergy 2017, 72, 177–182. [Google Scholar] [CrossRef]
- Néel, A.; Henry, B.; Barbarot, S.; Masseau, A.; Perrin, F.; Bernier, C.; Kyndt, X.; Puechal, X.; Weiller, P.-J.; Decaux, O.; et al. Long-Term Effectiveness and Safety of Interleukin-1 Receptor Antagonist (Anakinra) in Schnitzler’s Syndrome: A French Multicenter Study. Autoimmun. Rev. 2014, 13, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- De Koning, H.D. Schnitzler’s Syndrome: Lessons from 281 Cases. Clin. Transl. Allergy 2014, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Ahronowitz, I.; Harp, J.; Shinkai, K. Etiology and Management of Pyoderma Gangrenosum: A Comprehensive Review. Am. J. Clin. Dermatol. 2012, 13, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Ashchyan, H.J.; Butler, D.C.; Nelson, C.A.; Noe, M.H.; Tsiaras, W.G.; Lockwood, S.J.; James, W.D.; Micheletti, R.G.; Rosenbach, M.; Mostaghimi, A. The Association of Age With Clinical Presentation and Comorbidities of Pyoderma Gangrenosum. JAMA Dermatol. 2018, 154, 409. [Google Scholar] [CrossRef] [Green Version]
- Machan, A.; Azendour, H.; Frikh, R.; Hjira, N.; Boui, M. The Dilemma of Treating Pyoderma Gangrenosum Associated with Monoclonal Gammopathy of Undetermined Significance. Dermatol. Online J. 2020, 26. [Google Scholar] [CrossRef]
- Rongioletti, F.; Merlo, G.; Cinotti, E.; Fausti, V.; Cozzani, E.; Cribier, B.; Metze, D.; Calonje, E.; Kanitakis, J.; Kempf, W.; et al. Scleromyxedema: A Multicenter Study of Characteristics, Comorbidities, Course, and Therapy in 30 Patients. J. Am. Acad. Dermatol. 2013, 69, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Beyens, A.; Boel, A.; Symoens, S.; Callewaert, B. Cutis Laxa: A Comprehensive Overview of Clinical Characteristics and Pathophysiology. Clin. Genet. 2021, 99, 53–66. [Google Scholar] [CrossRef]
- Hu, Q.; Reymond, J.-L.; Pinel, N.; Zabot, M.-T.; Urban, Z. Inflammatory Destruction of Elastic Fibers in Acquired Cutis Laxa Is Associated with Missense Alleles in the Elastin and Fibulin-5 Genes. J. Invest. Dermatol. 2006, 126, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández de Larrea, C.; Rovira, M.; Mascaró, J.M., Jr.; Torras, A.; Solé, M.; Lloreta, J.; Serra, N.; Cibeira, M.T.; Bladé, J. Generalized Cutis Laxa and Fibrillar Glomerulopathy Resulting from IgG Deposition in IgG-Lambda Monoclonal Gammopathy: Pulmonary Hemorrhage during Stem Cell Mobilization and Complete Hematological Response with Bortezomib and Dexamethasone Therapy. Eur. J. Haematol. 2009, 82, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Jachiet, M.; Harel, S.; Saussine, A.; Battistella, M.; Rybojad, M.; Asli, B.; Bengoufa, D.; Mahevas, T.; Bessis, D.; Galicier, L.; et al. Cutis Laxa Associated with Monoclonal Gammopathy: 14 New Cases and Review of the Literature. J. Am. Acad. Dermatol. 2018, 79, 945–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, J.T.; D’Agati, V.D.; Sherman, W.H.; Grossman, M.E. Acquired Cutis Laxa Associated with Heavy Chain Deposition Disease Involving Dermal Elastic Fibers. JAMA Dermatol. 2014, 150, 1192. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Pon, K.; Bargman, J.; Ghazarian, D. Generalized Cutis Laxa Associated with Heavy Chain Deposition Disease. J. Cutan. Med. Surg. 2003, 7, 390–394. [Google Scholar] [CrossRef]
- Thompson, C.A.; Kyle, R.; Gertz, M.; Heit, J.; Pruthi, R.; Pardanani, A. Systemic AL Amyloidosis with Acquired Factor X Deficiency: A Study of Perioperative Bleeding Risk and Treatment Outcomes in 60 Patients. Am. J. Hematol. 2010, 85, 171–173. [Google Scholar] [CrossRef] [Green Version]
- Tiede, A. Diagnosis and Treatment of Acquired von Willebrand Syndrome. Thromb. Res. 2012, 130, S2–S6. [Google Scholar] [CrossRef]
- Franchini, M.; Mannucci, P.M. Acquired von Willebrand Syndrome: Focused for Hematologists. Haematologica 2020, 105, 2032–2037. [Google Scholar] [CrossRef]
- Castillo, J.J.; Gustine, J.N.; Meid, K.; Dubeau, T.; Severns, P.; Xu, L.; Yang, G.; Hunter, Z.R.; Treon, S.P. Low Levels of von Willebrand Markers Associate with High Serum IgM Levels and Improve with Response to Therapy, in Patients with Waldenström Macroglobulinaemia. Br. J. Haematol. 2019, 184, 1011–1014. [Google Scholar] [CrossRef] [Green Version]
- Kos, C.A.; Ward, J.E.; Malek, K.; Sanchorawala, V.; Wright, D.G.; O’Hara, C.; Connors, L.; Skinner, M.; Seldin, D.C. Association of Acquired von Willebrand Syndrome with AL Amyloidosis. Am. J. Hematol. 2007, 82, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Djunic, I.; Elezovic, I.; Ilic, V.; Milosevic-Jovcic, N.; Bila, J.; Suvajdzic-Vukovic, N.; Antic, D.; Vidovic, A.; Tomin, D. The Effect of Paraprotein on Platelet Aggregation: Paraprotein and Platelet Aggregation. J. Clin. Lab. Anal. 2014, 28, 141–146. [Google Scholar] [CrossRef] [PubMed]
- DiMinno, G.; Coraggio, F.; Cerbone, A.M.; Capitanio, A.M.; Manzo, C.; Spina, M.; Scarpato, P.; Dattoli, G.M.; Mattioli, P.L.; Mancini, M. A Myeloma Paraprotein with Specificity for Platelet Glycoprotein IIIa in a Patient with a Fatal Bleeding Disorder. J. Clin. Investig. 1986, 77, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garibaldi, D.C.; Gottsch, J.; de la Cruz, Z.; Haas, M.; Green, W.R. Immunotactoid Keratopathy: A Clinicopathologic Case Report and a Review of Reports of Corneal Involvement in Systemic Paraproteinemias. Surv. Ophthalmol. 2005, 50, 61–80. [Google Scholar] [CrossRef]
- Batalini, F.; Econimo, L.; Quillen, K.; Sloan, J.M.; Sarosiek, S.; Brauneis, D.; Havasi, A.; Stern, L.; Dember, L.M.; Sanchorawala, V. High-Dose Melphalan and Stem Cell Transplantation in Patients on Dialysis Due to Immunoglobulin Light-Chain Amyloidosis and Monoclonal Immunoglobulin Deposition Disease. Biol. Blood Marrow Transplant. 2018, 24, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Rögnvaldsson, S.; Steingrímsson, V.; Turesson, I.; Björkholm, M.; Landgren, O.; Kristinsson, S.Y. Peripheral Neuropathy and Monoclonal Gammopathy of Undetermined Significance: A Population-Based Study Including 15,351 Cases and 58,619 Matched Controls. Haematologica 2020, 105, 2679–2681. [Google Scholar] [CrossRef]
- Notermans, N.C.; Franssen, H.; Eurelings, M.; Van der Graaf, Y.; Wokke, J.H. Diagnostic Criteria for Demyelinating Polyneuropathy Associated with Monoclonal Gammopathy. Muscle Nerve 2000, 23, 73–79. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-Controlled Trial of Rituximab in IgM Anti-Myelin-Associated Glycoprotein Antibody Demyelinating Neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef]
- Léger, J.-M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.-M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. Placebo-Controlled Trial of Rituximab in IgM Anti-Myelin-Associated Glycoprotein Neuropathy. Neurology 2013, 80, 2217–2225. [Google Scholar] [CrossRef]
- Lunn, M.P.; Nobile-Orazio, E. Immunotherapy for IgM Anti-Myelin-Associated Glycoprotein Paraprotein-Associated Peripheral Neuropathies. Cochrane Database Syst. Rev. 2016, 10, CD002827. [Google Scholar] [CrossRef] [Green Version]
- Nobile-Orazio, E.; Bianco, M.; Nozza, A. Advances in the Treatment of Paraproteinemic Neuropathy. Curr. Treat. Options Neurol. 2017, 19, 43. [Google Scholar] [CrossRef]
- Colchester, N.T.H.; Allen, D.; Katifi, H.A.; Burt, T.; Lown, R.N.; Pinto, A.A.; Duncombe, A.S. Chemoimmunotherapy with Rituximab, Cyclophosphamide and Prednisolone in IgM Paraproteinaemic Neuropathy: Evidence of Sustained Improvement in Electrophysiological, Serological and Functional Outcomes. Haematologica 2020, 106, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton Tyrosine Kinase Inhibitor Ibrutinib Improves Anti-MAG Antibody Polyneuropathy. Neurol. Neuroimmunol. Neuroinflam. 2020, 7, e720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, J.S.; Saperstein, D.S.; Gronseth, G.; Amato, A.A.; Barohn, R.J. Distal Acquired Demyelinating Symmetric Neuropathy. Neurology 2000, 54, 615–620. [Google Scholar] [CrossRef]
- Chaudhry, H.M.; Mauermann, M.L.; Rajkumar, S.V. Monoclonal Gammopathy-Associated Peripheral Neuropathy: Diagnosis and Management. Mayo Clin. Proc. 2017, 92, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Mecoli, C.A.; Talbot, C.C.; Fava, A.; Cheadle, C.; Boin, F.; Wigley, F.M.; Hummers, L.K. Clinical and Molecular Phenotyping in Scleromyxedema Pretreatment and Posttreatment With Intravenous Immunoglobulin. Arthritis Care Res. 2020, 72, 761–767. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the Vicious Cycle within the Multiple Myeloma Ecosystem: Blocking JAM-A on Bone Marrow Endothelial Cells Restores Angiogenic Homeostasis and Suppresses Tumor Progression. Haematologica 2020, 106, 1943–1956. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef]
- Lamb, M.J.; Smith, A.; Painter, D.; Kane, E.; Bagguley, T.; Newton, R.; Howell, D.; Cook, G.; de Tute, R.; Rawstron, A.; et al. Health Impact of Monoclonal Gammopathy of Undetermined Significance (MGUS) and Monoclonal B-Cell Lymphocytosis (MBL): Findings from a UK Population-Based Cohort. BMJ Open 2021, 11, e041296. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Bjorkholm, M.; Andersson, T.M.-L.; Eloranta, S.; Dickman, P.W.; Goldin, L.R.; Blimark, C.; Mellqvist, U.-H.; Wahlin, A.; Turesson, I.; et al. Patterns of Survival and Causes of Death Following a Diagnosis of Monoclonal Gammopathy of Undetermined Significance: A Population-Based Study. Haematologica 2009, 94, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, T.; Offord, C.P.; Kyle, R.A.; Dingli, D. Schnitzler Syndrome: An under-Diagnosed Clinical Entity. Haematologica 2013, 98, 1581–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rögnvaldsson, S.; Love, T.J.; Thorsteinsdottir, S.; Reed, E.R.; Óskarsson, J.Þ.; Pétursdóttir, Í.; Sigurðardóttir, G.Á.; Viðarsson, B.; Önundarson, P.T.; Agnarsson, B.A.; et al. Iceland Screens, Treats, or Prevents Multiple Myeloma (IStopMM): A Population-Based Screening Study for Monoclonal Gammopathy of Undetermined Significance and Randomized Controlled Trial of Follow-up Strategies. Blood Cancer J. 2021, 11, 94. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affected Organ | Disease |
|---|---|
| Skin | Type 1 cryoglobulinemia Schnitzler syndrome Pyoderma gangrenosum Scleromyxedema Acquired generalized cutis laxa |
| Neurologic M-protein-related diseases | IgM MGUS neuropathy IgG/IgA MGUS neuropathy |
| Ocular | Paraproteinemic keratopathy |
| M-protein-related bleeding disorders | Acquired von Willebrand syndrome Impaired platelet aggregation |
| Disease | Underlying Mechanism | M-Protein Isotype | Treatment |
|---|---|---|---|
| Type 1 cryoglobulinemia | Monoclonal immunoglobulin crystallization. Cold exposure is a trigger to induce aggregation of cryoglobulins (skin) or other unknown factors (kidney, nerves). | IgG, IgM | Glucocorticoids Alkylating agents (i.e., cyclophosphamide) PE Rituximab (IgM type) Anti-myeloma therapy (non-IgM types) |
| Schnitzler syndrome | Inflammasome upregulation leads to IL-1β and IL-18 release. IgM deposits in the skin of patients with rash (possible autoantibody effect). Suspected genetic predisposition: NLRP3 mutation. | IgM, (rarely IgG) | Anti-IL1 (anakinra) Oral prednisone Rituximab or ibrutinib Anti-myeloma therapy (non-IgM) |
| Pyoderma gangrenosum | Interaction between monoclonal IgA with its receptors that leads to cytokine release and pro-inflammatory mediators (IL-6, EGF, MCP-1). Abnormal activation of neutrophils. | IgA, (rarely IgM) | Topical or oral prednisone Anti-TNF (infliximab) Steroid-sparing drugs (cyclosporine A, mycophenolate, tacrolimus) Anti-myeloma therapy if refractoriness |
| Scleromyxedema | High expression of TGF-β, and collagen-1a might increase proliferation of fibroblasts. Reduced levels of pro-inflammatory mediators are seen after IVIG therapy. | IgG | IVIG for non-severe symptoms Anti-myeloma therapy for refractory or severe symptoms |
| Acquired cutis laxa | Elastic fiber destruction by phagocytosis after monoclonal immunoglobulin deposition Elastic fiber destruction mediated by complement. | IgG | Anti-myeloma therapy |
| Disease | Underlying Mechanism | M-Protein Isotype | Treatment |
|---|---|---|---|
| Platelet aggregation disorder | Aberrant deposition of monoclonal immunoglobulin on platelet surface targets (glycoprotein IIIa, GP1b).Immunologic destruction of VWF (autoantibody activity). | IgG | Anti-myeloma therapy |
| Keratopathy | Crystalline monoclonal immunoglobulin deposits or non-organized light-chains deposits on corneal surface. Overproduction of abnormal immunoglobulin conformation, impaired enzymatic degradation, and high tropism for organ deposition. | Heavy or light chains | Anti-myeloma therapy |
| Peripheral neuropathy | Monoclonal IgM targets HNK-1 epitope on MAG glycoprotein causing demyelinating lesions (autoantibody activity). Other potential targets: gangliosides (GM1, GM2, GM3, GD1a, GD1b, GT1b), and paraglobosides. | IgM | Anti-MAG/ganglioside: Rituximab No antibodies or non-IgM neuropathy: IVIG, prednisone, anti-myeloma agents |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, D.F.; Rosiñol, L.; Cibeira, M.T.; Bladé, J.; Fernández de Larrea, C. Treatment of Patients with Monoclonal Gammopathy of Clinical Significance. Cancers 2021, 13, 5131. https://doi.org/10.3390/cancers13205131
Moreno DF, Rosiñol L, Cibeira MT, Bladé J, Fernández de Larrea C. Treatment of Patients with Monoclonal Gammopathy of Clinical Significance. Cancers. 2021; 13(20):5131. https://doi.org/10.3390/cancers13205131
Chicago/Turabian StyleMoreno, David F., Laura Rosiñol, María Teresa Cibeira, Joan Bladé, and Carlos Fernández de Larrea. 2021. "Treatment of Patients with Monoclonal Gammopathy of Clinical Significance" Cancers 13, no. 20: 5131. https://doi.org/10.3390/cancers13205131
APA StyleMoreno, D. F., Rosiñol, L., Cibeira, M. T., Bladé, J., & Fernández de Larrea, C. (2021). Treatment of Patients with Monoclonal Gammopathy of Clinical Significance. Cancers, 13(20), 5131. https://doi.org/10.3390/cancers13205131